

Abstract
The interaction between α2-autoreceptors and receptors for angiotensin (AT1) and bradykinin (B2) was studied in mouse isolated atria. The preparations were labelled with [3H]-noradrenaline and then superfused with desipramine-containing medium and stimulated electrically.
Angiotensin II (10−11–10−7 M), angiotensin III (10−10–10−6 M) and bradykinin (10−11–10−7 M) enhanced the evoked overflow of tritium when preparations were stimulated with conditions that led to marked α2-autoinhibition (120 pulses at 3 Hz), but not when stimulated with conditions that led to little α2-autoinhibition (20 pulses at 50 Hz).
Blockade of α-adrenoceptors by phentolamine (1 or 10 μM) reduced or abolished the effect of angiotensin II and bradykinin on the overflow response to 120 pulses at 3 Hz.
Addition of the δ-opioid agonist [D-Ser2]-leucine enkephalin-Thr (DSLET, 0.1 μM), or of neuropeptide Y (0.1 μM), together with phentolamine, restored the effect of angiotensin II and bradykinin.
The β-adrenoceptor agonist terbutaline (10−9–10−4 M) enhanced the evoked overflow of tritium irrespective of the degree of autoinhibition.
The experiments show that (i) a marked prejunctional facilitatory effect of angiotensin and bradykinin in mouse isolated atria requires prejunctional α2-autoinhibition; (ii) in the absence of α2-autoinhibition, activation of other prejunctional Gi/o protein-coupled reeptors, namely opioid and neuropeptide Y receptors, restores a marked effect of angiotensin II and bradykinin; and (iii) the facilitatory effect of terbutaline is not dependent upon the degree of α2-autoinhibition. The findings indicate that the major part of the release-enhancing effect elicited through prejunctional Gq/11 protein-coupled receptors is due to disruption of an ongoing, α2-autoreceptor-triggered Gi/o protein mediated inhibition.
Keywords: α2-autoreceptors, autoinhibition, angiotensin receptors, β-adrenoceptors, bradykinin receptors, cross-talk, δ-opioid receptors, mouse atria, neuropeptide Y receptors, noradrenaline release
Introduction
As in many other tissues, angiotensin II increases the average release of noradrenaline per sympathetic nerve action potential in the heart (Starke et al., 1969; for review see Fuder & Muscholl, 1995). Bradykinin also modulates cardiac sympathetic neurotransmission, but its effects are less uniform. Bradykinin increases the release of noradrenaline in mouse (Chulak et al., 1998), rat (Chulak et al., 1995; Rump et al., 1997a) and human atria (Rump et al., 1997a), rat ventricle (Vaz-da-Silva et al., 1996) and synaptosomes of the guinea-pig heart (Seyedi et al., 1997). However, bradykinin inhibits noradrenaline release in the rabbit heart, perhaps because in that tissue, stimulation of prostaglandin synthesis by bradykinin and subsequent inhibition of noradrenaline release by prostaglandins prevails (Starke et al., 1977; see also Chulak et al., 1998).
It was early observed that angiotensin II lost its effect in rabbit hearts pretreated with phenoxybenzamine, which by itself increased the release of noradrenaline by blocking presynaptic α2-autoreceptors; the explanation at that time was that release was already maximal after phenoxybenzamine alone (Starke & Schümann, 1972). Brasch et al. (1995) found a similar phenomenon in guinea-pig atria: angiotensin II failed to increase the stimulation-induced release of noradrenaline when it had already been increased by the α2-adrenoceptor antagonist idazoxan. However, angiotensin also failed to increase release when a special nerve stimulation protocol was used which, for unknown reasons and without any use of drugs, prevented the development of α2-autoinhibition. The authors concluded that ongoing α2-autoinhibition is a prerequisite for the effect of angiotensin, perhaps because ‘the intracellular signal transduction pathways of α2-adrenoceptors and-AT1-receptors are linked at some stage, perhaps via a common G-protein or by a protein kinase' (Brasch et al., 1995). In contrast to these studies, angiotensin became more, rather than less effective after blockade of α2-adrenoceptors in the rabbit pulmonary artery (Costa & Majewski, 1988).
Cross-talk between prejunctional receptors is not new, but the majority of studies have focused on interactions between receptors coupled to Gi/o proteins, such as α2-autoreceptors and opioid receptors (for review see Hertting et al., 1990; Schlicker & Göthert, 1998). Presynaptic angiotensin receptors couple to Gq/11 proteins (Musgrave et al., 1991; Chulak et al., 1995). It was of interest, therefore, to investigate further the interaction between presynaptic α2-autoreceptors and angiotensin receptors, especially in view of the discrepancy mentioned above. The present study was undertaken in mouse atria where the prejunctional angiotensin receptor has been characterized as AT1 (Cox et al., 1999). The interaction between α2-autoreceptors and bradykinin B2-receptors was also of interest because prejunctional B2-receptors also couple to Gq/11 (Chulak et al., 1995).
