

Abstract
We have studied the inhibitory autoreceptor control of acetylcholine (ACh) release from rat motor nerve endings using an electrophysiological technique to quantify evoked ACh release in isolated hemidiaphragm muscles. Quantal ACh release (m) was estimated from the ratio of amplitudes of nerve evoked endplate currents and spontaneously occurring miniature endplate currents.
The nicotinic ACh receptor agonist cytisine (1 μM) decreased m at 0.5 Hz by around 20% but had no effect on m at 50 Hz. Changing the extracellular Ca2+ concentration from 1.8 mM to either 0.45 or 3.6 mM abolished the effect of cytisine on m at 0.5 Hz. The nicotinic ACh receptor antagonist hexamethonium (200 μM) increased m at 0.5 Hz by 15–20%.
The effects of cytisine and hexamethonium on m at 0.5 Hz were blocked by 10 μM verapamil, which itself significantly increased m. However, the effects of cytisine and hexamethonium on m at 0.5 Hz were not sensitive to 10 μM of the calmodulin antagonist, W-7. This concentration of W-7 attenuates effects on ACh release mediated by facilitatory prejunctional nicotinic ACh autoreceptors.
Our present observations are suggestive of actions of cytisine and hexamethonium to activate and inhibit respectively negative-feedback prejunctional nicotinic ACh autoreceptors. Further, they strengthen the case for the existence of two separate and independent autoregulatory mechanisms for the control of ACh release from motor nerve terminals and give a preliminary insight into the cellular mechanism involved in the autoinhibition of ACh release.
Keywords: Neuromuscular junction, endplate current, acetylcholine release, miniature endplate current, autoreceptor, verapamil, autoinhibition, cytisine, motor nerve terminal, hexamethonium
Introduction
At the mammalian skeletal muscle neuromuscular junction systems exist whereby the neurotransmitter acetylcholine (ACh) can act on prejunctional autoreceptors to either enhance or depress its own evoked released (reviewed in Prior et al., 1995). Evidence suggests that autoinhibition occurs predominantly at low frequencies of activation, is dependent on the extracellular calcium ion concentration ([Ca2+]o) and is probably mediated by a prejunctional nicotinic ACh receptor of a neuronal subtype (Tian et al., 1994; 1997). Thus, the neuronal-type selective nicotinic ACh receptor antagonists hexamethonium and methyllycaconitine, by blocking endogenous autoinhibition, increase ACh release, but only at low frequencies of stimulation and not in the presence of a reduced [Ca2+]o (Tian et al., 1997). Conversely, the muscle-type selective nicotinic ACh receptor antagonist vecuronium decreases ACh release at high frequencies of stimulation by blocking endogenous autofacilitation but has no effect on evoked ACh release at low frequencies of stimulation (Tian et al., 1994).
To date, the existence of nicotinic ACh receptor mediated autoinhibition in motor nerve terminals is based solely on the facilitatory actions of selected antagonists. However, implicit in the autoinhibition hypothesis is that low concentrations of a neuronal-type selective nicotinic ACh receptor agonist should be capable of inhibiting evoked ACh release in a frequency and [Ca2+]o-dependent manner. Further, if this were demonstrable then the possible involvement of intracellular messenger systems in the autoinhibition could be examined using appropriate agents. Therefore, the objectives of the present study were 2 fold. Firstly, to provide further evidence for the existence of prejunctional autoinhibitory nicotinic ACh receptors at the neuromuscular junction through the demonstration of an inhibitory effect of a neuronal-type selective nicotinic ACh agonist on evoked ACh release. Secondly, to gain insight into the intracellular mechanisms that might underlie the autoinhibitory effect of ACh at the neuromuscular junction.
To achieve the first objective, we determined the effects of the neuronal-type nicotinic ACh agonist cytisine on ACh release, evoked at 0.5 and 50 Hz, from rat motor nerve terminals. For the second objective, two issues were identified. An important part of identifying the cellular mechanisms involved in autoinhibition would be to demonstrate the independence of this process from high-frequency associated autofacilitation. Since the calmodulin antagonist W-7 blocks autofacilitation (Singh & Prior, 1998), it appeared logical to determine the calmodulin-sensitivity of autoinhibition. Additionally, given the [Ca2+]o-dependence of the facilitatory effect of hexamethonium on ACh release (Tian et al., 1997), we were interested in determining the role of nerve terminal membrane Ca2+-fluxes in autoinhibition. We studied the actions of verapamil on autoinhibition because verapamil-sensitive Ca2+-fluxes are known to exist in mammalian motor nerve terminal membranes (Anderson & Harvey, 1987; Penner & Dreyer, 1986) that are not the primary trigger for evoked release (Uchitel et al., 1992). Therefore, these Ca2+-fluxes might possess a role in the modulation of ACh release by inhibitory autoreceptors.
Methods
Muscle preparation
Male Sprague-Dawley rats (150–200 g) were killed according to U.K. Home Office guidelines by dislocation of the cervical vertebrae followed by exsanguination. Both hemidiaphragm muscle preparations, along with 15–20 mm of their associated phrenic nerve, were removed from the animals immediately following sacrifice. Once isolated, each muscle preparation was dissected clear of connective tissue and fixed to the Sylgard (Dow-Corning) base of a 3–5 ml tissue bath continuously perfused (10–12 ml min−1) with physiological solution at 32°C (see below for composition). To immobilize the muscle for electrophysiological recording, a ‘cut-muscle' preparation was used (Barstad & Lilliheil, 1968). Transverse sectioning of the muscle fibres 2–3 mm either side of the main intramuscular nerve branches initially reduced the resting membrane potential in the motor endplate region to around −40 mV. This was usually sufficient to prevent generation of muscle fibre action potentials and the associated twitch of the fibres. The cut-muscle approach was preferred to the use of Mg2+ (to reduce ACh release), a nicotinic ACh receptor antagonist (to reduce postjunctional sensitivity to ACh) or μ-conotoxin (to selectively abolish muscle action potentials), since all these pharmacological treatments affect the motor nerve terminal and would therefore have compromised our quantitative estimates of evoked ACh release.
