

Abstract
A possible mechanism for the action of the KATP channel opener diazoxide on the improvement of energy metabolism of ischaemic/reperfused hearts was examined.
Isolated, perfused rat hearts were subjected to 40 min ischaemia followed by 60 min reperfusion. Diazoxide at concentrations of 3 to 30 μM was present in the perfusion buffer for the last 15 min of pre-ischaemia.
Treatment of the perfused heart with diazoxide enhanced the post-ischaemic recovery of rate-pressure product, attenuated the post-ischaemic rise in left ventricular end-diastolic pressure, and suppressed the release of creatine kinase and purine nucleosides and bases from the reperfused heart. Treatment of the heart with diazoxide also restored myocardial ATP and creatine phosphate and attenuated the decrease in mitochondrial oxygen consumption rate after reperfusion. This attenuation was maintained at the end of ischaemia as well as at the end of reperfusion.
In another set of experiments, myocardial skinned bundles were incubated for 30 min under hypoxic conditions in the presence and absence of diazoxide, and then the mitochondrial oxygen consumption rate was determined. Hypoxia induced a decrease in the mitochondrial oxygen consumption rate of the skinned bundles to approximately 40% of the pre-hypoxic value. In contrast, treatment of the bundles with 30 μM diazoxide preserved the normal mitochondrial oxygen consumption rate during hypoxia. This effect was abolished concentration-dependently by the combined treatment with either the KATP channel blocker glibenclamide or 5-hydroxydecanoate.
These results suggest that diazoxide is capable of attenuating ischaemia/reperfusion injury of isolated perfused hearts due to preservation of mitochondrial function during ischaemia.
Keywords: Diazoxide, glibenclamide, 5-Hydroxydecanoate, high-energy phosphate, ischaemia/reperfusion injury, KATP channel blocker, KATP channel opener, mitochondrial KATP channel, mitochondria
Introduction
ATP-sensitive potassium channel (KATP channel) openers, such as diazoxide, cromakalim, nicorandil, and pinacidil, were first identified as anti-anginal or anti-hypertensive drugs. The mechanism for their action is attributed to the opening of KATP channels, followed by inhibition of Ca2+ influx in smooth muscles, which then leads to vasodilatation.
Recently, there is increasing evidence that potassium channel openers are not only vasodilators but also protectors of heart muscle from injury. Several researchers have proposed that cardiac KATP channels may be involved in cardioprotection against ischaemia/reperfusion-induced injury (McPherson et al., 1993; Grover, 1994; Liang, 1997; Gross, 1999). In particular, opening of KATP channels has been shown to reduce infarct size, to mimic ischaemic preconditioning, and to enhance post-ischaemic recovery of cardiac contractile force (McPherson et al., 1993; Hearse, 1995; Grover, 1997; Schwarz et al., 1997). In addition, numerous studies have suggested that KATP channel blockers such as glibenclamide abolish the cardioprotective effects of KATP channel openers.
The primary mechanism responsible for these observations is thought to be the opening of sarcolemmal KATP channels. This action would be followed by shortening of action potential duration (APD), and a reduction in calcium loading in myocytes, whose events would be cardioprotective. However, several studies indicate that abbreviation of APD is not necessary for the protection conferred by potassium channel openers (Grover et al., 1995a,1995b). In addition, 5-hydroxydecanoate (5-HD), which is believed to be a KATP channel blocker, did not block the cromakalim-induced sarcolemmal KATP currents, but did block the anti-ischaemia action (McCullough et al., 1991). These experimental results suggest that the protective effects of potassium channel openers are exerted by a mechanism that is unrelated to the opening of sarcolemmal KATP channels.
Recently, a possible action of KATP channel openers on mitochondrial function was proposed. Diazoxide and cromakalim were shown to improve post-ischaemic functional recovery in isolated rat hearts and to stimulate KATP channel activity in purified mitochondria (Garlid, 1996; Garlid et al., 1997). It was also reported that sarcolemmal KATP channels were substantially insensitive to diazoxide, whereas they were activated by cromakalim to a degree similar to that found for mitochondrial KATP channels. In addition, 5-HD completely abolished the diazoxide-induced protective effect and inhibition of mitochondrial K+ flux (Garlid et al., 1997; Liu et al., 1998). Thus, it is possible to consider that diazoxide protected ischaemic hearts by opening of mitochondrial KATP channels rather than sarcolemmal KATP channels and that 5-HD appears to be a selective blocker of mitochondrial KATP channels.
It has been shown that mitochondrial KATP channels present on the inner membrane regulate mitochondrial volume and energetics (Inoue et al., 1991; Paucek et al., 1992; Garlid et al., 1996; Holmuhamedov et al., 1998). Regulation of volume changes is important for metabolic control in mitochondria (Halestrap, 1989). Thus, the effects of KATP channel openers on the mitochondria may be linked indirectly to improvement of cardiac energy production. However, the mechanism by which opening of mitochondrial KATP channels is linked to cardioprotection remains unclear. In the present study, we sought to determine whether or not diazoxide may protect cardiac mitochondrial function against ischaemia/reperfusion injury through its action on the energy-producing ability of mitochondria.
