

Abstract
Some studies suggest that β-adrenoceptor-mediated vasorelaxation is in part mediated through nitric oxide (NO) release. We wished to determine the contribution of the L-arginine / NO system to vasodilatation in response to β-adrenoceptor stimulation with isoprenaline or cyclic adenosine-3′,5′-monophosphate (cyclic AMP) elevation with forskolin and dibutyryl cyclic AMP in vivo, using a rabbit femoral artery constant perfusion model.
Baseline femoral artery pressure was similar in rabbits receiving isoprenaline, forskolin or dibutyryl cyclic AMP. Isoprenaline, forskolin and dibutyryl cyclic AMP each decreased femoral artery pressure in a dose-dependent manner. The doses (mol kg−1) of isoprenaline, forskolin and dibutyryl cyclic AMP which decreased pressure by 10% from baseline, expressed as a negative logarithm (−log ED10) were: 10.0±0.2, 9.5±0.1 and 4.9±0.1 respectively (P<0.0001 for each). Use of β-adrenoceptor subtype-selective antagonists showed that the vascular response to isoprenaline was purely due to stimulation of the β2-adrenoceptor subtype.
Injection of 1 μmol kg−1 NG-nitro-L-arginine methyl ester (L-NAME) did not alter baseline pressure. However, it abolished the pressure response to isoprenaline (P<0.0001), and significantly attenuated the pressure responses to forskolin and dibutyryl cyclic AMP: −log ED10 values for forskolin and dibutyryl cyclic AMP, in the presence of L-NAME, were 7.9±0.1 and 3.5±0.3 respectively (P<0.0001 for each, as compared with values in the absence of L-NAME).
These results indicate that β2-adrenergic stimulation and cylic AMP elevation activate the L-arginine/NO system in rabbit femoral artery in vivo, and that NO generation contributes importantly to the changes in vascular tone induced by agents which modulate β-adrenoceptors or cyclic AMP.
Keywords: β-Adrenergic receptors, cyclic AMP, vascular endothelium, vasodilatation, nitric oxide
Introduction
Stimulation of β-adrenoceptors on vascular smooth muscle results in vasorelaxation, through activation of adenylyl cyclase and a consequent increase in cyclic adenosine-3′,5′-monophosphate (cyclic AMP) intracellularly (Kuriyama et al., 1982). Vascular endothelial cells may also express β-adrenoceptors (Steinberg et al., 1984; Buxton et al., 1987; Summers et al., 1987; Howell et al., 1988; Molenaar et al., 1988; Lefroy et al., 1993), and there is increasing evidence that these may contribute to β-adrenergic vasorelaxation, although the relative contributions of endothelial and smooth muscle β-adrenoceptors to vasodilatation may vary between different vascular beds and also between different species (Rubanyi & Vanhoutte, 1985; Kamata et al., 1989; Gardiner et al., 1991; Gray & Marshall, 1992; Blankesteijn & Thien, 1993; Graves & Poston, 1993; Parent et al., 1993; Rebich et al., 1995; Dawes et al., 1997; Priest et al., 1997). We have recently demonstrated, in cultured human umbilical vein endothelial cells, that stimulation of β-adrenoceptors with isoprenaline increases uptake of L-arginine, the precursor for nitric oxide (NO) generation, and activates NO synthase (NOS) activity; furthermore, forskolin (a direct activator of adenylyl cyclase) and dibutyryl cyclic AMP (a non-hydrolysable membrane-permeable analogue of cyclic AMP) also increase L-arginine uptake and NOS activity in these cells (Ferro et al., 1999). However, the relevance of these findings to vasodilatation in vivo remains unclear.
We therefore investigated the effects on vascular tone of local infusion of isoprenaline, forskolin and dibutyryl cyclic AMP in the rabbit femoral artery in vivo, with and without inhibition of NOS using the L-arginine analogue NG-nitro-L-arginine methyl ester (L-NAME). To date, there are no published studies describing the contribution of the L-arginine/NO system to vasodilatation induced by β-adrenoceptor stimulation or cyclic AMP elevation in this vessel.