For evoked responses, the motor nerve was stimulated at either 0.5 Hz for 3 min or at 50 Hz for 2 s to generate around 100 individual endplate currents (epcs). Nerve stimulation was via a pair of silver wire electrodes over which the phrenic nerve was fixed. Pulse of 0.05–0.1 ms duration and supramaximal voltage (typically 10–20 V) were delivered by a Grass S88 stimulator linked to a Grass SIU5 stimulus isolation unit. Spontaneously occurring miniature endplate currents (mepcs) were also recorded from the same cells for 1 min immediately prior to the epcs.
Electrophysiological technique
Epc and mepc were recorded from motor endplates using a standard two intracellular microelectrode voltage clamp technique (Dionne & Stevens, 1975; Prior et al., 1993). Microelectrodes were prepared from borosilicate glass (Clarks Electromedical Instruments, Pangbourne, U.K.) using a Sutter P30 micropipette puller. Voltage-recording electrodes (5–10 MΩ) were filled with 3 M KCl and current-passing electrodes (2–5 MΩ) filled with 2 M potassium citrate. The gain and bandwidth of the voltage-clamp amplifier (Axon Instruments, Axoclamp 2B) were adjusted for optimum performance and adequacy of voltage-clamping was assessed from the voltage escape during evoked responses, less than 1% of the holding potential (i.e. 0.5 mV) being deemed acceptable. In any individual fibre studied, both epcs and mepcs were recorded with the same voltage-clamp operating parameters. All recordings were made at a holding potential of −50 mV. This depolarized potential was used to eliminate the problems associated with the reactivation of muscle Na+ channels in fibres where the endplate is clamped close to the normal resting membrane potential (Prior et al., 1993). All currents were recorded on FM-tape (Racal Store 4DS, d.c.–5 kHz) for off-line computer analysis.
Data analysis and statistics
For analysis, current signals were replayed from the FM-tape, digitized at 25 kHz (Lab-PC+laboratory interface, National Instruments, Newbury, U.K.) and stored on the hard disk of a microcomputer (Viglen 486DX). Sampling and analysis were performed using the Strathclyde Electrophysiological Software suite of programs (Dempster, 1988; 1993). All currents were individually displayed so that artefacts could be eliminated from the analysis. For each epc recording, between 80 and 100 epcs were individually analysed for peak amplitude and time course as described elsewhere (Prior et al., 1993). For epcs at 50 Hz the first 20 signals were excluded from the analysis to avoid the initial rundown of epc amplitudes. Data obtained from the individual epcs were averaged to give mean values for each experimental record. For mepcs, because of the relatively low signal-to-noise of individual current signals, we have previously determined that the best estimates of amplitude and time course are obtained from a composite averaged signal (Tian et al., 1994; 1997). Therefore, all mepcs from a single experimental recording (typically 20–40 mepcs) were aligned to the mid-point of their rising phase and subjected to sequential averaging of data points to give a single averaged mepc signal. This average mepc was then analysed for peak amplitude and time course in an identical fashion as to the epcs.
Evoked quantal ACh release was measured as the epc quantal content (m) and this was calculated for each experimental record as the ratio of mean epc amplitude and the amplitude of the averaged mepc. For each condition studied, 7–10 identical individual experiments were performed and the data from these were averaged to give the presented mean and standard error of the mean values. Since, in each fibre studied, both control and drug-treated data was collected, it was possible to perform statistical analysis using a paired Student's t-test (P<0.05, two-tailed being taken as an indication of a significant difference). Using paired data has the advantage of reducing the influence of inter-fibre variability on the statistical tests, allowing detection of small effects of the drug treatment.
Drugs and solutions
All isolated muscle preparations were maintained (at 32°C) in a solution of the following composition (mM): NaCl 118, KCl 5, KH2PO4 1.2, MgSO4 1, NaHCO3 25, CaCl2 0.45–3.6, and glucose 11. The solution was continuously bubbled with a gas mixture of 5% CO2 in O2 to provide oxygenation and to maintain solution pH at 7.2–7.4. Solutions of hexamethonium, verapamil and W-7 (N-(6-aminohexyl)-5-chloro-1-napthalenesulphonamide.HCl) were made fresh each day from powder (1 mM in distilled water). These were diluted in physiological solution to the desired working concentration as required. A stock solution of cytisine (10 mM) was made in distilled water and stored in 100 μl aliquots at −20°C for up to 3 months. Cytisine, hexamethonium and verapamil were purchased from Sigma Chemical Co. (Poole, Dorset, U.K.) and W-7 was purchased from Tocris Cookson (Bristol, U.K.).
In all experiments, cytisine, hexamethonium, verapamil and W-7 were each used at a single concentration. The concentrations of hexamethonium (200 μM) and W-7 (10 μM) have been previously shown to be effective in modulating ACh release from rat motor nerve terminals (Tian et al., 1994; 1997). The concentration of cytisine (1 μM) was selected, following pilot studies, to minimize complications arising from effects of the compound on muscle-type nicotinic ACh receptors (see below). The concentration of verapamil (10 μM) was one that previous studies have shown is maximal for block of verapamil-sensitive motor nerve terminal Ca2+-currents (Penner & Dreyer, 1986) without significant effects on either postjunctional ACh receptors (Edeson et al., 1988) or prejunctional Na+-channels (Penner & Dreyer, 1986).