Methods
Animals
Male Wistar rats, weighing 230–285 g, were used in the present study. The animals were conditioned at 23±1°C with a constant humidity of 55±5%, a cycle of 12 h light and 12 h darkness, and were given free access to food and tap water according to the Guide for the Care and Use of Laboratory Animals as promulgated by the National Research Council.. The protocol of this study was approved by The Committee of Animal Use and Welfare of Tokyo University of Pharmacy and Life Science.
Agents
Diazoxide, glibenclamide, and 5-hydroxydecanoate were purchased from Sigma Chemical Co. (St Louis, MO, U.S.A.).
Perfusion of hearts
The rats were anaesthetized with diethyl ether. The hearts were rapidly isolated, transferred to a Langendorff apparatus, and perfused at a constant perfusion pressure of 70 mmHg with Krebs-Henseleit bicarbonate buffer of the following composition (mM): NaCl, 120; KCl, 4.8; KH2PO4, 1.2; MgSO4, 1.2; CaCl2, 1.25; NaHCO3, 25; and glucose, 11. The perfusion buffer, at 37°C, was equilibrated with a gas mixture of 95% O2 and 5% CO2 to pH 7.4. A latex balloon, with an uninflated diameter of 3.7 mm and connected to a pressure transducer (TP-200, Nihonkohden, Tokyo, Japan), was inserted into the left ventricular cavity through the mitral opening and secured by ligation. A pressure 5-mmHg of the initial left ventricular end-diastolic pressure (LVEDP) was loaded onto the perfused heart. Left ventricular developed pressure (LVDP), a convenient marker of cardiac contractile function, was monitored by a pressure transducer connected to a carrier amplifier (AP-621G, Nihonkohden) throughout the experiment. Heart rate (HR) was measured by means of a heart rate counter AT-601G, Nihonkohden, Tokyo). The flow rate of the perfusate was monitored by an electromagnetic blood flow meter (MFV-3100, Nihonkohden, Tokyo) connected just proximal to the aortic cannula. Haemodynamic parameters were recorded on a thermal pen recorder (WT-645G, Nihonkohden, Tokyo). The rate-pressure product (RPP) was determined by multiplying HR by LVDP.
After a 30-min equilibration, an additional 15-min perfusion was carried out, and then the perfusion was stopped (ischaemia). The heart was immediately submerged in an organ bath filled with the Krebs-Henseleit bicarbonate buffer in which the 11 mM glucose had been replaced with 11 mM Tris/HCl. This buffer had been previously equilibrated with a gas mixture of 95% N2 and 5% CO2 to pH 7.4 and maintained at 37°C during the experiment to avoid hypothermia-induced cardioprotection. After 40 min of ischaemia, the buffer in the organ bath was drained, and the hearts were reperfused for 60 min at 37°C with the Krebs-Henseleit bicarbonate buffer equilibrated with a gas mixture of 95% O2 and 5% CO2. The hearts were allowed to beat spontaneously throughout the experiments. For the purpose of comparison, hearts were perfused for 100 min under normoxic conditions, the time equal to that for ischaemic plus reperfusion (normoxic group).
Treatment of the perfused hearts with different concentrations of diazoxide (3 to 30 μM) was carried out by perfusing the hearts with the agent in Krebs-Henseleit bicarbonate buffer for the last 15 min of pre-ischaemia. Alternatively, diazoxide at 30 μM was perfused from the onset of reperfusion to the end of reperfusion. One preparation out of seven in the case of 10 or 30 μM diazoxide-treated hearts was excluded from data analysis due to sustained ventricular tachycardia during reperfusion. Diazoxide was dissolved in dimethylsulfoxide (DMSO) and diluted in the Krebs-Henseleit buffer. The final concentration of DMSO in the Krebs-Henseleit buffer was 0.04% (v v−1). Vehicle control perfusions were performed with the same concentration of DMSO alone. We previously confirmed that this concentration of DMSO had no effect on cardiac function under our experimental conditions.
Examination of perfusate
The perfusate eluted from the heart was collected to determine creatine kinase (CK) activity according to the method of Bergmeyer et al. (1970). The perfusate was also used for determination of purine nucleosides and bases (ATP metabolites) such as adenosine, inosine and hypoxanthine by the HPLC method described previously (Takeo et al., 1988).
Determination of myocardial energy metabolites
After appropriate sequences of perfusion, the hearts were quickly frozen with liquid nitrogen. The frozen ventricle was pulverized and mixed with 0.3 N HClO4 and 0.25 mM EDTA under liquid nitrogen cooling. The extract was centrifuged at 8000×g for 15 min at 4°C, and the resulting supernatant was sampled for determination of myocardial ATP, ADP, AMP, and creatine phosphate (CP). Myocardial adenine nucleotides were measured by the HPLC method described previously (Takeo et al., 1996). Myocardial CP was converted to ATP by the enzymatic method of Lowry & Passonneau (1972). Energy charge was calculated from the formula (ATP+0.5×ADP)/(ATP+ADP+AMP).
