

Abstract
The ability of endothelin-1 (ET-1) to modulate the cyclic nucleotides, guanosine 3′ 5′ cyclic monophosphate (cyclic GMP) and adenosine 3′ 5′ cyclic monophosphate (cyclic AMP) was assessed in the main elastic pulmonary elastic artery (4–5 mm i.d.) and the small muscular pulmonary arteries (150–200 μm i.d.) of the rat.
ET-1 caused an increase in cyclic GMP in the larger vessels but had no effect in the smaller arteries. The increase in cyclic GMP was not dependent on an intact endothelium and was inhibited by the ETA-receptor antagonist FR139137 (1 μM).
ET-1 caused a decrease in cyclic AMP in the main pulmonary arteries, an effect that was partially blocked by FR139317 but not influenced by the ETB-receptor antagonist BQ-788 (1 μM) or removal of the vascular endothelium. In contrast, ET-1 caused an increase in cyclic AMP in the small vessels, an effect that was blocked by BQ-788 but unaffected by FR139317.
In the main pulmonary arteries, ET-1 caused enhanced incorporation of radiolabelled ADP-ribose by cholera toxin into Gi2 in the main pulmonary artery, an indicator of its receptor-mediated activation.
In summary, we have shown that in the small muscular pulmonary artery of the rat, (where ETB mediated vasoconstriction prevails), there is an ETB-mediated increase in cyclic AMP with no net effect on cyclic GMP levels. In the large arteries, (where vasoconstriction is mediated via the ETA receptor), there is an ETA-mediated increase in cyclic GMP (endothelium independent) and an ETA-mediated (endothelium independent) decrease in cyclic AMP.
Keywords: Pulmonary artery, endothelin-1, cyclic AMP, cyclic GMP, G protein, cyclic nucleotides
Introduction
The endothelins (ETs) are a family of potent vasoactive peptides of 21 amino acids originally isolated from the culture supernatant from a primary culture of porcine aortic endothelial cells (Yanigisawa et al., 1988). These peptides, which exert their effects via G protein coupled receptors, have been implicated in the pathophysiology of a variety of human disease states including myocardial infarction, atherosclerosis and pulmonary hypertension (Remuzzi & Benigni, 1993; Stewart et al., 1991; Giaid et al., 1993). Molecular cloning studies have identified two endothelin receptor subtypes, ETA, which shows selectivity for ET-1 over ET-3 and ETB which is non-isopeptide selective (Miller et al., 1993), although splice variant forms of ETB-receptors have been reported (Shyamala et al., 1994; Elshourbagy et al., 1996). We have previously demonstrated that in the large pulmonary arteries of the rat, vasoconstriction to ET-1 is mediated via an ETA-receptor whereas in the pulmonary resistance vessels from rat, rabbit and man, vasoconstriction is mediated through an ETB-like receptor (MacLean et al., 1994; 1996; McCulloch & MacLean, 1995; McCulloch et al., 1998; Docherty & MacLean, 1998).
ET receptors belong to the ‘G protein coupled receptor' superfamily of heptahelical receptors that couple to a variety of guanine nucleotide binding proteins, directly modulating various second messenger molecules (Sokolovsky, 1995). The intracellular signalling pathways mediating pulmonary vascular response to ETs have still to be fully defined. Agonist occupation of the ETA receptor subtype in the vasculature results in an activation of phosphoinositidase C, stimulating inositol phosphate formation (Marsden et al., 1989). Subsequent studies have shown the ETs to interact with other second messenger systems. ET-1-mediated activation of phospholipase C through both ETA and ETB receptor subtypes has been observed in a variety of vascular cells and tissues in vitro (Resink et al., 1988; Eguchi et al., 1992; Ohlstein et al., 1995). This activation, mediated through a Gq protein, results in the formation of inositol-1,4,5-trisphosphate which has been demonstrated to release calcium from intracellular stores (Berridge, 1993), a process normally refractory to pertussis toxin (PT). However, there is ample evidence in the literature to implicate the involvement of PT-sensitive mechanisms in ET-1-mediated signalling. ET-1 can increase cyclic GMP levels through the peptide-induced release of nitric oxide and subsequent activation of soluble guanylyl cyclase in a variety of cells such as cultured bovine endothelial cells (Hirata et al., 1993) and rat aorta (Fujitani et al., 1993). ET receptors can also couple to adenylyl cyclase and induce cyclic AMP formation, activation occurring through ETA receptors in cultured vascular smooth muscle cells and inhibition through ETB receptors in endothelial cells (Eguchi et al., 1993a). It is currently unclear how ET-1 effects net cyclic GMP and cyclic AMP production in the pulmonary circulation. This circulation is unusual in that it is normally fully vasodilated, and therefore pulmonary vascular tone would be greatly increased by decreased accumulation of cyclic nucleotides. Indeed, we have shown that in the chronic hypoxic, pulmonary hypertensive rat, there are decreased cyclic GMP and cyclic AMP levels in the pulmonary circulation (MacLean et al., 1996). This is a consequence, in part, of an increase in phosphodiesterase activity (MacLean et al., 1997) but given that there is an increased influence of ET-1 in this rat model (Li et al., 1994; Tjen-A-Looi et al., 1996; McCulloch et al., 1998), we wished to identify the effect of ET-1 on cyclic nucleotide levels.
The aim of the present study was assess the net effect of ET-1 on cyclic nucleotide levels in rat pulmonary arteries and, subsequently, to examine further the G protein involved in ET-mediated decreased cyclic AMP which may contribute to the contractile effect of ET-1. In the light of the evidence that there is regional receptor heterogeneity, we also wished to investigate if there were regional differences in ET-1 cyclic nucleotide handling.
Preliminary data from these studies have been presented previously (Mullaney et al., 1997)
Methods
In studying the signal transduction pathways of ET-1 in the pulmonary artery, with an aim of correlating this with previously observed functional effects, it is preferable to use intact arteries rather than primary cultures where phenotypic changes occur, especially with respect to the ET receptor (Owe-Young et al., 1999). For this reason we employed homogenates from the main pulmonary arteries and the small muscular pulmonary arteries used in our previous functional studies (MacLean et al., 1994). In order to correlate the net effect on cyclic nucleotides with maximum contraction we chose to study a concentration of ET-1 (0.1 μM) which we had previously shown to induce maximum contraction in the vessels under study (MacLean et al., 1996; MacLean & McCulloch, 1998; McCulloch et al., 1998).