Methods
Materials
CGP 20712A was graciously provided by Ciba-Geigy Ltd (Basel, Switzerland), and ICI 118551 by Zeneca Pharmaceuticals (Macclesfield, Cheshire, U.K.). All other drugs and chemicals were from Sigma-Aldrich Company Ltd (Poole, Dorset, U.K). They were prepared on the day of study in normal saline, and kept at 4°C until use, at which time they were re-warmed to 37°C. New Zealand White rabbits were from the Nanjing Experimental Animals Centre of China.
Protocol
Rabbits were anaesthetized with intraperitoneal sodium pentobarbitone 25 mg kg−1, following which both femoral arteries were dissected from the inguinal ligament to the distal 1/3 of the leg. One femoral artery was cannulated with a catheter connected to a pressure transducer (Gould P50), for monitoring of systemic blood pressure. Following administration of heparin 600 IU kg−1, the femoral artery on the contralateral side was ligated 0.5 cm below the inguinal ligament, and a catheter was inserted into a branch of the medial saphenous artery on this side, which was connected to another pressure transducer for monitoring of intra-arterial pressure. Two further catheters (of internal diameter 2 mm) were introduced into this femoral artery, one proximal to the ligature and one distal to it; the two catheters were connected to the import and export tubes of a constant infusion rate pump respectively, which was used to regulate the flow of blood from the proximal to the distal parts of the femoral artery. Drugs were infused through the import tube by means of a Y-connector. The intra-arterial pressure on the ligated side was adjusted to the mean systemic blood pressure by regulating the flow rate, which was kept constant for a further 30 min prior to all experiments.
Integrity of endothelium-dependent responses
In order to confirm that the cannulation procedure did not abolish endothelium-dependent vasorelaxation, an initial set of experiments (n=7) was performed to examine changes in vascular tone in response to shear stress. For this purpose, on the ligated side, flow rate was abruptly increased to 140% of the baseline value, following which intra-arterial pressure was monitored for a further 20 min. Endothelial integrity was confirmed by a sudden increase in measured pressure, followed by a gradual drift back to baseline (due to shear stress-induced endothelial NO release) which was inhibited by prior bolus administration of 1 μmol kg−1 L-NAME.
β-adrenoceptor- and cyclic AMP-mediated responses
The femoral artery on the ligated side was injected with incremental doses of isoprenaline (10−12–10−7 mol kg−1), forskolin (10−11–10−7 mol kg−1) or dibutyryl cyclic AMP (10−7–10−3 mol kg−1), or with equal volumes of saline. Femoral intra-arterial pressure and systemic blood pressure responses were monitored throughout. All drugs were administered in 0.5 ml boluses; repeated bolus doses of 0.5 ml saline were found not to alter perfusion pressure (data not shown). Following washout and return of haemodynamic indices to baseline, experiments were repeated 5 min following intrafemoral bolus injection of 1 μmol kg−1 L-NAME. Experiments were also performed to ascertain the subtype selectivity of the β-adrenergic responses to isoprenaline. For this purpose, CGP 20712A, a β1-adrenoceptor selective antagonist (Kaumann, 1986), and ICI 118551, a β2-adrenoceptor selective antagonist (Bilski et al., 1983; Lemoine et al., 1985), were used. Following injections of isoprenaline and subsequent washout, repeat injections of bolus doses of isoprenaline were performed as before, this time 5 min following intrafemoral bolus injection of either 30 nmol kg−1 CGP 20712A or 10 nmol kg−1 ICI 118551 (three rabbits received CGP 20712A and three received ICI 118551), and intra-arterial pressure was monitored. Following repeat washout and return of pressure to baseline, further injections of isoprenaline were administered, in the presence of the other subtype-selective antagonist, and intra-arterial pressure measured once again. Repeat administrations of isoprenaline alone were not associated with tachyphylaxis, and the order of administration of the β-adrenoceptor subtype-selective antagonists did not affect the results obtained.
Statistical analysis
Changes in intra-arterial pressure in response to shear stress, isoprenaline, forskolin and dibutyryl cyclic AMP, in the absence or presence of L-NAME or β-adrenoceptor subtype antagonists, were analysed by two-way ANOVA. Log ED10 values in the absence or presence of L-NAME were compared by paired Student's t-test. Statistical significance was taken in all cases as P<0.05 (two-sided). All data are expressed as mean±s.e.mean.