Mitochondrial oxygen consumption rate
The mitochondrial oxygen consumption rate was determined by the method of Sanbe et al. (1993), which is a modification of the method of Saks et al. (1989). After appropriate sequences of perfusion, the vehicle- and diazoxide-treated hearts were quickly removed from the perfusion apparatus. Myocardial bundles, 0.3 to 0.4 mm in diameter and 3 to 4 mm in length, were prepared from the left ventricular free wall by use of a McIlwain Tissue Chopper (Mickle Lab. Engineering Co., NY, U.S.A.) and transferred into relaxing medium A of the following composition (mM): EGTA, 10; MgSO4, 3; taurine, 20; dithiothreitol, 0.5; imidazole, 20; potassium 2-(N-morpholino)-ethanesulphonate, 160; ATP, 5; CP, 15 (pH 7.0). The bundles were incubated for 20 min in 1 ml of medium A containing 75 μg ml−1 saponin. After incubation, the bundles (skinned bundles) were washed for 10 min in fresh medium A to remove the saponin. All procedures were carried out at 4°C. The oxygen consumption rate of skinned bundles was determined by means of a Clark-type electrode connected to an Oxygraph (Central Kagaku, Tokyo) containing skinned bundles in 1.0 ml of medium B (medium A without ATP and CP but supplemented with 0.5% bovine serum albumin) at 30°C with continuous and gentle stirring. The basal oxygen consumption rate was measured following the addition of 5 mM glutamate, 3 mM malate, and 3 mM KH2PO4. Total oxygen consumption rate was measured after further addition of 1 mM ADP and 7.5 mM creatine. The maximal velocity of oxygen consumption rate (Vmax) of skinned bundles was taken as the difference between total and basal oxygen consumption rates. After determination of the oxygen consumption rate, the skinned bundles were solubilized with 0.5 ml of 2 N NaOH for 30 min at 60°C, and then the protein concentration was determined according to the method of Lowry et al. (1951). The mitochondrial oxygen consumption rate was expressed as nano-atoms of oxygen consumed per min per mg protein.
Hypoxic incubation of skinned bundles
To determine whether diazoxide directly affects mitochondrial function or not, we prepared skinned bundles of the left ventricular free wall from non-perfused hearts. The bundles were placed into 2 ml of medium B in a tightly sealed chamber at 30°C for 30 min. The hypoxic condition was induced by saturation of the chamber with 100% N2 at the flow rate of 20 ml min−1 as described previously (Hayashi et al., 1995). After a 30-min hypoxic or normoxic incubation, the skinned bundles were quickly transferred to the glass cell, and then their oxygen consumption rates were determined as described above. Diazoxide or vehicle (0.1% (v v−1) DMSO) was added to the incubation medium in the chamber before addition of the skinned bundles. A combination of either diazoxide+glibenclamide or diazoxide+5-HD was also examined in the same manner. The final concentration of DMSO in the medium B was 0.1%. We previously confirmed that this concentration of DMSO had no effect on oxygen consumption rates under hypoxic or normoxic conditions for at least 120 min.
Statistics
The results were expressed as the means±s.e.mean. The statistical significance of differences in HR, coronary flow, LVDP, RPP, and LVEDP just prior to 40 min ischaemia and at the end of reperfusion between the hearts treated with diazoxide and those treated with vehicle was evaluated by analysis of variance (ANOVA), followed by Bonferroni's or Dunnett's multiple comparison. Unpaired Student's t-test was performed for comparison between two groups. Differences with a probability of less than 5% were considered to be statistically significant (P<0.05).
Results
Cardiac function of perfused hearts
The effects of diazoxide on pre-ischaemic and post-ischaemic cardiac function and coronary flow are shown in Table 1, and the time course of changes in RPP of ischaemic/reperfused hearts treated with 3 to 30 μM diazoxide is shown in Figure 1. The basal (initial) values for HR, coronary flow, LVDP, RPP, and LVEDP were similar in the vehicle- and diazoxide-treated groups. Diazoxide had little effect on the heart rate before ischaemia. At that time, however, the drug at 10 and 30 μM significantly increased coronary flow with a concomitant increase in RPP. RPP declined to zero within 2.5 min after the onset of ischaemia. Thereafter, it remained at that value during ischaemia. During reperfusion, bradycardia and contractile dysfunction were observed in vehicle-treated hearts, suggesting the genesis of severe ischaemic/reperfusion injury. The RPP of the heart recovered to approximately 23% of the pre-ischaemic value by the end of the reperfusion period. In contrast, RPPs of the hearts treated with 10 and 30 μM diazoxide were significantly recovered to approximately 64 and 86%, respectively, of the pre-ischaemic value at the end of reperfusion. Coronary reflow of the vehicle-treated heart was reduced at the end of reperfusion, and the reflow was significantly improved by only 30 μM diazoxide. The LVEDP of the vehicle-treated heart began to rise at 10 min after the onset of ischaemia and reached its peak level at approximately 20 min of ischaemia. It was further increased upon reperfusion; the maximum level having been reached at 5 min after the onset of reperfusion. Although the LVEDP gradually declined during reperfusion, this high level of LVEDP was sustained throughout reperfusion. In contrast, treatment with various concentrations of diazoxide attenuated the rise in LVEDP during reperfusion in a concentration-dependent manner, but not during ischaemia. When perfused hearts were treated with 30 μM diazoxide only during reperfusion, the recovery of RPP was not enhanced at the end of the reperfusion period (6.6±2.0 mmHg min−1×10−3 for vehicle-treated heart versus 6.6±1.0 mmHg min−1×10−3 for 30 μM diazoxide-treated heart, n=5 each).
Table 1.