Methods
Preparation and protocols
Adult male NMRI mice were killed by exsanguination. The heart was placed in physiological salt solution (PSS), which had been bubbled with 95% CO2 and 5% O2, and stored on ice. The wall of each pair of atria was cut into six to eight pieces. Pieces of atria (12 to 14) were incubated in 2 ml of PSS containing [3H]-noradrenaline (0.1 μM) for 30 min at 37°C. Twelve preparations were then superfused in parallel in twelve superfusion chambers with PSS at a flow rate of 1.2 ml min−1. The preparations were subjected to electrical field stimulation by square wave pulses (p) of 1 ms width and 80 mA current strength, yielding a voltage drop of 45 V cm−1 between the electrodes of each chamber. A ‘priming' stimulation period (180 p at 3 Hz) was applied at t=30 min (t=0 min being the beginning of superfusion). At t=54 min, the preparations were subjected to either six (S1 to S6) or three (S1 to S3) periods of electrical field stimulation (either 20 p at 50 Hz, or 36, 60 or 120 p at 3 Hz), delivered 18 min apart. Consecutive 2-min samples of the superfusate were collected from t=50 min onwards. At the end of the experiment the tissue was dissolved in 0.5 ml of Soluene (Packard, Frankfurt am Main, Germany) and tritium was determined in the superfusate samples and preparations.
Concentration-response curves to the peptides and to terbutaline were determined in experiments with six stimulation periods; increasing concentrations were introduced after S1, 12 min before S2–S6). The effect of single concentrations of drugs was usually determined by introducing them after S2, 12 min before S3, and keeping them in for the remainder of the experiment (with three or six stimulation periods). Other details are explained in the Results section.
Drugs and radiochemicals
Unless stated otherwise, the PSS used for superfusion had the following composition (mM): NaCl 118, KCl 4.8, CaCl2 2.5, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11, ascorbic acid 0.57, ethylenediaminetetraacetic acid disodium salt 0.03 and desipramine 0.001. The PSS for incubation with [3H]-noradrenaline contained no desipramine and 0.2 mM CaCl2 (see Limberger et al., 1992).
The following drugs were used: angiotensin II (human), angiotensin III (human), bradykinin, [D-Ser2]-leucine enkephalin-Thr (DSLET), desipramine hydrochloride, rauwolscine hydrochloride, (±)-terbutaline hemisulphate (Sigma, Deisenhofen, Germany); D-Arg[Hyp3,Thi5,D-Tic7,Oic8]bradykinin (Hoe 140; gift from Hoechst, Frankfurt am Main, Germany); neuropeptide Y (NPY; human; Bachem, Heidelberg, Germany); losartan (gift from Merck, Darmstadt, Germany); phentolamine hydrochloride (gift from Ciba-Geigy, Basel, Switzerland); (−)-[2,5,6-3H]-noradrenaline (New England Nuclear, Dreieich, Germany, specific activity 46.8–62.3 Ci mmol−1).
Analysis of data
The outflow of tritium was calculated as a fraction of the tritium content of the tissue at the onset of the respective collection period, per min. The overflow of tritium evoked by electrical field stimulation was calculated as the total tritium outflow during the collection period in which stimulation was applied and during the next collection period thereafter, minus the estimated basal outflow. Basal outflow was assumed to decline linearly from the collection period before stimulation to the second collection period after stimulation. The evoked overflow was expressed as a percentage of the tritium content of the tissue at the time of stimulation. Overflow ratios (Sn/S1) were then determined for each period of stimulation. Percentage changes of Sn/S1 ratios caused by a drug added after S1 were calculated for each preparation, taking as reference value the average corresponding Sn/S1 ratio in otherwise identical control experiments, in the absence of the drug. Effects of drugs added after S1 on basal tritium outflow were calculated in the same manner, based on samples collected immediately before stimulation.
Data are expressed as means±standard errors of the mean (s.e.mean); n denotes the number of preparations. The statistical significance of differences between groups was determined by Mann-Whitney test with Bonferroni correction. In all cases probability levels less than 0.05 (P<0.05) were taken to indicate significant differences.
Results
The basal efflux of tritium was similar to that previously reported (Cox et al., 1999). Electrical field stimulation with either 120 p at 3 Hz or 20 p at 50 Hz greatly increased tritium outflow. The electrically evoked overflow of tritium elicited by the first periods of stimulation, expressed as per cent of tissue tritium, is shown in Table 1.
Table 1.
Electrically evoked overflow of tritium from mouse atria (stimulation period S1)

Effect of angiotensin II, angiotensin III and bradykinin on trains leading to marked autoinhibition: 120 p at 3 Hz
Phentolamine, when introduced 12 min before the third of six periods of stimulation by 120 p at 3 Hz (S3), increased the evoked overflow of tritium by 358±50% (S3; similar at S4–S6; n=6), indicating marked autoinhibition of transmitter release.