Results
In order to confirm that endothelium-dependent vasodilatory responses were intact, the effect of increased shear stress on vascular tone was investigated in rabbit femoral arteries (n=7 rabbits), since shear stress is known to activate endothelial NO generation (Rubanyi et al., 1986; Cooke et al., 1991; Kuchan & Frangos, 1994). To achieve this, the rate of blood flow was increased abruptly by 40% above baseline, and the consequent change in intra-arterial pressure recorded. In all femoral arteries tested, this caused a transient sudden increase in measured pressure, followed by a slow gradual decline of pressure back to baseline over the succeeding 20 min (Figure 1). The decline in pressure was due to a sustained release of NO secondary to an increase in wall stress in the vessel, as evidenced by the inhibition of this decline when 1 μmol kg−1 L-NAME was co-administered (Figure 1). Thus, we confirmed that femoral artery ligation and cannulation did not abolish endothelium- and NO-dependent vasorelaxation in our model.
Figure 1.
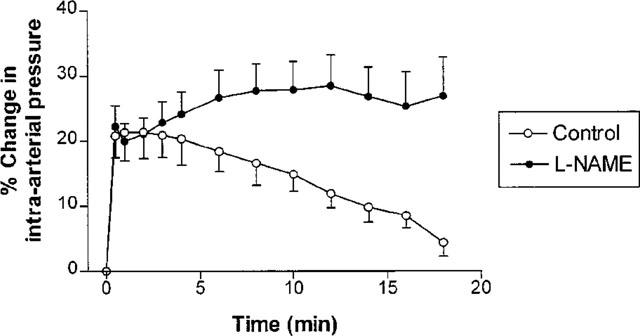
Percentage changes in rabbit femoral artery pressure (mean±s.e.mean) from baseline, as a function of time following an abrupt increase in perfusion rate to 140% of baseline (n=7). Pressure responses are shown before and after treatment with 1 μmol kg−1 L-NAME. The initial rise in pressure was the same in the absence or presence of L-NAME; however, L-NAME abolished the subsequent slow decline in pressure (P<0.0001).
Incremental doses of isoprenaline, forskolin or dibutyryl cyclic AMP were administered into the perfusate (n=6 rabbits for each drug), each dose being administered in 0.5 ml saline. All rabbits received one of these agents only. Each of these drugs induced a dose-dependent decrease in intra-arterial pressure, indicating vasodilatation (Figure 2). The change in pressure induced by isoprenaline was abolished (Figure 2a), and that induced by either forskolin or dibutyryl cyclic AMP was significantly inhibited (Figure 2b,c), by prior administration of 1 μmol kg−1 L-NAME. By contrast, 1 μmol kg−1 L-NAME alone caused no change in measured pressure at baseline (70.5±4.4 mmHg before vs 71.2±5.5 mmHg after administration of L-NAME, P>0.05), suggesting no (or very little) basal NO generation in this vessel in our system. The doses of isoprenaline, forskolin and dibutyryl cyclic AMP required to decrease intra-arterial pressure by 10% (ED10), in the absence or presence of 1 μmol kg−1 L-NAME, are displayed in Table 1. The observed abolition in isoprenaline responses following administration of L-NAME could not be explained by tachyphylaxis to the drug, since repeated administration of isoprenaline alone into rabbit femoral artery (n=6 rabbits) did not give rise to a measurable change in vascular responsiveness (Figure 3).
Figure 2.
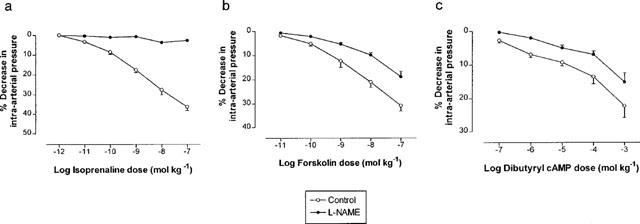
Percentage changes in rabbit femoral artery pressure (mean±s.e.mean) from baseline, in response to increasing doses of injected (a) isoprenaline, (b) forskolin or (c) dibutyryl cyclic AMP. In each case, dose-response curves are displayed before and after treatment with 1 μmol kg−1 L-NAME (n=6 for each). Treatment with L-NAME significantly blunted the reduction in measured intra-arterial pressure in response to each drug (P<0.0001 in each case).
Table 1.
Dose of isoprenaline, forskolin and dibutyryl cylic AMP required to decrease rabbit femoral artery pressure by 10% (ED10)

Figure 3.