Effects of diazoxide on pre-ischaemic and post-ischaemic cardiac function and coronary flow in isolated rat hearts
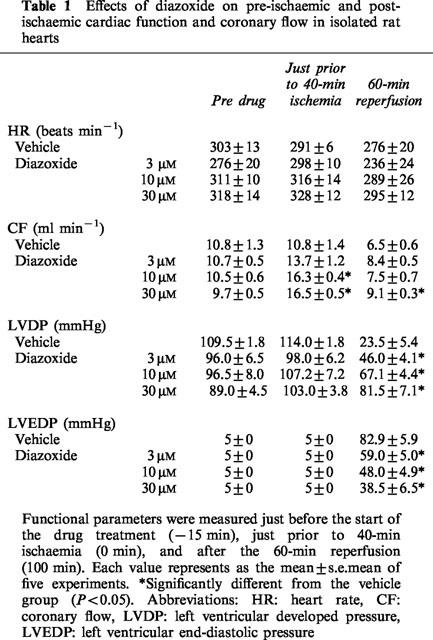
Figure 1.

Time course of changes in rate pressure product of the ischaemic/reperfused untreated heart and that treated with 3, 10, or 30 μM diazoxide. Each value represents the mean±s.e.mean of five experiments. Treatment with diazoxide was conducted for the last 15 min of pre-ischaemia (Agent) or only during reperfusion.
Since treatment with 30 μM diazoxide resulted in the maximum recovery of RPP and LVEDP, we employed 30 μM diazoxide in the subsequent experiments to further characterize the pharmacological profiles of the agent.
Release of CK and ATP metabolites
To determine the release of CK from perfused hearts, we collected the perfusate from the hearts and measured the CK activity in the perfusate (left panel in Figure 2). During the 15-min period of pre-ischaemic perfusion, CK activity in the perfusate was less than 4 nmol NADPH min−1 g−1 wet tissue regardless of the presence or absence of diazoxide (n=6 each). CK activity in the perfusate of the heart perfused for 100 min under normoxic conditions was less than 6 nmol NADPH min−1 g−1 wet tissue regardless of treatment with or without 30 μM diazoxide. CK activity in the perfusate from the vehicle-treated heart markedly increased during reperfusion, and treatment with diazoxide attenuated this release.
Figure 2.

Creatine kinase activity (left panel) and ATP metabolites (right panel) in the perfusate eluted from ischaemic/reperfused hearts treated with 30 μM diazoxide prior to ischaemia. Each value represents the mean±s.e.mean of six experiments. #Significantly different from the corresponding normoxic group (P<0.05). *Significantly different from the corresponding vehicle ischaemic/reperfused group (P<0.05).
The amount of ATP metabolites released during reperfusion was also determined (right panel in Figure 2). The release of ATP metabolites was less than 0.1 μmol g−1 wet tissue during pre-ischaemia (n=6). No appreciable release of ATP metabolites was observed in hearts perfused for 100 min under normoxic conditions (n=6). The release of ATP metabolites was markedly increased during reperfusion following ischaemia. Treatment with diazoxide partially suppressed the release of ATP metabolites during reperfusion (to approximately 78% of the value for the vehicle-treated heart, n=6, P<0.01).
Myocardial energy metabolites
Myocardial energy metabolites such as ATP, ADP, AMP and CP were determined in the heart treated with diazoxide to examine the myocardial energy profile (Table 2). Myocardial ATP and CP contents at the end of the pre-ischaemia were 24.9±0.7 and 35.2±1.0 μmol g−1 dry tissue, respectively (n=6). There were no significant differences in the metabolite contents at 100 min of normoxia as compared with the pre-ischaemic values (at 0 min). Myocardial ATP and CP contents at the end of the ischaemia were approximately 6 and 8% of the pre-ischaemic values, respectively. Reperfusion of the ischaemic heart resulted in little restoration of myocardial ATP and CP contents (to approximately 21 and 27% of the pre-ischaemic values, respectively). Treatment with 30 μM diazoxide for the last 15 min of the pre-ischaemia did not alter the pre-ischaemic value of these metabolites, nor did it prevent the decreases in myocardial ATP and CP during ischaemia. During reperfusion, however, myocardial ATP and CP contents were restored to approximately 46 and 78% of their pre-ischaemic values, respectively, by treatment with 30 μM diazoxide (n=6). The myocardial ADP content declined after sustained ischaemia and recovered after reperfusion in vehicle-treated hearts, and treatment with diazoxide did not alter this profile. Forty minutes of ischaemia markedly increased AMP levels by 36 and 28 fold of the pre-ischaemic values in vehicle- and diazoxide-treated hearts. After reperfusion, the myocardial AMP content was reversed in both hearts. The energy charge significantly decreased after ischaemia and recovered during reperfusion in both vehicle- and diazoxide-treated hearts. However, the energy charge returned to a greater degree in the diazoxide group than in the vehicle group after 60 min of reperfusion, as shown in Table 2.
Table 2.