Six-week-old male Wistar rats were killed by an overdose of sodium pentobarbitone and the lungs removed and placed in cold Krebs. The main pulmonary artery (4–5 mm i.d.) and the intralobar resistance arteries (approximately 150 μm i.d.) were dissected out, cleaned of the surrounding parenchyma and either used in assays designed to measure the intracellular levels of second messengers or to prepare plasma membrane fractions. Some main pulmonary artery preparations were denuded of the vascular endothelium by gentle rubbing of the intimal surface.
Preparation of plasma membrane fractions
The tissue from 4–5 animals was snap frozen in liquid N2 and ground, whilst still frozen, with a mortar and pestle. The powdered tissue was then resuspended in a small volume of homogenization buffer (Tris-HCl 10 mM, EDTA 0.1 mM, pH 7.5) and subjected to 50 strokes with a Teflon on glass homogenizer. Membranes were prepared using differential centrifugation as described previously (Mullaney et al., 1996), aliquotted in appropriate volumes and stored at −80°C.
Preparation of samples for cyclic nucleotide assays
Freshly dissected tissue was allowed to equilibrate for 30 min at 37°C in Krebs-bicarbonate solution (in mM): NaCl 119; KCl 4.7; MgSO4; 0.6; CaCl2 1.2; NaHCO3 25; glucose 11.1, bubbled with 16% O2 / 6% CO2 balance N2. Whole pieces of tissue (typically 3–6 mg wet weight) were incubated for 15 min at 37°C in Krebs-bicarbonate solution containing the general phosphodiesterase inhibitor isobutylmethylxanthine (1 mM) and drug where appropriate. The incubation was terminated by removing the tissue into 4% (w v−1) perchloric acid where it was homogenized with a Teflon on glass hand homogenizer (25 strokes) and left overnight at 4°C to extract the cyclic nucleotides. Next day the samples were sonicated for 15 min, centrifuged at 3000 r.p.m. for 10 min and the supernatant neutralized with KOH/HEPES.
Assay of intracellular cyclic AMP concentration
Cyclic AMP levels were measured essentially by the method described by Brown et al. (1972). Samples were incubated for 2 h at 4°C with a fixed amount of [3H]-cyclic AMP (approximately 5000 c.p.m. per assay tube) and cyclic AMP binding protein (regulatory subunit of protein kinase A partially purified from bovine adrenal cortex) in 50 mM Tris-HCl (pH 7.4), 4 mM EDTA. A standard curve was constructed with dilutions of unlabelled cyclic AMP (0–320 pmol ml−1). The incubation was terminated by addition of 2% (w v−1) Norit GSX activated charcoal, 1% (w v−1) bovine serum albumin to precipitate unbound cyclic AMP and the [3H]-cyclic AMP remaining in the supernatant was assessed by liquid scintillation counting.
Assay of intracellular cyclic GMP concentration
Cyclic GMP levels were measured by radioimmunoassay as described by Cailla et al. (1976). Samples were incubated overnight at 4°C with a fixed amount of [125I]-cyclic GMP (approximately 6500 c.p.m. per assay tube) and antiserum 487 (which was raised against 2′-O-succinyl cyclic GMP) in 50 mM Tris-HCl (pH 7.4). There is no cross-reactivity of this antibody with cyclic AMP. A standard curve was constructed with dilutions of unlabelled cyclic GMP (0–200 pmol ml−1). The incubation was terminated by addition of activated charcoal solution (Tris-HCl 50 mM (pH 7.4), EDTA 5 mM, 0.2% (w v−1) neomycin sulphate, 0.5% (w v−1) bovine serum albumin, 0.062% (w v−1) dextran T70, 0.6% (w v−1) Norit GSX activated charcoal), the samples centrifuged at 12,000 r.p.m. for 5 min and the clear supernatant counted on a gamma counter.
We tested the hypothesis that any differences in cyclic nucleotide levels, between the large and small pulmonary arteries, was due to the time taken at dissection by randomly varying the order in which the vessels were dissected.
Cholera toxin-catalyzed [32P]-ADP-ribosylation
[32P]-ADP-ribosylation of membranes prepared from rat main pulmonary artery was performed as described in Mullaney et al. (1996). Briefly, plasma membranes (20 mg) were incubated in a final volume of 50 ml containing 3 mM [32P]-NAD (approximately 4×106 c.p.m.) sodium phosphate buffer, pH 7.5 (250 mM), thymidine (20 mM), ATP (1 mM), arginine hydrochloride (20 mM) in the presence or absence of appropriate ligands for 60 min at 37°C. The assays were performed with pre-activated cholera toxin present at 50 mg ml−1 and in the absence of exogenous GTP in order to maximize agonist-induced ADP-ribosylation. Cholera toxin was pre-activated by incubation with an equal volume of 100 mM DTT for 1 h at room temperature. Samples were solubilized and immunoprecipitated with antiserum SG1. SG1 was raised against the C terminal decapeptide of Gi. This antisera identifies Gi1 and Gi2 equally but does not cross-react with other G proteins (Mitchell et al., 1989). There is no evidence for the presence of Gi1 in the vasculature. The radiolabelled polypeptides were separated by SDS–PAGE and dried gels were exposed to a phosphor storage plate for 24 h and then analysed with a FUJIX BAS1000 image analyser.
Data analysis
For each experimental group processed at one time, the cyclic nucleotide levels were calculated in the presence and absence of drug intervention. Each intervention was repeated to ‘n' and the values meaned. Statistical comparisons of the means of groups of data were made by one-way analysis of variance (ANOVA) with P<0.05 considered significant. In some figures, the data is displayed as a ‘percentage change' from ‘control' although the statistical analysis was carried out as described above.