Results
Effect of cytisine on mepcs
Cytisine (1 μM) had no effect on the frequency of occurrence of mepcs, the decay time constant of mepcs (mepc tau) or mepc amplitude (Figure 1 and Table 1). However, at 10 μM the compound produced decreases in both mepc amplitude and the decay time constant of mepcs (Figure 1 and Table 1). In addition, 10 μM cytisine significantly (and reversibly) depolarized the motor endplate of intact hemidiaphragm muscle fibres from 72.7±2.6 to 68.6±2.6 mV (n=7, P<0.05 two tailed paired Student's t-test). These effects of 10 μM cytisine are all indicative of actions of the compound on the motor endplate muscle-type nicotinic ACh receptors. Therefore, to avoid any effects of cytisine on this class of receptor, either post- or prejunctionally, from compromising our analysis of the autoinhibitory phenomenon, in all studies in which m was determined cytisine was used at the lower concentration of 1 μM.
Figure 1.
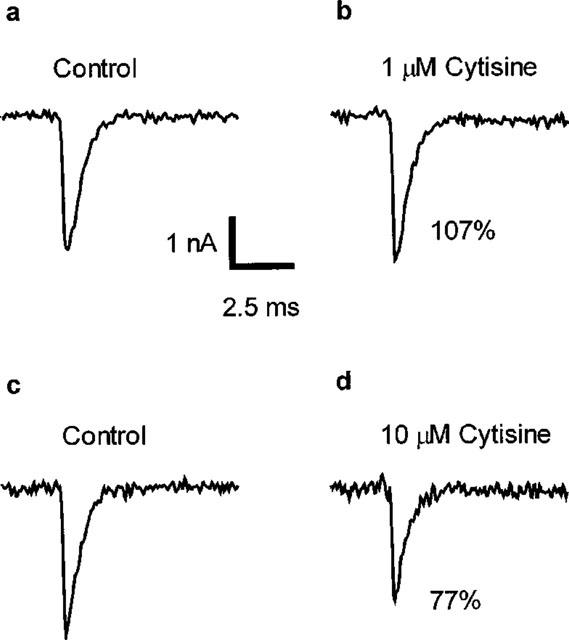
Examples of the effects of 1 and 10 μM cytisine on mepcs recorded from rat isolated hemidiaphragm muscle. Each trace is an average of approximately 40 individual mepc recorded before (a,c) or after (b,d) the application of 1 μM (a,b) or 10 μM (c,d) cytisine to the muscle fibre. In each case the control and drug-treated mepcs were recorded from the same muscle fibre. For all records, holding potential was −50 mV and [Ca2+]o was 1.8 mM. Annotated values are the amplitude of the mepcs in cytisine (b,d) expressed as a percentage of their respective control (a,c). The apparent slight increase in amplitude following 1 μM cytisine was not a consistent observation, there being on average no overall change. However, the approximate 20% reduction in mepc amplitude produced by 10 μM cytisine was reproducible and, across all cells studied, was statistically significant (see Table 1). Calibration bars for all traces: vertical, 1 nA; horizontal, 2.5 nA.
Table 1.
Effects of cytisine on mepcs in the rat isolated hemidiaphragm muscle
Frequency-dependent effect of cytisine
The effect of 1 μM cytisine on m was assessed at both 0.5 and 50 Hz with a [Ca2+]o of 1.8 mM. At 50 Hz, 1 μM cytisine had no effect on epc amplitude (Figure 2d,e) and consequently, given its lack of effect on mepc amplitudes at this concentration, m was unaffected (Figure 2f). However, at 0.5 Hz, epc amplitude was reduced by an average of around 20% (Figure 2b,e) leading to a corresponding reduction of approximately 20% in the size of m (Figure 2f).
Figure 2.
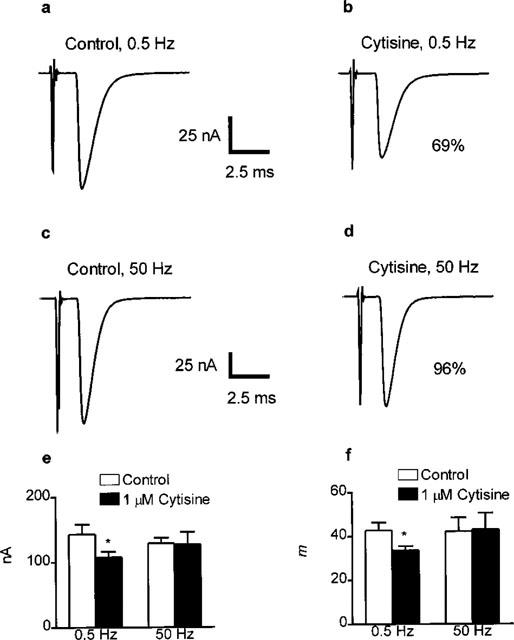
Examples of the effects of 1 μM cytisine on representative epcs at 0.5 Hz (a,b) or 50 Hz (c,d) and on average epc amplitude (e) and epc quantal content (f) recorded from rat isolated hemidia-phragm muscle fibres. Each trace in a–d is an average of approximately 80 individual epc recorded before (a,c) or after (b,d) the application of 1 μM cytisine to the muscle fibre. In each case the control and drug-treated epcs were recorded from the same muscle fibre. For all records, holding potential was −50 mV and [Ca2+] was 1.8 mM. Annotated values are the amplitude of the epcs in cytisine (b,d) expressed as a percentage of their respective control (a,c). Calibration bars for all traces: vertical 25 nA; horizontal, 2.5 nA. Panels e and f show bar chart of the average effects of 1 μM cytisine on epc amplitude and epc quantal content respectively. Data are mean and s.e.mean of values from ten (0.5 Hz) or eight (50 Hz) individual determinations. Asterisks indicate significant differences (P<0.05 two-tailed paired Student's t-test, cytisine versus control). Note that at 50 Hz, 1 μM cytisine affects neither epc amplitude nor m. Conversely, at 0.5 Hz, both parameters are reduced by around 20% by this concentration of cytisine.