Effects of diazoxide (30 μM) on high-energy phosphate content and energy charge of perfused hearts

Mitochondrial oxygen consumption rate of perfused hearts
The mitochondrial oxygen consumption rate of the left ventricular muscle of the hearts treated with diazoxide was determined next (Figure 3). The rate for pre-ischaemic hearts was 58.56±2.14 n atom O min−1 mg−1 protein (n=5). There were no significant differences in the mitochondrial oxygen consumption rate of perfused hearts under normoxic conditions regardless of treatment or not with diazoxide. The mitochondrial oxygen consumption rate of the vehicle-treated heart under ischaemic conditions was significantly lower than that of the normoxic heart (approximately 43% of the value for the normoxic heart, n=5). A further decline in the mitochondrial oxygen consumption rate was observed upon reperfusion (approximately 21% of the value for the normoxic heart, n=5). In contrast, treatment with diazoxide preserved the mitochondrial oxygen consumption rate at the ends of both ischaemia and reperfusion (approximately 90 and 89% of the value for the normoxic heart, respectively, n=5 each).
Figure 3.

Mitochondrial oxygen consumption rate of the left ventricular skinned bundles isolated from hearts at the end of ischaemia (at 40 min, left panel) and at the end of reperfusion (at 100 min, right panel) of hearts treated with 30 μM diazoxide. Each value represents the mean±s.e.mean of five experiments. #Significantly different from the corresponding normoxic group (P<0.05). *Significantly different from the corresponding vehicle ischaemic/reperfused hearts group (P<0.05).
Mitochondrial oxygen consumption rate under hypoxic conditions
To determine whether or not diazoxide may protect the mitochondrial oxygen consumption capacity from hypoxic injury, we prepared skinned bundles from the left ventricular free wall of normal rats and incubated them under hypoxic conditions. At first, to determine the experimental conditions of hypoxic incubations of skinned bundles, we measured the mitochondrial oxygen consumption rate after 15 to 120 min of hypoxia. The rate was reduced in a time-dependent manner between 0 and 30 min of hypoxia (data not shown). Thus, we employed 30-min hypoxia in subsequent experiments. Under this condition, we measured the mitochondrial oxygen consumption rate in the absence and in the presence of 3 to 30 μM diazoxide (Figure 4). After the 30-min hypoxic incubation, the mitochondrial oxygen consumption rate was decreased to approximately 40% of the value for the normoxic skinned bundles (n=6). When the skinned bundles were incubated in the presence of 1 to 30 μM diazoxide, the hypoxia-induced decrease in mitochondrial oxygen consumption rate was attenuated in a concentration-dependent manner (n=3 to 6).
Figure 4.

Effects of different concentrations of diazoxide on the mitochondrial oxygen consumption rate of myocardial skinned bundles subjected to 30-min normoxia or hypoxia. Each value represents the mean±s.e.mean of 3–6 experiments. #Significantly different from the corresponding normoxic group (P<0.05). *Significantly different from the corresponding 0 μM diazoxide (P<0.05).
The preservation of mitochondrial oxygen consumption capacity by treatment with 30 μM diazoxide was significantly abolished by the combined treatment with either 1 to 10 μM glibenclamide (n=3 to 4, left panel in Figure 5) or 10 or 30 μM 5-HD (n=3 to 5, right panel in Figure 5).
Figure 5.

Effects of various concentrations of glibenclamide (left panel) and 5-hydroxydecanoate (5-HD, right panel) on the mitochondrial oxygen consumption rate of myocardial skinned bundles subjected to 30-min hypoxia in the presence of 30 μM diazoxide. For the purpose of comparison, the rates for 30 min normoxia and 30-min hypoxia in the absence and in the presence of 30 μM diazoxide are depicted. Each value represents the mean±s.e.mean of 3–6 experiments. #Significantly different from the normoxic group (P<0.05). *Significantly different from the vehicle group (P<0.05). †Significantly different from the diazoxide-treated group (P<0.05).
Discussion
In the present study, we observed that treatment with diazoxide during pre-ischaemia markedly enhanced the post-ischaemic contractile recovery of ischaemic/reperfused hearts. The beneficial effects of diazoxide on cardiac function are comparable to those reported by other investigators (Garlid et al., 1997). The improvement was associated with restoration of myocardial high-energy phosphates and attenuation of the release of CK as well as ATP metabolites. There are controversial reports regarding the correlation between the recovery of function and intracellular ATP levels following myocardial ischaemia/reperfusion. Docherty et al. (1999) reported that there was a correlation between intracellular ATP levels and functional recovery following ischaemia/reperfusion and that this correlation, while significant, was quite modest. In contrast, Neely & Grotyohann (1984) suggested that there was no relation between intracellular ATP levels and functional recovery following ischaemia/reperfusion. Thus, the cause and effect relationship between recovery of post-ischaemic contractile function and myocardial energy levels still remains unclear. Despite such uncertainty, it is well recognized that cardiac contraction basically requires myocardial high-energy phosphates (Katz, 1977). Thus, appreciable levels of high-energy phosphates in the diazoxide-treated, reperfused heart may be substantially beneficial for the recovery of myocardial contractility of the reperfused heart.
The attenuation of the release of CK and ATP metabolites suggests that an ischaemia-induced increase in the membrane permeability of macromolecules such as CK protein across cell membranes and/or induction of cardiac cell necrosis in the reperfused heart was suppressed by treatment with diazoxide. The finding also suggests that relatively small restoration of myocardial ATP by treatment with the agent may be due to the loss of ATP metabolites from the reperfused heart, since ATP metabolites, such as adenosine and inosine, are substrates for the salvage synthesis of ATP in hearts. In contrast, the myocardial CP content of the heart treated with diazoxide was restored to 78% of the pre-ischaemic value during reperfusion. These results suggest that the ability to produce energy in mitochondria may be appreciably preserved in the ischaemic heart treated with diazoxide.