Materials
[32P]-NAD+ (800 Ci mmol−1) and [125I]-cyclic GMP (1180 Ci mmol−1) were obtained from DuPont / New England Nuclear. [5′,8-3H]-cyclic AMP (30–60 Ci mmol−1) were purchased from Amersham International plc. Cholera toxin and isobutylmethylxanthine were obtained from Sigma. ET-1 was from Thistle Peptides Ltd, Scotland, BQ-788 (N-cis-2,6-dimethylpiperidinocarbonyl L-g-MeLeu-D-Trp (COOCH3)-D-Nle) was from Peptide International and FR139317 (N-CO-L-Leu-D-1-Me-Trp-D-3(2-Pyridyl) Ala-OH) was from Neosystem Laboratoire. Antiserum 487 (which was raised against 2′-O-succinyl cyclic GMP) was a gift from Dr I. Morton, Western Infirmary, Glasgow and antiserum SG1 (raised against a synthetic peptide (KENLKDCGLF) which corresponds to the C-terminal decapeptide of transducin and recognizes Gi2) was a gift from Professor G. Milligan, IBLS, University of Glasgow.
Results
The effect of ET-1 on intracellular cyclic GMP and cyclic AMP levels in rat pulmonary arteries
In the main pulmonary arteries ET-1 (0.1 μM) caused an 88% increase in cyclic GMP (Table 1). In an independent series of experiments studying the concentration-dependency of this effect, the maximum effect of ET-1 on cyclic GMP levels was observed at 10 nM (Figure 1). In the small muscular arteries basal levels of cyclic GMP were 54% lower than in the larger arteries. This observation was consistent regardless of whether or not the main pulmonary arteries were dissected out first or second. Incubation with ET-1 did not appreciably change the intracellular levels of this cyclic nucleotide in small pulmonary arteries (Table 1).
Table 1.
The effects of 0.1 μM ET-1 on cyclic GMP and cyclic AMP levels in rat main pulmonary arteries and small muscular pulmonary arteries
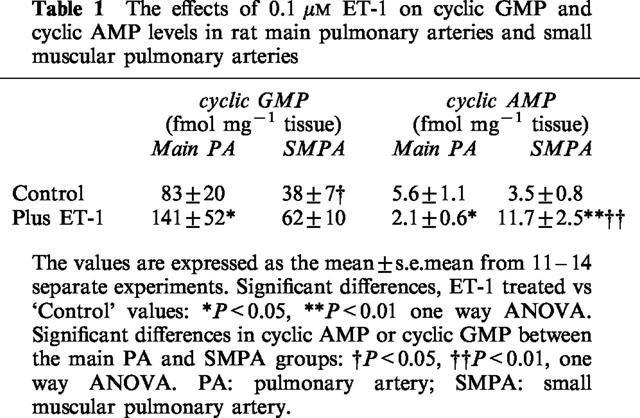
Figure 1.
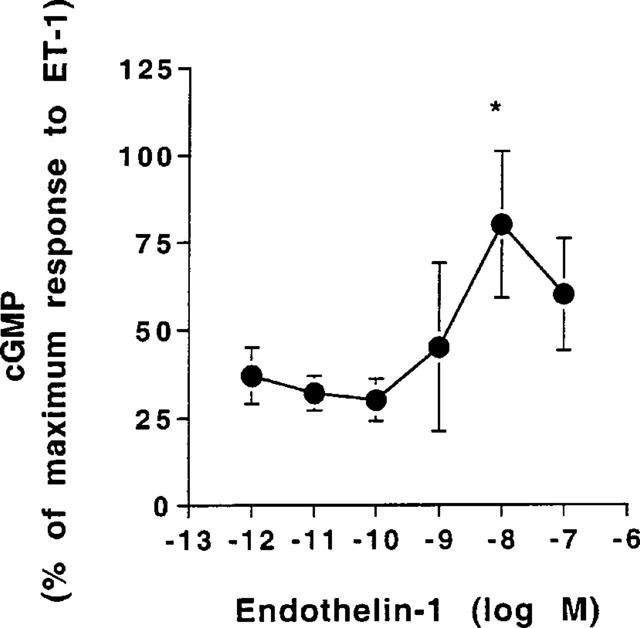
Effect of inceasing concentrations of ET-1 on cyclic GMP levels in main extralobar artery. The results are expressed as percentage of maximum stimulation and the values are the mean±s.e.mean from six separate experiments. Significant difference from 1 pM ET-1: *P<0.05, one-way ANOVA.
Comparison of basal cyclic AMP levels between the large and small vessels revealed there to be little difference in the tissue levels of this nucleotide (Table 1). This was found to be the case regardless of the time taken at dissection. ET-1 caused a 67% decrease in cyclic AMP in the large arteries. An independent series of experiments studying the concentration-dependency (1 pM–0.1 μM) of this effect showed that this effect was only significant at 0.1 μM ET-1.
In the small muscular pulmonary arteries, 0.1 μM ET-1 caused a 240% increase in cyclic AMP (Table 1).
The effect of the ETA receptor antagonist, FR139317 on ET-1 modulation of cyclic nucleotide levels
Table 2 illustrates the effect of FR139317 (1 μM) on ET-1-mediated changes in cyclic nucleotide levels. FR139317 partially reversed ET-1-mediated decrease in cyclic AMP in the main arteries by ∼30% and blocked the ability of ET-1 to elevate cyclic GMP in the main artery. In contrast, FR139317 had no significant effect on the ability of ET-1 to increase cyclic AMP in the small muscular pulmonary arteries.
Table 2.
The effect of FR139317 on changes induced by ET-1 on cyclic nucleotide levels in rat main pulmonary arteries and small muscular pulmonary arteries

The effect of the ETB receptor antagonist, BQ-788 on ET-1 modulation of cyclic nucleotide levels
Table 3 illustrates the effects of BQ-788 (1 μM) on ET-1-mediated changes in cyclic nucleotide levels. BQ-788 had no significant effect on the ability of ET-1 to decrease cyclic AMP in the main pulmonary artery whilst it inhibited the ability of ET-1 to raise intracellular cyclic AMP in the small muscular pulmonary arteries.
Table 3.