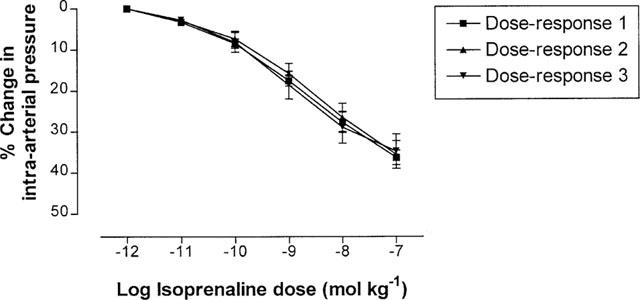
Percentage changes in rabbit femoral artery pressure (mean±s.e.mean) from baseline, in response to increasing doses of isoprenaline, when re-tested three successive times in the same vessel (dose-responses 1, 2 and 3 respectively). No tachyphylaxis was observed in pressure responses to isoprenaline (n=6).
In a single separate experiment, we also examined the effect of another NOS inhibitor, NG-monomethyl-L-arginine (L-NMMA), on femoral artery pressure responses to isoprenaline. In this experiment, isoprenaline again elicited a dose-dependent decrease in intra-arterial pressure, and following injection of L-NMMA 100 nmol kg−1 the isoprenaline response was once again abolished (Figure 4), in agreement with our findings using L-NAME.
Figure 4.
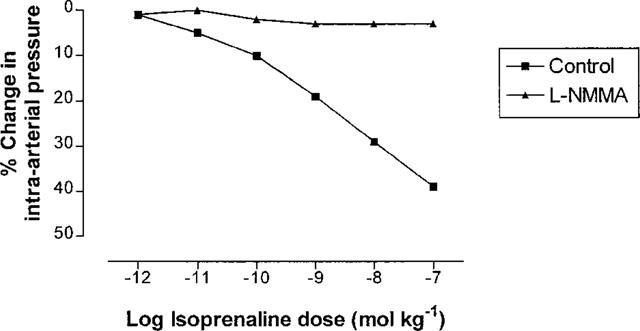
Percentage changes in rabbit femoral artery pressure from baseline, in response to increasing doses of injected isoprenaline, before and after treatment with 100 nmol kg−1 L-NMMA (one experiment).
In order to determine the β-adrenoceptor subtype(s) involved in the response to isoprenaline, experiments were also performed with the selective β1- and β2-adrenoceptor antagonists CGP 20712A and ICI 118551 respectively. In initial experiments, injection of CGP 20712A in increasing doses (10–90 nmol kg−1) had no effect on vascular responses to isoprenaline (Figure 5a), until at the highest dose used (120 nmol kg−1) cardiac asystole occurred due to excessive β-blockade (presumably β1 in type). By contrast, injection of ICI 118551 in increasing doses (2.5–10 nmol kg−1) caused a progressive inhibition of vascular isoprenaline responses (Figure 5b), and at the highest dose used (20 nmol kg−1) systemic blood pressure began to increase. We therefore chose to use concentrations of 30 nmol kg−1 for CGP 20712A and 10 nmol kg−1 for ICI 118551 in further experiments to examine the subtype selectivity of femoral artery β-adrenergic responses to isoprenaline.
Figure 5.
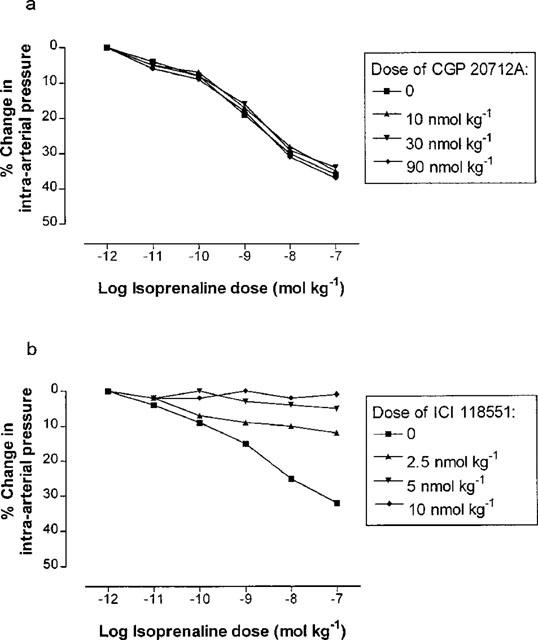
Percentage changes in rabbit femoral artery pressure from baseline, in response to increasing doses of injected isoprenaline, following treatment with different doses of β-adrenoceptor subtype-selective antagonists (n=1 in each case). (a) Dose-response curves to isoprenaline following administration of CGP 20712A (a β1-adrenoceptor-selective blocker) 0, 10, 30 and 90 nmol kg−1. (b) Dose-response curves to isoprenaline following administration of ICI 118551 (a β2-adrenoceptor-selective blocker) 0, 2.5, 5 and 10 nmol kg−1.