Recently, it has been shown that there is an isoform of the KATP channel on the mitochondrial inner membrane, where the channels regulate mitochondrial electron transport, volume, and energetics (Inoue et al., 1991; Paucek et al., 1992; Garlid et al., 1996; Holmuhamedov et al., 1998). Several investigators have suggested that mitochondrial KATP channels may be involved in cardioprotection against ischaemia/reperfusion injury (Jovanovic et al., 1998; Liu et al., 1998). In addition, Garlid et al. (1996; 1997) showed that diazoxide is 2000 fold more selective for mitochondrial than for sarcolemmal KATP channels. Therefore, it appears that diazoxide may protect ischaemic hearts through promoting mitochondrial rather than sarcolemmal function. Thus, we determined whether or not diazoxide may protect the cardiac mitochondrial function against ischaemia/reperfusion injury. In the present study, we used a method to determine mitochondrial functional capacity in skinned bundles (Saks et al., 1989). This method is efficient to study the quantitative relationship between parameters of mitochondrial respiration and cardiac function in each preparation. At the end of the ischaemia, the oxygen consumption rate of the myocardial skinned bundles decreased, whereas this decrease was partially restored by reperfusion. Diazoxide preserved this mitochondrial function of the heart during ischaemia as well as during reperfusion. Basically, the experimental conditions for measurement of mitochondrial oxygen consumption rate are the same as those for that of mitochondrial oxidative phosphorylation activity (Saks et al., 1989). Thus, these results suggest that diazoxide is capable of preserving mitochondrial oxidative phosphorylation activity during ischaemia and reperfusion.
Although we observed that treatment with diazoxide attenuated the ischaemia-induced decrease in mitochondrial activity, it remained to be determined whether diazoxide may directly affect the mitochondria or not. Therefore, this possibility was addressed in another set of experiments. Skinned myocardial bundles were prepared from the left ventricular free wall of normal rats and then exposed to 30-min hypoxia in the presence and absence of diazoxide. Incubation of the skinned bundles under hypoxic conditions resulted in a decrease in mitochondrial oxygen consumption rate, which was attenuated by diazoxide in a concentration-dependent manner. This effect of diazoxide was abolished in the presence of either KATP channel blocker glibenclamide or 5-HD in a concentration-dependent manner. Glibenclamide is known to block KATP channels on both cell membranes and mitochondrial inner membranes (Paucek et al., 1992; Szewczyk et al., 1995; Garlid et al., 1996). In contrast, 5-HD does not affect cromakalim-induced shortening of action potential duration or the cromakalim-induced decrease in outward whole-cell potassium currents (McCullough et al., 1991). Both events are considered to be related to ionic currents present in the cell membrane. In contrast, Garlid et al. (1997) showed that 5-HD inhibited the ability of diazoxide to open reconstituted mitochondrial KATP channels in a relatively selective manner. Thus, 5-HD appears to affect mitochondrial KATP channels rather than sarcolemmal KATP channels. The above results suggest that diazoxide preserves mitochondrial oxygen consumption rate under hypoxic conditions, probably through opening mitochondrial KATP channels.
In the present study, when perfused hearts were treated with diazoxide only during reperfusion, the recovery of RPP was not enhanced. In addition, it was at the end of ischaemia that the mitochondrial oxygen consumption rate of the vehicle-treated heart was decreased. These findings positively indicate that the mitochondrial oxygen consumption capacity of the heart had already decreased under ischaemic conditions prior to reperfusion injury and suggest that the presence of diazoxide during the ischaemic period is necessary to elicit the improvement in the recovery of cardiac function. If so, ischaemia/reperfusion-induced damage to cardiac function and metabolism, at least in part, might be initiated and/or promoted by the impairment in mitochondrial function during the ischaemic period, but not during reperfusion.
It is still unclear how opening of mitochondrial KATP channels during the ischaemic period might protect against the following reperfusion damage. During the steady state, respiration is balanced by K+ influx into mitochondria through the K+/H+ anti-porter. One possibility is that the opening of mitochondrial KATP channels may cause a net influx of K+ and partially dissipate the mitochondrial membrane potential (Garlid, 1996). This may be beneficial against ischaemia-induced mitochondrial dysfunction, because partial dissipation of the electrical gradient decreases the paradoxical Ca2+ influx into mitochondria (Liu et al., 1998; Holmuhamedov et al., 1999). It is likely that preservation of mitochondrial function during the ischaemic period may contribute to the better recovery of myocardial high-energy phosphate and myocardial ventricular function particularly at an early period of reperfusion. However, an increased oxygen consumption rate does not necessarily result in an increased ATP synthesis (Liu et al., 1996). This discrepancy remains to be addressed.
An increase in coronary flow during pre-ischaemic period was seen in the heart treated with diazoxide. Since this agent was shown to dilate peripheral vascular smooth muscles (Nakai, 1994; Xia & Han, 1994; Quayle et al., 1997), this effect may be anticipated to lead to recovery of post-ischaemic cardiac function. To determine this possibility, we examined the relationship between coronary flow at the end of pre-ischaemia and recovery of RPP at the end of reperfusion in hearts treated with 3 to 30 μM diazoxide. There was no significant relationship between these two parameters (r=0.384, n=15, P>0.05). It is, therefore, unlikely that this increase in coronary flow contributes much to the cardioprotective action of diazoxide.