Calcium-dependent effect of cytisine
The facilitatory effect of the nicotinic ACh receptor antagonist hexamethonium on m at a low frequency of stimulation is known to be dependent on [Ca2+]o (Tian et al., 1997). Thus we might expect the inhibitory effect of cytisine on m at 0.5 Hz also to be dependent on [Ca2+]o We therefore determined the effect on 1 μM cytisine on mepc and epc amplitudes, and hence m, in conditions where [Ca2+]o was 2 fold greater (3.6 mM) and 4 fold less (0.45 mM) than normal. Cytisine (1 μM) had no effect on mepc amplitude irrespective of the [Ca2+]o used (Figure 3a). However, epc amplitude, and hence m, was only reduced when the [Ca2+]o was 1.8 mM. No effect of cytisine on either epc amplitude (Figure 3b) or m (Figure 3c) was detected when the [Ca2+]o was lowered to 0.45 mM or raised to 3.6 mM.
Figure 3.
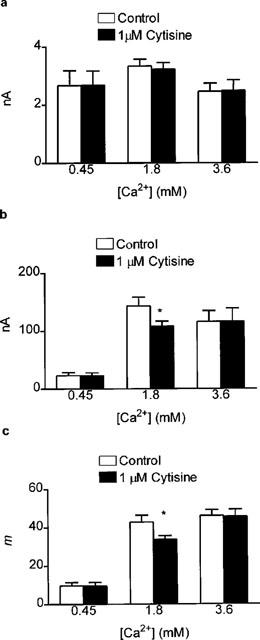
Bar graph showing the effects of 1 μM cytisine on mepc amplitude (a), epc amplitude (b) and epc quantal content (c) in the rat isolated hemidiaphragm muscle. Epcs were recorded at 0.5 Hz and three different [Ca2+]o were used as indicated in the figure. Data for 1.8 mM [Ca2+]o are the same as in Figure 2 and are included for comparison. Note that 1 μM cytisine had no effect on mepc amplitude, irrespective of the [Ca2+]o. However, 1 μM cytisine reduced epc amplitude (and hence m) only when the [Ca2+]o was 1.8 mM. All data are mean and s.e.mean of 6–10 individual determinations. Asterisks indicate significant differences (P<0.05, two-tailed Student's t-test, cytisine versus control).
Sensitivity of cytisine action to W-7 and verapamil
To investigate the potential intracellular signalling system involved in autoinhibition we studied the ability of two compounds, verapamil and W-7, to modulate the effects of cytisine and hexamethonium on m at 0.5 Hz. Exposure of neuromuscular preparations to verapamil (10 μM) alone had no effect on mepc amplitude or frequency of occurrence (Table 2) but produced an approximate 20% increase in epc amplitude, and therefore a similar effect on m (Table 2). This itself suggests the possibility that there exists a degree of endogenous autoinhibition that is sensitive to a prejunctional action of the compound. As expected, neither cytisine nor hexamethonium had an effect on mepc frequency, irrespective of the presence or absence of verapamil or W-7 (Figure 4).
Table 2.
Effect of verapamil (10 μM) on epc and mepc amplitudes and on quantal ACh release in the rat isolated hemidiaphragm muscle
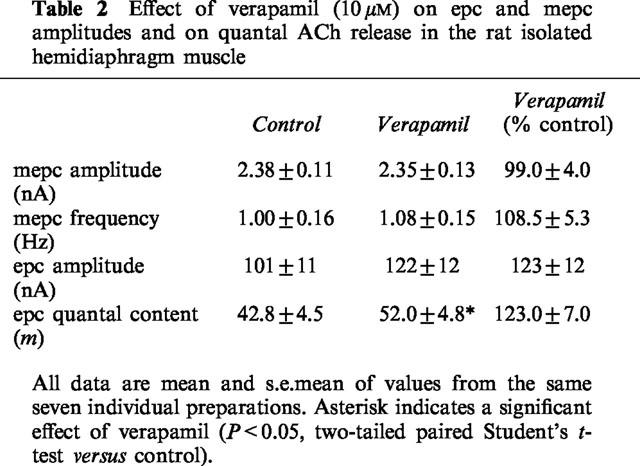
Figure 4.