Pressure changes in response to isoprenaline were unchanged following the administration of CGP 20712A 30 nmol kg−1 (n=6). However, isoprenaline pressure responses were abolished by ICI 118551 10 nmol kg−1 (n=6), indicating that the responses to isoprenaline were mediated solely through stimulation of β2-adrenoceptors (Figure 6).
Figure 6.
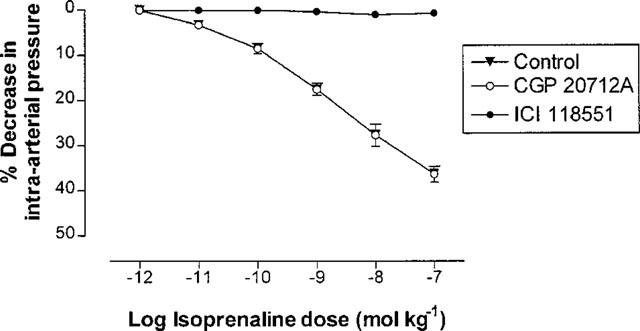
Percentage changes in rabbit femoral artery pressure (mean±s.e.mean) from baseline, in response to increasing doses of injected isoprenaline, before and after administration of 30 nmol kg−1 CGP 20712A or 10 nmol kg−1 ICI 118551. Isoprenaline responses were unaffected by CGP 20712A, but were abolished by ICI 118551 (P<0.0001).
Discussion
Previous studies have shown that β-adrenoceptor-mediated vasorelaxation may have an endothelium-dependent component in several species (Graves & Poston, 1993; Rubanyi & Vanhoutte, 1985; Kamata et al., 1989; Gardiner et al., 1991; Gray & Marshall, 1992; Blankesteijn & Thien, 1993; Parent et al., 1993; Rebich et al., 1995; Priest et al., 1997). However, few of these studies have looked at endothelial β-adrenergic function in vivo, and none have examined the possible role of the endothelial adenylyl cyclase / cyclic AMP system in inducing vasodilatation through NO generation in vivo. We have previously demonstrated that β-adrenoceptor stimulation activates adenylyl cyclase and cyclic AMP formation in cultured human umbilical vein endothelial cells in vitro, and that forskolin and dibutyryl cyclic AMP (which both increase intracellular cyclic AMP) activate the L-arginine / NO system in these cells (Ferro et al., 1999). The present results suggest that β-adrenoceptor stimulation or cyclic AMP elevation by other means induce vasodilatation in rabbit femoral artery in vivo, and that such vasodilatation is largely dependent on NO generation, presumably from the vascular endothelium. In the case of isoprenaline, vascular responses are abolished by co-administration of L-NAME, suggesting that, at least in this vessel, β-adrenergic vasodilatation is entirely due to NO release. By contrast, responses to forskolin or dibutyryl cyclic AMP are incompletely inhibited by L-NAME at the same dose (1 μmol kg−1). This is consistent with an endothelium-dependent component to vasorelaxation, caused by activation of the L-arginine / NO system in response to cyclic AMP elevation, although it is likely that there is also a substantial endothelium-independent component caused by direct elevation of cyclic AMP in vascular smooth muscle.
Our finding that the isoprenaline-induced decrease in femoral arterial tone is abolished by NOS inhibition is consistent with data derived from certain other blood vessels. Endothelial denudation has been found to abolish vasorelaxation of rat thoracic aortic rings to isoprenaline or to salbutamol (Gray & Marshall, 1992). Furthermore, we have recently demonstrated that isoprenaline-induced vasorelaxation is abolished, and forskolin- or dibutyryl cyclic AMP-induced vasorelaxation is diminished, by NOS inhibition in human umbilical vein in vitro (Ferro et al., 1999).