In conclusion, the present study has shown that diazoxide is capable of protecting the myocardium against ischaemia/reperfusion injury and enhancing the recovery of post-ischaemic myocardial contractile function in association with restoration of myocardial high-energy phosphates. The mechanism underlying this cardioprotective effect of diazoxide may be attributed to preservation of mitochondrial function during ischaemic period, probably via opening of mitochondrial KATP channels. The major findings in the present study are that the mitochondrial oxygen consumption capacity of the heart had already decreased at the end of ischaemia, prior to reperfusion injury, and that diazoxide protected the mitochondrial oxygen consumption capacity from this ischaemic injury.
Abbreviations
- APD
action potential duration
- CK
creatine kinase
- CP
creatine phosphate
- ECP
energy charge potential
- 5-HD
5-hydroxydecanoate
- KATP channel
ATP-sensitive potassium channel
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- RPP
rate-pressure product
References
- BERGMEYER H.U., RICH W., BUTHER H., SCHMIDT E., HILLMAN G., KREU F.H., STAMM D., LANG H., SZASZ G., LAUE D. Standardization of methods for estimation of enzyme activity in biological fluids. Z. Klin. Chem. Biochem. 1970;8:658–669. [Google Scholar]
- DOCHERTY J.C., KUZIO B., AILVESTER J.A., BOWES J., THIEMERMANN C. An inhibitor of poly(ADP-ribose) synthetase activity reduces contractile dysfunction and preserves high energy phosphate levels during reperfusion of the ischaemic rat heart. Br. J. Pharmacol. 1999;127:1518–1524. doi: 10.1038/sj.bjp.0702705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARLID K.D. Cation transport in mitochondria: the potassium cycle. Biochim. Biophys. Acta. 1996;1275:123–126. doi: 10.1016/0005-2728(96)00061-8. [DOI] [PubMed] [Google Scholar]
- GARLID K.D., PAUCEK P., YAROV-YAROVOY V., MURRAY H.N., DARBENZIO R.B., D'ALONZO A.J., LONGE N.J., SMITH M.A., GROVER G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- GARLID K.D., PAUCEK P., YAROV-YAROVOY V., SUN X., SCHINDLER P.A. The mitochondrial KATP channel as a receptor for potassium channel openers. J. Biol. Chem. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- GROSS G.J., FRYER R.M. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ. Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- GROVER G.J. Protective effects of ATP-sensitive potassium channel openers in experimental myocardial ischaemia. J. Cardiovasc. Pharmacol. 1994;24 Suppl:S18–S27. [PubMed] [Google Scholar]
- GROVER G.J. Pharmacology of ATP-sensitive potassium channel (KATP) openers in models of myocardial ischemia and reperfusion. Can. J. Physiol. Pharmacol. 1997;75:309–315. doi: 10.1139/cjpp-75-4-309. [DOI] [PubMed] [Google Scholar]
- GROVER G.J., D'ALONZO A.J., HESS T., SLEPH P.G., DARBENZIO R.B. Glybride-reversible cardioprotective effect of BMS-180448 is independent of action potential shortening. Cardiovasc. Res. 1995a;30:731–738. [PubMed] [Google Scholar]
- GROVER G.J., D'ALONZO A.J., PARHAM C.S., DARBENZIO R.B. Cardioprotection with the KATP opener cromakalim is not correlated with ischemic myocardial action potential duration. J. Cardiovasc. Pharmacol. 1995B;26:145–152. doi: 10.1097/00005344-199507000-00023. [DOI] [PubMed] [Google Scholar]
- HALESTRAP A.P. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim. Biophys. Acta. 1989;973:355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- HAYASHI M., NASA Y., TANONAKA K., SASAKI H., MIYAKE R., HAYASHI J., TAKEO S. The effects of long-term treatment with eicosapentaenoic acid and docosahexaenoic acid on hypoxia/reoxygenation injury of isolated cardiac cells in adult rats. J. Mol. Cell. Cardiol. 1995;27:2031–2041. doi: 10.1016/0022-2828(95)90024-1. [DOI] [PubMed] [Google Scholar]
- HEARSE D.J. Activation of ATP-sensitive potassium channels: a novel pharmacological approach to myocardial protection. Cardiovasc. Res. 1995;30:1–17. [PubMed] [Google Scholar]
- HOLMUHAMEDOV E.L., JOVANOVIC S., DZEJA P.P., JOVANOVIC A., TERZIC A. Mitochondrial ATP-sensitive K+ channels modulate cardiac function. Am. J. Physiol. 1998;275:H1567–H1576. doi: 10.1152/ajpheart.1998.275.5.H1567. [DOI] [PubMed] [Google Scholar]