The effect of BQ-788 on changes induced by ET-1 on cyclic nucleotide levels in rat main pulmonary arteries and small muscular pulmonary arteries

The effect of denuding the endothelium on ET-1 signalling in the main pulmonary artery
Endothelium removal had no significant effect on basal cyclic AMP (3.8±2.6 (controls) c.f. 5.6±1.1 (rubbed endothelium) pmol mg−1 tissue (n=5)). Endothelium removal did not alter basal cyclic GMP levels (80±15 fmol cyclic GMP mg−1 tissue (controls, n=4) vs 60.7±15.9 fmol cyclic GMP mg−1 tissue (rubbed endothelium, n=3)). Removal of the endothelium had no effect on the ability of ET-1 to increase cyclic GMP. Incubation of the rubbed tissue with the peptide resulted in an increase in cyclic GMP levels from to 60.7±15.9 to 97.0±11.9 fmol cyclic GMP mg−1 tissue (n=3). Removal of the endothelium had no effect in the percentage reduction of cyclic AMP elicited by ET-1. This was 43±15% in intact vessels and 36±14% in endothelium-denuded vessels (n=5). No attempt was made to remove the endothelium from the small arteries as experience dictates this damages and significantly reduces the vascular smooth muscle layer.
ET-1 enhances cholera toxin-catalyzed [32P]-ADP-ribosylation through activation of ETA-receptors in main extrapulmonary arteries
Thiol-activated cholera toxin was able to catalyze [32P]-ADP-ribosylation of both long (45 kDa) and short (42 kDa) forms of Gsa in membranes prepared from rat extrapulmonary arteries (data not shown). When the assay was performed in the absence of exogenous guanine nucleotides, labelling of a 40 kDa polypeptide was also observed. Upon subsequent immunoprecipitation with the specific antiserum SG1, this protein was identified as Gi2 (Figure 2). Incorporation of [32P]-ADP-ribose into Gi2 was markedly enhanced by addition of 0.1 μM ET-1 to the assay. FR139317 (1 μM) blocked the ET-1-mediated increase in radiolabel incorporation whilst BQ-788 (1 μM) had no effect (Figure 2).
Figure 2.
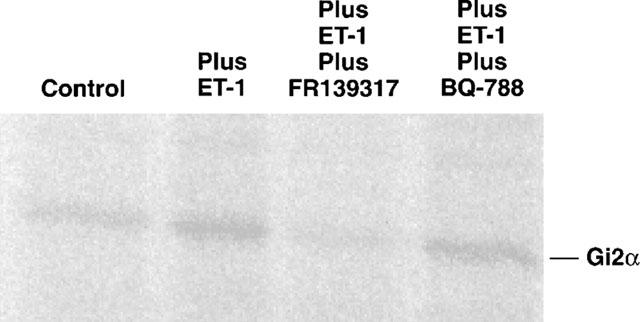
A phosphorimage showing the regulation of cholera toxin-catalyzed [32P]-ADP-ribosylation of Gi2 in membranes from main extrapulmonary artery. Membranes (100 mg) from rat extrapulmonary artery were incubated with [32P]-NAD and thiol-activated cholera toxin (50 mg ml−1) in the absence of ligand, with 0.1 μM ET-1, 1 μM FR139317 or 1 μM BQ-788. Samples were solubilized, immunoprecipitated with antiserum SG1, resolved by SDS–PAGE (10% w v−1 acrylamide) and subjected to autoradiography. This is a representative image of a series of three. In all, densitometrically, there was no difference between the ‘plus ET-1 plus FR139317' band and the ‘control band', neither was there any difference between the ‘plus ET-1 plus BQ-788' band and the ‘plus ET-1' band.
Discussion
Clear differences in ET receptor distribution exist between species. For example, in large pulmonary arteries, the ETA-subtype is dominant in the human whist the ETB-subtype is more prevalent in rabbits (Fukuroda et al., 1994). In the main pulmonary artery of the rat, the ETA-receptor is responsible for ET-1-mediated vasoconstriction (MacLean et al., 1994; Higashi et al., 1997). ET receptor heterogeneity is also observed between different areas of the pulmonary circulation. In the rat, we have previously shown that progression through the pulmonary arterial system is accompanied by an apparent switch from ETA-receptor-mediated contraction in the main pulmonary arteries to the ETB-like receptor-mediated contraction in the resistance vessels (MacLean et al., 1994; McCulloch & MacLean, 1995; McCulloch et al., 1998).
The object of the study described here was to establish the net effect of ET-1 on cyclic nucleotides in the rat pulmonary arterial circulation using a whole vessel preparation, and to correlate our previous findings with respect to the functional effects of ET-1 in the same vessels. In summary, we have shown that in the small muscular pulmonary artery of the rat, where ETB-mediated vasoconstriction prevails there is an ETB-mediated increase in cyclic AMP with no net effect on cyclic GMP levels. In the large arteries, where vasoconstriction is mediated via the ETA receptor, there is an ETA-mediated increase in cyclic GMP (endothelium independent) and an ETA (endothelium independent) decrease in cyclic AMP.
The ETB-mediated increase in cyclic AMP in the small pulmonary arteries was somewhat unexpected as previous studies on cultured endothelial cells have shown that ETB receptors are negatively coupled to adenylate cyclase (Eguchi et al., 1993a,1993b; Sokolovsky, 1994). ET-1 has been shown, via the ETA receptor, to stimulate cyclic AMP formation in rat vascular smooth muscle cells, Chinese hamster ovary cells and cultured embryonic bovine tracheal cells (Eguchi et al., 1993a; Aramori & Nakanishi, 1992; Oda et al., 1992). Hence this is the first report of an ETB-mediated increase in cyclic AMP in vasculature. This occurred in the small muscular pulmonary arteries where ETB-mediated vasoconstriction prevails. Elevated cyclic AMP would cause a vasodilator effect and may serve as a physiological brake upon the ET-1-induced vasoconstriction. This would be functionally important as this study also shows that there is no ET-1 mediated elevation of cyclic GMP in these vessels, consistent with our inability to demonstrate ET-1-induced, nitric oxide dependent vasodilation in these vessels from either rat or rabbit (Docherty & MacLean, 1998).
ET-1 can induce prostacyclin production in rat lungs (Barnard et al., 1991) and therefore the rise in cyclic AMP could be secondary to prostacyclin production. ET-1- and ETB-mediated vasodilation is not, however, inhibited by indomethacin in this vascular bed (Hasunuma et al., 1990; Eddahibi et al., 1993; Lal et al., 1996). Thus, it is unlikely that the rise in cyclic AMP is entirely due to prostacyclin production.