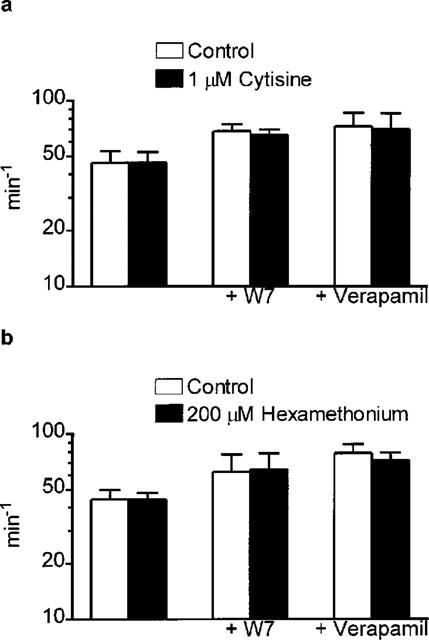
Bar graph showing the effects of 1 μM cytisine (a) and 200 μM hexamethonium (b) on the frequency of occurrence of mepcs in the rat isolated hemidiaphragm preparation in the absence and presence of W-7 or verapamil (both 10 μM). Note that neither cytisine nor hexamethonium affected mepc frequency under any of the conditions studied. Data are mean and s.e.mean of values from 7–10 individual determinations. All data P>0.05, two-tailed paired Student's t-test, control versus cytisine or hexamethonium.
As already stated, in the absence of verapamil or W-7, 1 μM cytisine depressed epc amplitude (Figure 5c) with no effect on mepc amplitude (Figure 5a). However, hexamethonium (200 μM) depressed mepc amplitude (Figure 5b) with no effect on epc amplitude (Figure 5d) Thus cytisine increased and hexamethonium decreased m (Figure 5e,f). These are consistent with previous published data and indicate that cytisine can activate and hexamethonium can block the autoinhibitory nicotinic ACh receptors on the motor nerve terminals. In the presence of W-7, cytisine (1 μM) had no statistically significant effect on either mepc or epc amplitudes (Figure 5a,c). However, W-7 did not prevent the inhibitory effect of cytisine on m (Figure 5e) – there was no statistically significant difference between the effects of cytisine on m in the presence and absence of cytisine (P>0.05, unpaired Student's t-test). In contrast, in the presence of verapamil, cytisine was without effect on mepc and epc amplitudes (Figure 5a,c) and also m (Figure 5e). Hexamethonium (200 μM) reduced mepc amplitudes irrespective of the presence of W-7 or verapamil (Figure 5b) but only reduced epc amplitudes in the presence of verapamil (Figure 5d). Hence, verapamil, but not W-7, attenuated the facilitatory effect of hexamethonium on m (Figure 5f)
Figure 5.
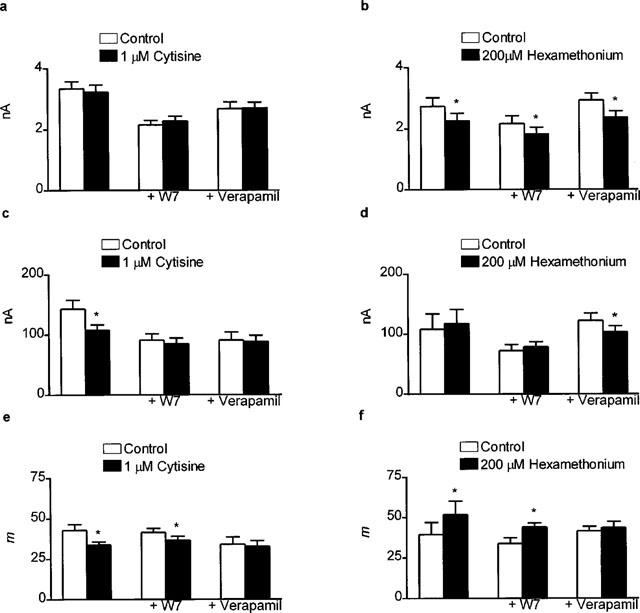
Bar graph showing the effects of 1 μM cytisine (a,c,e) and 200 μM hexamethonium (b,d,f) on the amplitude of mepcs (a,b), the amplitude of epcs (c,d) and the epc quantal content (e,f) at 0.5 Hz in the rat isolated hemidiaphragm preparation in the absence and presence of W-7 or verapamil (both 10 μM). [Ca2+]o was 1.8 mM in all experiments. Data for cytisine alone are the same as in Figure 3 and are included for comparison. Data are mean and s.e.mean of values from 7–10 individual determinations. Asterisks indicate a significant effect of cytisine or hexamethonium on mepc amplitude, epc amplitude or epc quantal content (P<0.05, two-tailed paired Student's t-test, control versus cytisine or hexamethonium). Note that the effects of cytisine and hexamethonium on m were unaffected by the presence of W-7. However, in the presence of verapamil, both effects were abolished.
Discussion
Cytisine is most effective in activating nicotinic ACh receptors containing a β4-subunit (Luetje & Patrick, 1991; Covernton et al., 1994; Chavez-Noriega et al., 1997). However, the ability of 10 μM cytisine to depolarize the endplates of intact muscle fibres suggests that, in agreement with previous observations (Yost & Winegar, 1997), the compound is an agonist at postjunctional muscle-type nicotinic ACh receptors. Thus the decrease in the amplitudes of mepcs produced by 10 μM cytisine could be due to fast-desensitization of postjunctional muscle-type nicotinic ACh receptors as previously described for other cholinergic agonists (Wray, 1981; Pennefather & Quastel, 1982; Singh & Prior, 1998). Alternatively, this decrease might result from a concurrent competitive antagonist action of cytisine on the muscle-type nicotinic ACh receptors, as has been seen in β2-containing neuronal-type nicotinic ACh receptors (Papke & Heinemann, 1994). Finally, the slight decrease in the decay time constants of mepcs suggests a small degree of open ion channel block, However, at 1 μM, cytisine had no effect on either mepc amplitude or time constant of decay, suggesting no action on postjunctional muscle-type nicotinic ACh receptors at this lower concentration. In addition, 1 μM cytisine had no effect on m at 50 Hz. This suggests that, unlike DMPP, this concentration of cytisine lacks an action on the prejunctional muscle-type nicotinic ACh receptors which mediate a facilitatory control over ACh release (Tian et al., 1994; Singh & Prior, 1998). In contrast to its lack of an effect at 50 Hz, cytisine (1 μM) reduced m at 0.5 Hz by around 20%. This supports the suggested existence of prejunctional nicotinic ACh receptors that inhibit evoked ACh release and that have been reported to operate under conditions of low release (Wilson & Thomsen, 1991; 1992; Tian et al., 1994; 1997). The relative pre- and postjunctional activities of cytisine reported here supports the previous published conjecture that the autoinhibitory prejunctional nicotinic ACh receptor is of a neuronal sub-type (Tian et al., 1994; 1997). The possibility that the autoinhibitory receptors are muscle-type nicotinic ACh receptors of the extrajunctional (foetal) isoform is unlikely since the intrinsic activity of cytisine at these is 6–8 fold less than at the postjunctional (adult) muscle-type nicotinic ACh receptors (Yost & Winegar, 1997).