- HOLMUHAMEDOV E.L., WANG L., TERZIC A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 1999;519:347–360. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INOUE I., NAGASE H., KISHI K., HIGUTI T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- JOVANOVIC A., JOVANOVIC S., LORENZ E., TERZIC A. Recombinant cardiac ATP-sensitive K+ channel subunits confer resistance to chemical hypoxia-reoxygenation injury. Circulation. 1998;98:1548–1555. doi: 10.1161/01.cir.98.15.1548. [DOI] [PubMed] [Google Scholar]
- KATZ A.M. Energetics of muscle Physiology of Heart 1977Raven Press: New York; 25–34.ed. Katz, A.M. pp [Google Scholar]
- LIANG B.T. Protein kinase C-mediated preconditioning of cardiac myocytes: role of adenosine receptor and KATP channel. Am. J. Physiol. 1997;273:H847–H853. doi: 10.1152/ajpheart.1997.273.2.H847. [DOI] [PubMed] [Google Scholar]
- LIU B., ALAOUI-TALIBI Z.E., CLANACHAN A.S., SHULZ R., LOPASCHUK G.D. Uncoupling of contractile function from mitochondrial TCA cycle activity and MVO2 during reperfusion of ischaemic heart. Am. J. Physiol. 1996;270:H72–H80. doi: 10.1152/ajpheart.1996.270.1.H72. [DOI] [PubMed] [Google Scholar]
- LIU Y., SATO T., O'ROURKE B., MARBAN E. Mitochondrial ATP-dependent potassium channels. Novel effectors of cardioprotection. Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., PASSONNEAU J.R. A collection of metabolite assays A Flexible System of Enzymatic Analysis 1972Academic Press: New York; 120–152.eds. Lowry, O.H., Passonneau, J.R. pp [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;261:6300–6306. [PubMed] [Google Scholar]
- MCCULLOUGH J.R., NORMANDIN D.E., CONDER M.L., SLEPH P.G., DZWONCZYK S., GROVER G.J. Specific block of the anti-ischemic actions of cromakalim by sodium 5-hydroxydecanoate. Circ. Res. 1991;69:949–958. doi: 10.1161/01.res.69.4.949. [DOI] [PubMed] [Google Scholar]
- MCPHERSON C.D., PIERECE G.N., COLE W.C. Ischemic cardioprotection by ATP-sensitive K+ channels involves high-energy phosphate preservation. Am. J. Physiol. 1993;265:H1809–H1818. doi: 10.1152/ajpheart.1993.265.5.H1809. [DOI] [PubMed] [Google Scholar]
- NEELY J.R., GROTYOHANN L.W. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischaemic heart. Circ. Res. 1984;55:816–824. doi: 10.1161/01.res.55.6.816. [DOI] [PubMed] [Google Scholar]
- NAKAI T. Effect of diazoxide on serum and tissue electrolyte levels in rats with deoxycorticosterone acetate-induced hypertension. J. Pharmacol. Sci. 1994;83:704–707. doi: 10.1002/jps.2600830522. [DOI] [PubMed] [Google Scholar]
- PAUCEK P., MIRONOVA G., MAHDI F., BEAVIA A.D., WOLDEGIORGIS G., GARLID K.D. Reconstitution and partial purification of the glibenclamide-dependent, ATP-dependent K+ channels from rat liver and beef mitochondria. J. Biol. Chem. 1992;267:26062–26069. [PubMed] [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. ATP-sensitive and inward-rectifying potassium channels in smooth muscle. Physiol. Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- SAKS V.A., KAPELKO V.I., KUPRIYANOV V.V., KUZNETSOV A.V., LAKOMKIN V.I., SHAROV V.G., JAVADOV S.A., SEPPET E.K., KAIRANE C. Quantitative evaluation of relationship between cardiac energy metabolism and post-ischemic recovery of contractile function. J. Mol. Cell. Cardiol. 1989;21 Suppl 1:67–78. doi: 10.1016/0022-2828(89)90839-0. [DOI] [PubMed] [Google Scholar]
- SANBE A., TANONAKA K., HANAOKA Y., KATOH T., TAKEO S. Regional energy metabolism of failing hearts following myocardial infarction. J. Mol. Cell. Cardiol. 1993;25:995–1013. doi: 10.1006/jmcc.1993.1113. [DOI] [PubMed] [Google Scholar]
- SCHWARZ E.R., WHYTE W.S., KLONER R.A. Ischemic preconditioning. Curr. Opin. Cardiol. 1997;12:475–481. [PubMed] [Google Scholar]
- SZEWCZYK A., WOJCIK G., NALECZ M.J. Potassium channel opener, RP66471, induces membrane depolarization of rat liver mitochondria. Biochem. Biophys. Res. Commun. 1995;207:126–132. doi: 10.1006/bbrc.1995.1162. [DOI] [PubMed] [Google Scholar]
- TAKEO S., MIYAKE K., TANONAKA K., TAKAGI N., TAKAGI K., KISHIMOTO K., SUZUKI M., KATSURAGI A., GOTO M. Beneficial effect of nebracetam on energy metabolism after microsphere-induced embolism in rat brain. Arch. Int. Pharmacodyn. 1996;331:232–245. [PubMed] [Google Scholar]
- TAKEO S., TANONAKA K., MIYAKE K., FUKUMOTO T. Role of ATP metabolites in induction of incomplete recovery of cardiac contractile force after hypoxia. Can. J. Cardiol. 1988;4:193–200. [PubMed] [Google Scholar]
- XIA Y., HAN C. Role of K+ channels in neuropeptide Y-induced vasoconstriction in rabbit cerebral basilar artery. Eur. J. Pharmacol. 1994;255:67–72. doi: 10.1016/0014-2999(94)90083-3. [DOI] [PubMed] [Google Scholar]