The current results may explain some previously reported unexpected observations in these vessels (McCulloch et al., 1998). We demonstrated that, despite there being an ETB-mediated vasoconstriction in these vessels, the ETB receptor antagonist BQ-788 failed to block the vasoconstrictor response to ET-1, while the mixed ETA/B antagonist bosentan actually potentiated the responses to ET-1. These phenomena may, partially, be due to blockade of ETB-receptor mediated increases in cyclic AMP.
An ETA (endothelium independent) decrease in cyclic AMP was observed in the main pulmonary arteries. This effect was only observed at 0.1 μM ET-1 and not observed to lower concentrations. This may be a consequence of the ability of ET-1 to also stimulate cyclic GMP and inositol-1,4,5-trisphosphate in these vessels and it is widely accepted that there is ‘crosstalk' between these second messengers which may depress effects of ET-1 on cyclic AMP metabolism at lower concentrations. For example, we have shown that elevated cyclic GMP levels can inhibit 5-HT1-stimulated contractions in bovine pulmonary arteries which are mediated by Gi protein-mediated decreased cyclic AMP levels (Sweeney et al., 1995).
The ETA antagonist FR139317 only partially reversed the decrease in cyclic AMP levels. Synergism between ETA- and ETB-receptor mediated vasoconstriction can occur in rabbit pulmonary arteries such that combined blockade of both receptors is required to inhibit the vasoconstrictor responses to ET-1 (Fukuroda et al., 1994). However, in the rat elastic pulmonary arteries there is no evidence for such synergism and ET-1-mediated vasoconstriction is soley mediated by the ETA receptor (MacLean et al., 1994; 1995). Hence, the inability of FR139317 to fully inhibit the decrease in cyclic AMP levels induced by ET-1 is more likely to be due to the chosen concentration of FR139317. Indeed this concentration of FR139317 (1 μM) does not fully block the vasoconstrictor response to 0.1 μM ET-1 in the main pulmonary artery of the rat (MacLean et al., 1995).
ET-1-induced reduction of cyclic AMP has been previously reported in endothelial cells and cardiac myocytes (Ladoux & Frelin, 1991; Hilal-Dandan et al., 1992), but has not been previously reported in the pulmonary artery smooth muscle. The previous studies indicated that this effect of ET-1 may be via coupling to a Gi protein. As ETA receptors mediate vasoconstriction in this area of the rat pulmonary vascular bed, the reduction in cyclic AMP could contribute to ET-1 mediated vasoconstriction. We wished, therefore to elucidate the mechanism behind such an effect by examining coupling to the Gi2 protein. We investigated the possibility that the ET-1 mediated decrease in cyclic AMP occurs as a consequence of direct inhibition of adenylyl cyclase through Gi2. Recent advances in methodology have allowed definition of the molecular interaction between a receptor and the G protein with which it interacts. We have developed an assay in which pertussis toxin-sensitive G proteins become substrates for cholera toxin-catalyzed [32P]-ADP-ribosylation when they interact with an agonist-activated receptor (Mullaney et al., 1996). When used together with a specific immunoprecipitating G protein antiserum, the assay provides the best means to elucidate the specificity of receptor-G protein coupling. Using specific antisera, we have recently characterized the G protein complement in rat pulmonary arteries and shown that they contain Gi2 (Mullaney & MacLean, unpublished observations). Incubation with ET-1 caused an increase in incorporation of radiolabel specifically into Gi2 in the main extralobar artery. This increase, which is indicative of receptor activation of Gi2, was blocked by FR139317 but unaffected by BQ-788. These results provide definitive evidence of direct activation of Gi2 by interaction with ETA receptors in the rat pulmonary artery, a phenomenon not previously described in the pulmonary artery.
A previous study demonstrated that chronic hypoxia abolished the ET-1-mediated reduction in cyclic AMP levels in rat main pulmonary artery (Mullaney et al., 1998). This would serve to counteract any increase in the vasoconstrictor effect of ET-1 and indeed, there is no enhancement of contraction of ET-1 in these vessels (MacLean et al., 1995) whereas there is an enhanced vasoconstriction to ET-1 in the smaller arteries mediated via the ETA receptor (McCulloch et al., 1998). We also previously showed that the potency of FR139317 increases in hypoxic conditions in the large pulmonary artery (MacLean et al., 1994). A loss of ETA-mediated vasoconstriction via reduced cyclic AMP may explain this phenomenon.
Basal levels of cyclic GMP were lower in the small muscular pulmonary arteries than in the large ET-1. This is consistent with there being abundant expression of endothelial nitric oxide synthase (eNOS) in the rat main pulmonary artery whereas eNOS is virtually absent from the small pulmonary arteries (Xue et al., 1994). ET-1 had no effect on cyclic GMP in the pulmonary resistance arteries. This is consistent with a previous study where we demonstrated that treatment with the nitric oxide synthase inhibitor, L-NW nitroarginine methyl ester (L-NAME) has no effect on the ability of ET-1 to constrict pulmonary resistance arteries (MacLean & McCulloch, 1998). In addition, we have never been able to demonstrate nitric-oxide dependent, ET-1-induced, vasodilation in these vessels (Docherty & MacLean, 1998). In contrast, incubation of main pulmonary arteries with ET-1 did result in a significant increase in cyclic GMP levels. This increased was preserved in vessels denuded of endothelium and inhibited by FR139317. Previous studies have confirmed that our procedures for endothelium removal are successful, inhibiting ACh-induced vasodilation (MacLean et al., 1994). Removal of the endothelium had no significant effect on either cyclic GMP or cyclic AMP basal levels, consistent with the major site of basal cyclic nucleotide generation being in the vascular smooth muscle.
Collectively, these observations suggest that ET-1 can activate, via the ETA receptor, guanylyl cyclase directly at the level of the smooth muscle. The lack of effect of endothelium removal and BQ-788 was surprising as, in the vascular endothelium, activation of the nitric oxide/cyclic GMP signalling pathway as a result of ETB-receptor stimulation plays a key role in the vasodilator actions of the peptide (Masaki et al., 1991). Non-endothelial, non nitric oxide-dependent, ETA receptor-mediated cyclic GMP production has, however, previously been intimated in rat cerebellar slices where it may be mediated via carbon monoxide production (Shraga-Levine et al., 1994).