Earlier studies showed that the nicotinic ACh receptor antagonist-induced facilitation in m at low frequencies of stimulation is sensitive to a change in the [Ca2+]o. Thus, Tian et al. (1997) observed that the increase in m produced by hexamethonium at 0.5 Hz in the presence of a [Ca2+]o of 2.0 mM was abolished when the [Ca2+]o was lowered to 0.5 mM. Therefore to equate the results with previous studies, the effects of cytisine were also investigated on lowering the [Ca2+]o to 0.45 mM. In agreement with the studies with nicotinic ACh receptor antagonists, changes in the amplitudes of epcs and m produced by 1 μM cytisine at 0.5 Hz were abolished when the [Ca2+]o was lowered to 0.45 mM. This implies clearly that the inhibitory nicotinic ACh receptor agonists and facilitatory nicotinic ACh receptor antagonists share a common mechanism for their effect on evoked ACh release from motor nerve terminals.
The reduction in m produced by 1 μM cytisine in the presence of 1.8 mM [Ca2+]o, instead of becoming more pronounced on raising the [Ca2+]o, was found to be abolished when the [Ca2+]o was raised to 3.6 mM. One explanation for this could be that although autoinhibition requires the presence of [Ca2+]o, there is a very narrow range of [Ca2+]o within which the system can remain effective. This would be seen if activation of the prejunctional neuronal-type ACh receptors shifts the relationship between [Ca2+]o and m to higher levels of [Ca2+]o, i.e. a rightward shift of the [Ca2+]o-m relationship. Since in mammalian motor nerve terminals m saturates above 3 mM [Ca2+]o (Cooke et al., 1973), a consequence of the rightward shift would be that at high and low [Ca2+]o there would be modest, if detectable, changes in m by cytisine while the largest effect of the compound would be seen in the mid-range of [Ca2+]o. Evidence that in these experiments the evoked release of ACh was near maximal, even at 1.8 mM [Ca2+]o, can be seen in the controls of Figure 3c; no statistically significant increase in m was seen when [Ca2+]o was raised from 1.8 to 3.6 mM (P>0.05, unpaired, two-tailed Student's t-test).
Previously, we have shown that the ability of DMPP to increase evoked ACh release and the ability of vecuronium to inhibit evoked ACh release (both at 50 Hz) represent activation and inhibition respectively of a prejunctional nicotinic ACh receptor mediating autofacilitation (Singh & Prior, 1998). In addition, the effects of DMPP and vecuronium are attenuated by W-7 implicating a calmodulin-dependent process in autofacilitation. In this study, it was observed that both the cytisine-induced reduction and the hexamethonium-induced facilitation in m (at 0.5 Hz) were unaffected by the presence of W-7. However, in the presence of verapamil, neither was there any cytisine-induced facilitation nor hexamethonium-induced depression in this parameter. This suggests that mechanism by which nicotinic ACh receptor activation depresses the release of ACh at low frequencies of stimulation is not the same as the mechanism by which release of ACh is enhanced through activation of nicotinic ACh receptors at high frequencies of stimulation. Therefore, the autofacilitatory and autoinhibitory effects of ACh on evoked ACh release from rat motor nerve terminals must be separate and individually identifiable processes involving unique and distinct intracellular second messenger systems. Finally, despite the lack of an effect of W-7 on the cytisine-induced depression of quantal ACh release, W-7 attenuated the effect of cytisine on epc amplitudes. This suggests that, in addition to its calmodulin-independent effect on ACh release, cytisine may have another unidentified calmodulin-dependent neuromuscular effect that can depress epc amplitude.
Verapamil attenuated the inhibitory effect of cytisine and the facilitatory effect of hexamethonium on m, suggesting that the compound blocks autoinhibition in motor nerve terminals. Further, in the absence of any other pharmacological agents, 10 μM verapamil increased m by around 10–20% suggesting the existence of some tonic activation of the autoinhibitory system by endogenous ACh. The question now arises as to what the underlying mechanism could be that is triggered by activation of a neuronal-type nicotinic ACh receptor, is dependent on [Ca2+]o and is inhibited by verapamil? Neuronal-type nicotinic ACh receptors have been shown to be more permeable to Ca2+ compared to their muscle-type counterparts (Decker & Dani, 1990; Sands & Barish, 1991; Mulle et al., 1992a; Vernino et al., 1992), their function can be modulated by [Ca2+]o (Vernino et al., 1992; Mulle et al., 1992b) and they can be blocked by verapamil at the concentration used here (Donnelly-Roberts et al., 1995; Villarroya et al., 1997). Thus a verapamil-sensitive Ca2+-flux through prejunctional neuronal-type nicotinic ACh receptors may be important in autoinhibition.