Use of the subtype-selective antagonists CGP 20712A and ICI 118551 enabled us also to determine the subtype selectivity of the β-adrenergic responses to isoprenaline. Our results indicate that isoprenaline-induced changes in vascular tone are mediated solely through stimulation of β2-adrenoceptors, in rabbit femoral artery in vivo.
Since L-NAME inhibits endogenous NO production, one would predict that its administration would cause vasoconstriction, if there is a substantial basal production of NO. Indeed, there is evidence that arterial endothelium can produce significant amounts of NO basally (Lüscher, 1991). Therefore, our finding of a blunted pressure decrease to isoprenaline, forskolin and dibutyryl cyclic AMP might be explained by a direct vasoconstrictive effect opposing the vasodilator effect of these compounds, irrespective of any putative inhibition of their NO-generating effects. However, this is unlikely, since L-NAME had no effect on measured intra-arterial pressure in the absence of these drugs, in our system.
In general, NOS inhibition in vivo constricts arteries but not veins, indicating that arteries generate important amounts of NO in the basal state. This is believed to occur through the physical activation of endothelial cells by pulsatile flow, and shear stress may also play a role (Moncada & Higgs, 1993). In our experimental model, pulsatile flow is abolished by ligation of the femoral artery. This may explain the lack of observed effect of L-NAME on basal intra-arterial pressure.
Graier et al. (1992) examined the possible role of cyclic AMP in NO formation in cultured porcine aortic endothelial cells; they found that, whilst forskolin, adenosine or isoprenaline (all of which stimulate cyclic AMP formation) alone did not increase NO synthesis, they amplified the formation of NO in response to bradykinin or ATP. This accords with the known ability of cyclic AMP to increase cyclic guanosine-3′,5′-monophosphate levels. By contrast, we have previously demonstrated that β-adrenoceptor stimulation with isoprenaline, or direct cyclic AMP elevation with either forskolin or dibutyryl cyclic AMP, activate NOS activity in human umbilical vein endothelial cells (Ferro et al., 1999). Our present findings indicate that NO generation is similarly increased by β-adrenergic stimulation or cyclic AMP elevation in rabbit femoral artery in vivo.
However, it is likely that these findings cannot be generalized to all blood vessels and all species. For example, Moncada et al. (1991) have found no evidence of endothelium dependence on isoprenaline-induced vasorelaxation in rat aorta in vitro or in the rat circulation in vivo, and two independent studies of isolated canine coronary arteries have also suggested that β-adrenoceptor-mediated vasorelaxation is endothelium-independent (Macdonald et al., 1987; Béa et al., 1994).
In conclusion, we have shown that β-adrenoceptor activation or cyclic AMP elevation by other means stimulates NO production in rabbit femoral artery in vivo, giving rise to vasodilatation. This NO is presumed to be of endothelial origin, although the present experiments cannot rule out a possible origin in vascular smooth muscle. The physiological consequences of vascular endothelial NO production in response to such stimuli are at present unknown, but it is possible that this process is important in the normal control of vessel tone by the sympathoadrenal system. Until recently, β-adrenoceptor- and cyclic AMP-mediated vasodilatation has been believed to occur solely through direct relaxation of vascular smooth muscle. Our study lends support to the increasing body of evidence that β-adrenoceptors and the adenylyl cyclase / cyclic AMP system can contribute to vasorelaxation, specifically through the generation of NO.
Acknowledgments
This work was supported by a Wellcome Trust International Research Development Award to B. Xu.