In these large pulmonary vessels, ET-1 induces vasoconstriction via the ETA receptor and hence ETA-mediated increases in cyclic GMP would oppose this effect and may serve as a physiological brake on ET-mediated vasoconstriction. The results of the current study may explain some previously unexplained phenomena that we have reported (MacLean et al., 1995). We described how 1 μM FR139317 and 0.1 μM FR139317 were equipotent against the vasoconstrictor effects of ET-1 in the rat main pulmonary artery. This may be due to 1 μM FR139317 blocking the ETA-mediated rise in cyclic GMP in addition to its inhibitory effect against ET activation of Gq- and Gi-protein linked receptors.
Hence, it is evident from this and other studies that ET-1 couples with multiple G protein subtypes in the pulmonary arterial bed. As Sokolovsky stated in 1995, apparent discrepancies in ET-1 pharmacology between preparations and species are probably attributable to the complex signalling pathways involved and the heterogeneous nature of the participating G proteins as well as the distribution of ET receptor subtypes (Sokolovsky, 1995). All these complexities exist in the pulmonary circulation which may explain many of the current controversies in ET receptor pharmacology, some of which are discussed here.
It should be noted that levels of cyclic nucleotides can be modulated by mechanisms other than by direct interaction with guanylyl or adenylyl cyclase. Multiple phosphodiesterase enzymes can hydrolyze these compounds to the corresponding 5′-nucleotide (Houslay & Milligan, 1997). Indeed, the decreased levels of cyclic nucleotides in the pulmonary arteries of rats exposed to chronic hypoxia can partly be explained by increased activity of phosphodiesterases (MacLean et al 1996; 1997). Our experiments were performed, however, in the presence of the general phosphodiesterase inhibitor IBMX at a concentration we have previously demonstrated to inhibit all PDE activity in rat pulmonary arteries (MacLean et al., 1997).
In conclusion we have shown that in the small muscular pulmonary artery of the rat, where ETB mediated vasoconstriction prevails there is an ETB-mediated increase in cyclic AMP. In the large arteries, where vasoconstriction is mediated via the ETA receptor, there is an ETA mediated increase in cyclic GMP (endothelium independent) and an ETA (endothelium independent) decrease in cyclic AMP. Hence, ET-1 couples with multiple G protein subtypes in the pulmonary circulation and these results may explain some of the previously reported regional complexities in the pharmacology of ET-1 in the pulmonary circulation.
Acknowledgments
This study was funded by the Wellcome Trust. The authors wish to thank Dr J.J. Morton, Glasgow Royal Infirmary for his aid in the cyclic GMP assays.
Abbreviations
- cyclic AMP
adenosine 3′ 5′ cyclic monophosphate
- cyclic GMP
guanosine 3′ 5′ cyclic monophosphate
- ET-1
endothelin-1
- PT
pertussis toxin
References
- ARAMORI I., NAKANISHI S. Coupling of two endothelin receptor subtypes to differing signal transduction in transfected Chinese hamster ovary cells. J. Biol. Chem. 1992;267:12468–12474. [PubMed] [Google Scholar]
- BARNARD J.W., BARMAN S.A., ADKINS W.K., LONGNECHER G.L., TAYLOR A.E. Sustained effects of endothelin-1 on rabbit, dog, and rat pulmonary circulations. Am. J. Physiol. 1991;261:H479–H486. doi: 10.1152/ajpheart.1991.261.2.H479. [DOI] [PubMed] [Google Scholar]
- BERRIDGE M.J. Inositol triphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- BROWN B.L., EKINS R.P., ALBANO J.D.M. Saturation assay for cyclic AMP using endogenous binding protein. Adv. Cyclic Nucleotide Res. 1972;2:25–29. [PubMed] [Google Scholar]
- CAILLA H.L., VANNIER C.J., DELAAGE M.A. Guanosine3′,5′-cyclic monophosphate assay at 10-15 mole level. Anal. Biochem. 1976;70:195–202. doi: 10.1016/s0378-5173(83)90100-x. [DOI] [PubMed] [Google Scholar]
- DOCHERTY C.C., MACLEAN M.R. EndothelinB receptors in rabbit pulmonary resistance arteries: effect of left ventricular dysfunction. J. Pharmacol. Exp. Ther. 1998;284:895–903. [PubMed] [Google Scholar]
- EDDAHIBI S., SPRINGALL D., MANNAN M., CARVILLE C., CHABRIER P.E., LEVAME M., RAFFESTIN B., POLAK J., ADNOT S. Dilator effect of endothelins in pulmonary circulation: changes associated with chronic hypoxia. Am. J. Physiol. 1993;265:L571–L580. doi: 10.1152/ajplung.1993.265.6.L571. [DOI] [PubMed] [Google Scholar]
- EGUCHI S., HIRATA Y., IHARA M., YANO M., MARUMO F. A novel ETA antagonist (BQ123) inhibits ET-1-induced phosphoinositide breakdown and DNA synthesis in rat vascular smooth muscle cells. FEBS Lett. 1992;302:243–246. doi: 10.1016/0014-5793(92)80451-l. [DOI] [PubMed] [Google Scholar]
- EGUCHI S., HIRATA Y., IMAI T., MARUMO F. Endothelin receptor subtypes are coupled to adenylate cyclase via different guanyl nucleotide-binding proteins in vasculature. Endocrinology. 1993a;132:524–529. doi: 10.1210/endo.132.2.7678793. [DOI] [PubMed] [Google Scholar]
- EGUCHI S., HIRATA Y., MARUMO F. Endothelin subtype B receptors are coupled to adenylate cyclase via inhibitory G protein in cultured bovine endothelial cells. J. Cardiovasc. Pharmacol. 1993b;22 suppl 8:S161–S163. doi: 10.1097/00005344-199322008-00043. [DOI] [PubMed] [Google Scholar]