Alternatively, activation of the prejunctional neuronal-type nicotinic ACh receptors may lead to the activation of nerve terminal verapamil-sensitive (L-type) VOCCs and that control autoinhibition. Verapamil-sensitive Ca2+-currents do exist in motor nerve terminals (Penner & Dreyer, 1986; Anderson & Harvey, 1987). However, a direct involvement of verapamil-sensitive VOCCs in the evoked release of ACh at the neuromuscular junction is mostly denied (Nachshen & Blaustein, 1979). It is more generally accepted that the channels responsible for the evoked release of ACh from mammalian motor nerve terminals are the ω-agatoxin-sensitive P-type VOCCs (Uchitel et al., 1992; Katz et al., 1996). However, it is not inconceivable that prejunctional L-type VOCCs could possess a modulatory role in ACh release. Whatever the mechanism underlying the putative verapamil-sensitive Ca2+-flux, it is unlikely that it would lead to a generalized rise in the intracellular Ca2+ concentration ([Ca2+]i) since an increase in [Ca2+]i should increase the frequency of occurrence of mepcs (Hubbard & Willis, 1962), which was not seen in the present study. However, a localized rise in [Ca2+]i may be sufficient enough to produce autoinhibition.
In conclusion, we have obtained evidence confirming that the autoinhibition of evoked ACh release from rat motor nerve terminals seen at low frequencies of stimulation is dependent upon the activity of a prejunctional nicotinic ACh receptor probably of a neuronal subtype. Additionally, autoinhibition represents a distinct phenomenon from the calmodulin-dependent autofacilitation that is also mediated by prejunctional nicotinic ACh receptors. It is possible that autoinhibition involves the activation of a verapamil-sensitive Ca2+-flux across the nerve terminal membrane. However, the exact nature of this Ca2+-flux and how its activity subsequently leads to depressed evoked ACh release remain to be determined.
Acknowledgments
The authors are indebted to Professor Ian G. Marshall and Drs J. Dempster and E.G. Rowan for their continued help and advice throughout the execution of these studies. Shila Singh was supported by a postgraduate scholarship from the Overseas Research Student Award Scheme.
Abbreviations
- epc
endplate current
- mepc
miniature endplate current
- m
endplate current quantal content
- VOCC
voltage-operated Ca2+ channel
References
- ANDERSON A.J., HARVEY A.L. ω-Conotoxin does not block the verapamil-sensitive calcium channels at mouse motor nerve terminals. Neurosci. Lett. 1987;82:177–180. doi: 10.1016/0304-3940(87)90125-x. [DOI] [PubMed] [Google Scholar]
- BARSTAD J.A.B., LILLIHEIL G. Transversally cut diaphragm preparation from the rat. Arch. Int. Pharmacodyn. Therap. 1968;175:373–390. [PubMed] [Google Scholar]
- CHAVEZ-NORIEGA L.E., CRONA J.H., WASHBURN M.S., URRUTIA A., ELLIOT K.J., JOHNSON E.C. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors hα2β2, hα2β4, hα3β2, hα3β4, hα4β2, hα4β4 and hα7 expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- COOKE J.D., OKAMOTO K., QUASTEL D.M.J. The role of calcium in depolarisation-secretion coupling at the motor nerve terminal. J. Physiol. 1973;228:459–497. doi: 10.1113/jphysiol.1973.sp010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVERNTON P.J.O., KOJIMA H., SIVILOTTI L.G., GIBB A.J., COLQUHOUN D. Comparison of neuronal nicotinic receptors in rat sympathetic neurones with subunit pairs expressed in Xenopus oocytes. J. Physiol. 1994;481:27–34. doi: 10.1113/jphysiol.1994.sp020416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DECKER E.R., DANI J.A. Calcium permeability of the nicotinic acetylcholine receptor: the single-channel calcium influx is significant. J. Neurosci. 1990;10:3413–3420. doi: 10.1523/JNEUROSCI.10-10-03413.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEMPSTER J.Computer analysis of electrophysiological signals Microcomputers in Physiology: a Practical Approach 1988Oxford: IRL Press; 51–93.Frazer, P.J., (ed) pp [Google Scholar]
- DEMPSTER J. Computer Analysis of Electrophysiological Signals. London: Academic Press; 1993. [Google Scholar]
- DIONNE V.E., STEVENS C.F. Voltage dependence of agonist effectiveness at the frog neuromuscular junction: resolution of a paradox. J. Physiol. 1975;251:245–270. doi: 10.1113/jphysiol.1975.sp011090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONNELLY-ROBERTS D.L., ARNERIC S.P., SULLIVAN J.P. Functional modulation of human ‘ganglionic-like' neuronal nicotinic acetylcholine receptors (nAChRs) by L-type calcium channel antagonists. Biochem. Biophys. Res. Commun. 1995;231:657–662. doi: 10.1006/bbrc.1995.2182. [DOI] [PubMed] [Google Scholar]
- EDESON R.O., MADSEN B.W., MILNE R.K., LE DAIN A.C. Verapamil, neuromuscular transmission and the nicotinic receptor. Eur. J. Pharmacol. 1988;151:301–306. doi: 10.1016/0014-2999(88)90812-6. [DOI] [PubMed] [Google Scholar]