Abbreviations
- cyclic AMP
cyclic adenosine-3′,5′-monophosphate
- L-NAME
NG-nitro-L-arginine methyl ester
- L-NMMA
NG-monomethyl-L-arginine
- NO
nitric oxide
- NOS
nitric oxide synthase
References
- BÉA M.-L., GHALEH B., GIUDICELLI J.-F., BERDEAUX A. Lack of importance of NO in β-adrenoceptor-mediated relaxation of large epicardial canine coronary arteries. Br. J. Pharmacol. 1994;111:981–982. doi: 10.1111/j.1476-5381.1994.tb14839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BILSKI A., HALLIDAY S.E., FITZGERALD J.D., WALE J.L. The pharmacology of a β2-selective adrenoceptor antagonist (ICI 118,551) J. Cardiovasc. Pharmacol. 1983;5:430–437. doi: 10.1097/00005344-198305000-00013. [DOI] [PubMed] [Google Scholar]
- BLANKESTEIJN W.M., THIEN T. Effect of NG-monomethyl-L-arginine on the β-adrenoceptor-mediated relaxation of rat mesenteric resistance arteries. Life Sci. 1993;52:PL135–PL139. doi: 10.1016/0024-3205(93)90178-6. [DOI] [PubMed] [Google Scholar]
- BUXTON B.F., JONES C.R., MOLENAAR P., SUMMERS R.J. Characterization and autoradiographic localization of β-adrenoceptor subtypes in human cardiac tissues. Br. J. Pharmacol. 1987;92:299–310. doi: 10.1111/j.1476-5381.1987.tb11324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOKE J.P., ROSSITCH E., ANDON N.A., LOSCALZO L., DZAU V.J. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J. Clin. Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWES M., CHOWIENCZYK P.J., RITTER J.M. Effects of inhibition of the L-arginine / nitric oxide pathway on vasodilation caused by β-adrenergic agonists in human forearm. Circulation. 1997;95:2293–2297. doi: 10.1161/01.cir.95.9.2293. [DOI] [PubMed] [Google Scholar]
- FERRO A., QUEEN L.R., PRIEST R.M., XU B., RITTER J.M., POSTON L., WARD J.P.T. Activation of nitric oxide synthase by β2-adrenoceptors in human umbilical vein endothelium in vitro. Br. J. Pharmacol. 1999;126:1872–1880. doi: 10.1038/sj.bjp.0702512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARDINER S.M., KEMP P.A., BENNETT T. Effects of NG-nitro-L-arginine methyl ester on vasodilator responses to acetylcholine, 5′-N-ethylcarboxamidoadenosine or salbutamol in conscious rats. Br. J. Pharmacol. 1991;103:1725–1732. doi: 10.1111/j.1476-5381.1991.tb09854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAIER W.F., GROSCHNER K., SCHMIDT K., KUKOVETZ W.R. Increases in endothelial cyclic AMP levels amplify agonist-induced formation of endothelium-derived relaxing factor (EDRF) Biochem. J. 1992;288:345–349. doi: 10.1042/bj2880345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAVES J., POSTON L. β-Adrenoceptor agonist mediated relaxation of rat isolated resistance arteries: a role for the endothelium and nitric oxide. Br. J. Pharmacol. 1993;108:631–637. doi: 10.1111/j.1476-5381.1993.tb12853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY D.W., MARSHALL I. Novel signal transduction pathway mediating endothelium-dependent β-adrenoceptor vasorelaxation in rat thoracic aorta. Br. J. Pharmacol. 1992;107:684–690. doi: 10.1111/j.1476-5381.1992.tb14507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOWELL R.E., ALBEDA S.M., DAISE M.L., LEVINE E.M. Characterization of β-adrenergic receptors in cultured human and bovine endothelial cells. J. Appl. Physiol. 1988;65:1251–1257. doi: 10.1152/jappl.1988.65.3.1251. [DOI] [PubMed] [Google Scholar]
- KAMATA K., MIYATA N., KASUYA Y. Involvement of endothelial cells in relaxation and contraction responses of the aorta to isoproterenol in naive and streptozotocin-induced diabetic rats. J. Pharmacol. Exp. Thera. 1989;249:890–894. [PubMed] [Google Scholar]
- KAUMANN A.J. The β1-adrenoceptor antagonist CGP 20,712A unmasks β2-adrenoceptors activated by (-) adrenaline in the rat sino-atrial node. Naunyn Schmiedeberg's Arch. Pharmacol. 1986;333:73–76. doi: 10.1007/BF00500096. [DOI] [PubMed] [Google Scholar]