- ELSHOURBAGY N.A., ADAMOU J.E., GAGNON A.W., WU H.L., PULLEN M., NAMBI P. Molecular characterization of a novel human endothelin receptor splice variant. J. Biol. Chem. 1996;271:25300–25307. doi: 10.1074/jbc.271.41.25300. [DOI] [PubMed] [Google Scholar]
- FUJITANI Y., UEDA H., OKADA T., URADE Y., KARAKI H. A selective agonist of endothelin type B receptor, IRL 1620, stimulates cyclic GMP increase via nitric oxide formation in rat aorta. J. Pharmacol. Exp. Ther. 1993;267:683–689. [PubMed] [Google Scholar]
- FUKURODA T., KOBIYASHI M., OZAKI S., YANO M., MIYAUCHI T., ONIZUKA M., SUGISHITA Y., GOTO K., NISHIKIBE M. Endothelin receptor subtypes in human versus rabbit pulmonary arteries. J. Appl. Physiol. 1994;76:1976–1982. doi: 10.1152/jappl.1994.76.5.1976. [DOI] [PubMed] [Google Scholar]
- FUKURODA T., OZAKI S., IHARA M., ISHIKAWA K., YANO M., NISHIKIBE M. Synergistic inhibition by BQ-123 and BQ-788 of endothelin-1-induced contractions of the rabbit pulmonary artery. Br. J. Pharmacol. 1994;113:336–338. doi: 10.1111/j.1476-5381.1994.tb16901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIAID M., YANAGISAWA M., LANGLEBEN D., MICHEL R.P., LEVY R., SHENNIB M., KIMURA S., MASAKI T., DUGUID W., STEWART D.J. Expression of endothelin in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993;328:1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- HASUNUMA K., RODMAN D.M., O'BRIEN R.F., MCMURTRY I.F. Endothelin 1 causes pulmonary vasodilation in rats. Am. J. Physiol. 1990;259:H48–H54. doi: 10.1152/ajpheart.1990.259.1.H48. [DOI] [PubMed] [Google Scholar]
- HIGASHI T., ISHIZAKI T., SHIGEMORI K., NAKAI T., MIYABO S., INUI T., YAMAMURA T. Pharmacological heterogeneity of constrictions mediated by endothelin receptors in rat pulmonary arteries. Am. J. Physiol. 1997;272:L287–L293. doi: 10.1152/ajplung.1997.272.2.L287. [DOI] [PubMed] [Google Scholar]
- HILAL-DANDAN R., URASAWA K., BRUNTON L.L. Endothelin inhibits adenylate cyclase and stimulates phosphoinositide hydrolysis in adult cardiac myocytes. J. Biol. Chem. 1992;267:10620–10624. [PubMed] [Google Scholar]
- HIRATA Y., EMORI T., EGUCHI S., KANNO K., IMAI T., OHTA K., MARUMO F. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J. Clin. Invest. 1993;91:1367–1373. doi: 10.1172/JCI116338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUSLAY M.D., MILLIGAN G. Tailoring cyclic AMP-signalling responses through isoform multiplicity. Trends Biochem. Sci. 1997;22:217–224. doi: 10.1016/s0968-0004(97)01050-5. [DOI] [PubMed] [Google Scholar]
- LAL H., WOODWARD B., WILLIAMS K.I. Investigation of the contributions of nitric oxide and prostaglandins to the actions of endothelins and sarafotoxin 6c in rat isolated perfused lungs. Br. J. Pharmacol. 1996;118:1931–1938. doi: 10.1111/j.1476-5381.1996.tb15627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LADOUX A., FRELIN C. Endothelins inhibit adenylate cyclase in brain capillary endothelial cells. Biochem. Biophys. Res. Comm. 1991;180:169–173. doi: 10.1016/s0006-291x(05)81271-9. [DOI] [PubMed] [Google Scholar]
- LI H., ELTON T.S., CHEN Y.F., OPARIL S. Increased endothelin receptor gene expression in hypoxic rat lung. Am. J. Physiol. 1994;266:L553–L560. doi: 10.1152/ajplung.1994.266.5.L553. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R. Endothelin: a mediator of pulmonary hypertension. Pulm. Pharmacol. Ther. 1998;11:125–132. doi: 10.1006/pupt.1998.0126. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R., MCCULLOCH K.M. Influence of applied tension and nitric oxide on responses to endothelins in rat pulmonary resistance arteries: effect of chronic hypoxia. Br. J. Pharmacol. 1998;123:91–99. doi: 10.1038/sj.bjp.0701682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACLEAN M.R., JOHNSTON E.D., MCCULLOCH K.M., POOLEY L., HOUSLAY M.D., SWEENEY G. Phosphodiesterase isoforms in the pulmonary arterial circulation of the rat: Changes in pulmonary hypertension. J. Pharm. Exp. Ther. 1997;283:619–624. [PubMed] [Google Scholar]
- MACLEAN M.R., MCCULLOCH K.M., BAIRD M. Endothelin ETA- and ETB- receptor mediated vasoconstriction in rat pulmonary arteries and arterioles. J. Cardiovasc. Pharmacol. 1994;23:838–845. doi: 10.1097/00005344-199405000-00022. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R., SWEENEY G., BAIRD M., MCCULLOCH K.M., HOUSLAY M., MORECROFT I. 5-hydroxytryptamine receptors mediating vasoconstriction in pulmonary arteries from control and pulmonary hypertensive rats. Br. J. Pharmacol. 1996;119:917–930. doi: 10.1111/j.1476-5381.1996.tb15760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSDEN P.A., DANTHULURI N.R., BRENNER B.M., BALLERMAN B.J., BROCK T.A. Endothelin action on vascular smooth muscle involves inositol trisphosphate and calcium mobilisation. Biochem. Biophys. Res. Commun. 1989;158:86–93. doi: 10.1016/s0006-291x(89)80180-9. [DOI] [PubMed] [Google Scholar]
- MASAKI T., KIMURA S., YANIGASAWA M., GOTO K. Molecular and cellular mechanisms of endothelin regulation. Implications for vascular function. Circulation. 1991;84:1457–1468. doi: 10.1161/01.cir.84.4.1457. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH K.M., MACLEAN M.R. Evidence for endothelinB receptor mediated contraction of human and rat pulmonary resistance arteries and the effect of pulmonary hypertension on endothelin receptor-mediated responses in the rat. J. Cardiovasc. Pharmacol. 1995;26 suppl 3:S169–S179. [PubMed] [Google Scholar]