- HUBBARD W.P., WILLIS W.D. Effects of depolarisation of motor nerve terminals upon the release of neurotransmitter by nerve impulses. J. Physiol. 1962;194:381–405. doi: 10.1113/jphysiol.1968.sp008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATZ E., FERRO P.A., WEISZ G., UCHITEL O.D. Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. J. Physiol. 1996;497:687–697. doi: 10.1113/jphysiol.1996.sp021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUETJE C.W., PATRICK J. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J. Neurosci. 1991;11:837–845. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLE C., CHOQUET D., KORN H., CHANGHEUX J.-P. Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron. 1992a;8:135–143. doi: 10.1016/0896-6273(92)90115-t. [DOI] [PubMed] [Google Scholar]
- MULLE C., LENA C., CHANGEUX J.-P. Potentiation of nicotinic receptor responses by external calcium in rat central neurones. Neuron. 1992b;8:937–945. doi: 10.1016/0896-6273(92)90208-u. [DOI] [PubMed] [Google Scholar]
- NACHSHEN D.A., BLAUSTEIN M.P. The effects of some organic ‘calcium antagonists' on calcium influx in presynaptic nerve terminals. Mol. Pharmacol. 1979;16:579–586. [PubMed] [Google Scholar]
- PAPKE R.L., HEINEMANN S.F. Partial agonist properties of cytisine on neuronal nicotinic receptors containing the β4-subunit. Mol. Pharmacol. 1994;45:142–149. [PubMed] [Google Scholar]
- PENNEFATHER P., QUASTEL D.M.J. Fast desensitization of the nicotinic receptor at the mouse neuromuscular junction. Br. J. Pharmacol. 1982;77:395–404. doi: 10.1111/j.1476-5381.1982.tb09311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENNER R., DREYER F. Two different presynaptic calcium currents in mouse motor nerve terminals. Pflugers Arch. 1986;406:190–197. doi: 10.1007/BF00586682. [DOI] [PubMed] [Google Scholar]
- PRIOR C., DEMPSTER J., MARSHALL I.G. Electrophysiological analysis of transmission at the skeletal neuromuscular junction. J. Pharmacol. Toxicol. Meth. 1993;30:1–17. doi: 10.1016/1056-8719(93)90002-v. [DOI] [PubMed] [Google Scholar]
- PRIOR C., TIAN L., DEMPSTER J., MARSHALL I.G. Prejunctional actions of muscle relaxants: synaptic vesicles and transmitter mobilization as sites of action. Gen. Pharmacol. 1995;26:659–666. doi: 10.1016/0306-3623(94)00246-j. [DOI] [PubMed] [Google Scholar]
- SANDS S.B., BARISH M.E. Calcium permeability of neuronal nicotinic acetylcholine receptor channels in PC12 cells. Brain Res. 1991;560:38–42. doi: 10.1016/0006-8993(91)91211-i. [DOI] [PubMed] [Google Scholar]
- SINGH S., PRIOR C. Prejunctional effects of the nicotinic ACh receptor agonist dimethylphenylpiperazinium at the rat neuromuscular junction. J. Physiol. 1998;511:451–460. doi: 10.1111/j.1469-7793.1998.451bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIAN L., PRIOR C., DEMPSTER J., MARSHALL I.G. Nicotinic antagonist-produced frequency-dependent changes in acetylcholine release from rat motor nerve terminals. J. Physiol. 1994;476:517–529. doi: 10.1113/jphysiol.1994.sp020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIAN L., PRIOR C., DEMPSTER J., MARSHALL I.G. Hexamethonium- and methyllycaconitine-induced changes in acetylcholine release from rat motor nerve terminals. Br. J. Pharmacol. 1997;122:1025–1034. doi: 10.1038/sj.bjp.0701481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCHITEL O.D., PROTTI D.A., SANCHEZ V., CHERKSEY B.D., SUGIMORI M., LLINAS R. P-type voltage-dependent calcium-channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc. Natl Acad. Sci. U. S. A. 1992;89:3330–3333. doi: 10.1073/pnas.89.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERNINO S., AMADOR M., LUETJE C.W., PATRICK J., DANI J.A. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- VILLARROYA M., DE LA FUENTE M.-T., LOPEZ M.G., GANDIA L., GARCIA A.G. Distinct effects of ω-toxins and various groups of Ca2+-entry inhibitors on nicotinic acetylcholine receptor and Ca2+ channels in chromaffin cells. Eur. J. Pharmacol. 1997;320:249–257. doi: 10.1016/s0014-2999(96)00902-8. [DOI] [PubMed] [Google Scholar]
- WILSON D.F., THOMSEN R.H. Nicotinic receptors on the rat phrenic nerve: evidence for negative feedback. Neurosci. Lett. 1991;132:163–166. doi: 10.1016/0304-3940(91)90292-2. [DOI] [PubMed] [Google Scholar]
- WILSON D.F., THOMSEN R.H. Effect of hexamethonium on transmitter release from rat phrenic nerve. Neurosci. Lett. 1992;143:79–82. doi: 10.1016/0304-3940(92)90237-2. [DOI] [PubMed] [Google Scholar]
- WRAY D. Prolonged exposure to acetylcholine: noise analysis and channel inactivation in cat tenuissimus. J. Physiol. 1981;310:37–56. doi: 10.1113/jphysiol.1981.sp013536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOST C.S., WINEGAR B.D. Potency of agonists and competitive antagonists on adult- and fetal-type nicotinic acetylcholine receptors. Cell. Mol. Neurosci. 1997;17:35–50. doi: 10.1023/A:1026325020191. [DOI] [PMC free article] [PubMed] [Google Scholar]