- KUCHAN M.J., FRANGOS J.A. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Physiol. 1994;266:C628–C636. doi: 10.1152/ajpcell.1994.266.3.C628. [DOI] [PubMed] [Google Scholar]
- KURIYAMA H., ITO Y., SUZUKI H., KITAMURA K., ITOH T. Factors modifying contraction-relaxation cycle in vascular smooth muscles. Am. J. Physiol. 1982;243:H641–H662. doi: 10.1152/ajpheart.1982.243.5.H641. [DOI] [PubMed] [Google Scholar]
- LEFROY D.C., DONNELLY L.E., MCEWAN J.R., MACDERMOT J. Phorbol ester enhances activation of adenylate cyclase in bovine aortic endothelial cells. Life Sci. 1993;54:87–94. doi: 10.1016/0024-3205(94)00778-0. [DOI] [PubMed] [Google Scholar]
- LEMOINE H., EHLE B., KAUMANN A.J. Direct labelling of β2-adrenoceptors: comparisons of binding potency of 3H-ICI 118,551 and blocking potency of ICI 118,551. Naunyn Schmiedeberg's Arch. Pharmacol. 1985;331:40–51. doi: 10.1007/BF00498850. [DOI] [PubMed] [Google Scholar]
- LÜSCHER T.F. Endothelium-derived nitric oxide: the endogenous nitrovasodilator in the human cardiovascular system. Eur. Heart J. 1991;12 Suppl. E:2–11. doi: 10.1093/eurheartj/12.suppl_e.2. [DOI] [PubMed] [Google Scholar]
- MACDONALD P.S., DUBBIN P.M., DUSTING G.J. β-Adrenoceptors on endothelial cells do not influence release of relaxing factor in dog coronary arteries. Clin. Exp. Pharmacol. Physiol. 1987;14:525–534. doi: 10.1111/j.1440-1681.1987.tb01508.x. [DOI] [PubMed] [Google Scholar]
- MOLENAAR P., MALTA E., JONES C.R., BUXTON B.F., SUMMERS R.J. Autoradiographic localization and function of β-adrenoceptors on the human internal mammary artery and saphenous vein. Br. J. Pharmacol. 1988;95:225–233. doi: 10.1111/j.1476-5381.1988.tb16568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., HIGGS A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- MONCADA S., REES D.D., SCHULZ R., PALMER R.M.J. Development and mechanism of a specific supersensitivity to nitrovasodilators after inhibition of vascular nitric oxide synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARENT R., AL-OBAIDI M., LAVALLÉE M. Nitric oxide formation contributes to β-adrenergic dilation of resistance coronary vessels in conscious dogs. Circ. Res. 1993;73:241–251. doi: 10.1161/01.res.73.2.241. [DOI] [PubMed] [Google Scholar]
- PRIEST R.M., HUCKS D., WARD J.P.T. Noradrenaline, β-adrenoceptor mediated vasorelaxation and nitric oxide in large and small pulmonary arteries of the rat. Br. J. Pharmacol. 1997;122:1375–1384. doi: 10.1038/sj.bjp.0701528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REBICH S., DEVINE J.O., ARMSTEAD W.M. Role of nitric oxide and cAMP in β-adrenoceptor-induced pial artery vasodilation. Am. J. Physiol. 1995;268:H1071–H1076. doi: 10.1152/ajpheart.1995.268.3.H1071. [DOI] [PubMed] [Google Scholar]
- RUBANYI G., VANHOUTTE P.M. Endothelium-removal decreases relaxations of canine coronary arteries caused by β-adrenergic agonists and adenosine. J. Cardiovasc. Pharmacol. 1985;7:139–144. doi: 10.1097/00005344-198501000-00023. [DOI] [PubMed] [Google Scholar]
- RUBANYI G.M., ROMERO J.C., VANHOUTTE P.M. Flow-induced release of endothelium-derived relaxing factor. Am. J. Physiol. 1986;250:H1145–H1149. doi: 10.1152/ajpheart.1986.250.6.H1145. [DOI] [PubMed] [Google Scholar]
- STEINBERG S.F., JAFFE E.A., BILEZIKIAN J.P. Endothelial cells contain beta adrenoceptors. Naunyn Schmiedeberg's Arch. Pharmacol. 1984;325:310–313. doi: 10.1007/BF00504374. [DOI] [PubMed] [Google Scholar]
- SUMMERS R.J., MOLENAAR P., STEPHENSON J.A., JONES C.R. Autoradiographic localization of receptors in the mammalian cardiovascular system. Clin. Exp. Pharmacol. Physiol. 1987;14:437–447. doi: 10.1111/j.1440-1681.1987.tb00995.x. [DOI] [PubMed] [Google Scholar]