- MCCULLOCH K.M., DOCHERTY C.C., MACLEAN M.R. Endothelin receptors mediating contraction of rat and human pulmonary resistance arteries: effect of chronic hypoxia in the rat. Br. J. Pharmacol. 1998;123:1621–1630. doi: 10.1038/sj.bjp.0701785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER R.C., PELTON J.T., HUGGINS J.P. Endothelins – from receptors to medicine. Trends Pharmacol. Sci. 1993;14:54–60. doi: 10.1016/0165-6147(93)90031-e. [DOI] [PubMed] [Google Scholar]
- MITCHELL F.M., GRIFFITHS S.L., SAGGERSON E.D., HOUSLAY M.D., KNOWLER J.T., MILLIGAN G. Guanine-nucleotide-binding proteins expressed in rat white adipose tissue: identification of both mRNAs and proteins corresponding to Gi1, Gi2 and Gi3. Biochem. J. 1989;262:403–408. doi: 10.1042/bj2620403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLANEY I., CARR I.C., MILLIGAN G. Analysis of inverse agonism at the δ opioid receptor after expression in Rat 1 fibroblasts. Biochem. J. 1996;315:227–234. doi: 10.1042/bj3150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLANEY I., VAUGHAN D.M., MACLEAN M.R. Effects of ET-1 on intracellular cyclic nucleotide levels in rat pulmonary arteries. Br. J. Pharmacol. 1997;122:P227. doi: 10.1038/sj.bjp.0703153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLANEY I., VAUGHAN D.M., MACLEAN M.R. Endothelin-1 modulation of cyclic AMP in rat pulmonary arteries: effect of chronic hypoxia. J. Cardiovasc. Pharmacol. 1998;31 suppl 1:S112–S114. doi: 10.1097/00005344-199800001-00034. [DOI] [PubMed] [Google Scholar]
- ODA K., FUJITANI Y., WATAKABE T., INUI T., OKADA T., URADE Y., OKUDA-ASHITAKA E., ITO S. Endothelin stimulates both cyclic AMP formation and phosphatidylinositol hydrolysis in cultured embryonic bovine tracheal cells. FEBS Lett. 1992;299:187–191. doi: 10.1016/0014-5793(92)80244-b. [DOI] [PubMed] [Google Scholar]
- OHLSTEIN E.H., DOUGLAS S.A., BROOKS D.P., HAY D.W.P., FEURSTEIN G.Z., RUFFOLO R.R.Functions mediated by peripheral endothelin receptors Endothelin receptors–from the gene to the human 1995CRC Press: Boca, Raton; 109–185.In: Ruffolo, R.R. (ed) [Google Scholar]
- OWE-YOUNG R., SCHYVENS C.G., QASABIAN R.A., CONIGRAVE A.D., MACDONALD P.S., WILLIAMSON D.J. Transcriptional down-regulation of the rabbit pulmonary artery endothelinB receptor during phenotypic modulation. Br. J. Pharmacol. 1999;126:103–110. doi: 10.1038/sj.bjp.0702280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REMUZZI G., BENIGNI A. Endothelins in the control of cardiovascular and renal function. Lancet. 1993;342:589–593. doi: 10.1016/0140-6736(93)91414-h. [DOI] [PubMed] [Google Scholar]
- RESINK T.J., SCOTT-BURDEN T., BUHLER F.R. Endothelin stimulates phospholipase C in cultured vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1988;157:1360–1368. doi: 10.1016/s0006-291x(88)81025-8. [DOI] [PubMed] [Google Scholar]
- SHRAGA-LEVINE Z., GALRON R., SOKOLOVSKY M. Cyclic GMP formation in rat cerebellar slices is stimulated by endothelins via nitric oxide formation and by sarafotoxins via formation of carbon monoxide. Biochem. 1994;33:14656–14659. doi: 10.1021/bi00253a002. [DOI] [PubMed] [Google Scholar]
- SHYAMALA V., MOULTHORP T.H.M., STRATTONTHOMAS J., TEKAMPOLSON P. 2 distinct human endothelin-B receptors generated by alternative splicing from a single gene. Cell. Mol. Biol. Res. 1994;40:285–296. [PubMed] [Google Scholar]
- STEWART D.J., LEVY R.D., CERNACEK P., LANGLEBEN D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease. Ann. Intern. Med. 1991;114:464–469. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- SOKOLOVSKY M. Endothelins and sarafotoxins: receptor heterogeneity. Int. J. Biochem. 1994;26:335–340. doi: 10.1016/0020-711x(94)90053-1. [DOI] [PubMed] [Google Scholar]
- SOKOLOVSKY M. Endothelin receptor subtypes and their role in transmembrane signaling mechanisms. Pharmacol. Ther. 1995;68:435–471. doi: 10.1016/0163-7258(95)02015-2. [DOI] [PubMed] [Google Scholar]
- SWEENEY G., TEMPLETON A.G.B., CLAYTON R.A., BAIRD M., SHERIDAN S., JOHNSTON E.D., MACLEAN M.R. Contractile responses to sumatriptan in isolated bovine pulmonary artery rings: relationship to tone and cyclic nucleotide levels. J. Cardiovasc. Pharmacol. 1995;26:751–760. doi: 10.1097/00005344-199511000-00012. [DOI] [PubMed] [Google Scholar]
- TJEN-A-LOOI S., EKMAN R., OSBORN J., KEITH I. Pulmonary vascular pressure effects by endothelin-1 in normoxia and chronic hypoxia: a longitudinal study. Am. J. Physiol. 1996;271:H2246–H2253. doi: 10.1152/ajpheart.1996.271.6.H2246. [DOI] [PubMed] [Google Scholar]
- XUE C., RENGASEMY A., LE CRAS T.D., KOBERNA P.A., DAILEY G.C., JOHNS R.A. distribution of NOS in normoxic vs. hypoxic rat lung: upregulation of NOS by chronic hypoxia. Am. J. Physiol. 1994;267:L667–L678. doi: 10.1152/ajplung.1994.267.6.L667. [DOI] [PubMed] [Google Scholar]
- YANAGISAWA M., KURIHARA H., KIMURA S., TOMOBE Y., KOBAYASHI M., MITSUI Y., YAZAKI Y., GOTO K., MASAKI T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]