Under these stimulation conditions, in the absence of phentolamine, angiotensin II (10−11–10−7 M), angiotensin III (10−10–10−6 M) and bradykinin (10−11–10−7 M) increased the evoked overflow of tritium in a concentration-dependent manner (Figure 1). The maximal enhancement produced by the angiotensins and bradykinin was about 80 and 100%, respectively. The EC50 values, determined as the concentrations causing half-maximal enhancement, i.e. 40% in the case of the angiotensins and 50% in the case of bradykinin, amounted to 0.17 nM for angiotensin II, 2.2 nM for angiotensin III and 0.04 nM for bradykinin (interpolated from the nearest points of the concentration-response curve, see also Cox et al., 1999). Addition before the third of three stimulation periods of either angiotensin II (0.1 μM) alone, bradykinin (0.1 μM) alone, or the two combined yielded similar enhancement of the overflow at S3, indicating that the effects of the peptides were not additive (angiotensin II 83±10% increase, n=4; bradykinin 92±12% increase, n=5; combination 117±10% increase, n=7).
Figure 1.
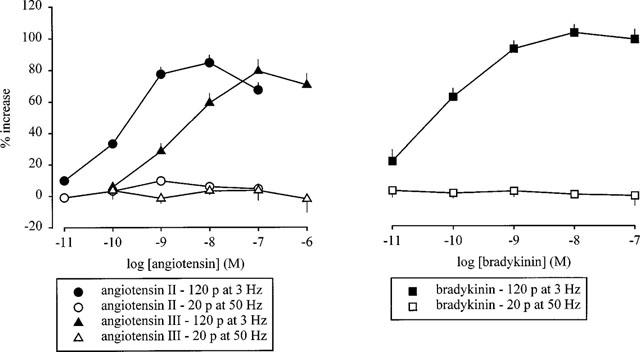
Effect of angiotensin II, angiotensin III (left panel) and bradykinin (right panel) on the overflow of tritium, evoked by stimulation trains leading either to marked or to little autoinhibition. The preparations were stimulated for 6 periods (S1–S6) by either 120 p at 3 Hz (marked autoinhibiton) or 20 p at 50 Hz (little autoinhibition). Angiotensin II, angiotensin III and bradykinin were introduced into the PSS in increasing concentrations, 12 min before S2–S6. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of the angiotensins and bradykinin. The vertical lines represent the s.e.mean from 4–30 preparations. Most of the experiments with angiotensin II and angiotensin III, 120 p at 3 Hz, are from Cox et al. (1999).
The facilitatory effect of the angiotensins and bradykinin was blocked respectively by the AT1-receptor antagonist losartan (0.1 μM) and the B2-receptor antagonist Hoe 140 (0.01 μM; data not shown, but see Cox et al. (1999) for angiotensin and Chulak et al. (1998) for bradykinin).
None of the drugs altered basal tritium efflux (data not shown).
Effect of angiotensin II, angiotensin III and bradykinin on trains leading to little autoinhibition: 20 p at 50 Hz
Stimulation with single, brief trains of 20 p at 50 Hz produced very little autoinhibition: phentolamine, when introduced 12 min before the third of six periods of stimulation, enhanced the overflow of tritium significantly, but only by 31±7% (S3; similar at S4–S6; n=4). When the preparations were stimulated under these conditions, neither angiotensin II, angiotensin III nor bradykinin altered the evoked overflow of tritium (Figure 1). Again none of the drugs tested altered basal tritium efflux (data not shown).
Effect of angiotensin II and bradykinin with α2-adrenoceptor blockade
Another approach to investigating the influence of prejunctional α2-adrenoceptors on the facilitatory effect of angiotensin II and bradykinin was to block α2-adrenoceptors during stimulation with normally autoinhibition-rich trains such as 120 p at 3 Hz. Phentolamine (1 or 10 μM), when present throughout superfusion, increased the overflow (S1) elicited by stimulation with 120 p at 3 Hz from, on average, 0.98 to 3.40% and 3.70%, respectively (Table 1), in accord with its effect when added before S3 (see above). Phentolamine (1 μM) markedly reduced the facilitatory effect of both angiotensin II and bradykinin on the overflow response to 120 p at 3 Hz (Figure 2). A 10 fold higher concentration of phentolamine abolished the effect of angiotensin II (Figure 2). Most of the small effects of angiotensin and bradykinin remaining in the presence of phentolamine (1 μM) were abolished by losartan (0.1 μM) and Hoe 140 (0.01 μM), respectively (Figure 2). To eliminate a possible non-selective effect of phentolamine, the selective α2-adrenoceptor antagonist rauwolscine was also used. Like phentolamine, rauwolscine (1 μM), when present throughout superfusion, increased the overflow at S1 (2.49±0.38%, n=10). The facilitatory effect of bradykinin was reduced by rauwolscine (1 μM; data not shown, n=6). None of the receptor antagonists when introduced alone, or in combination with each other, had any effect on basal tritium efflux (data not shown).
Figure 2.
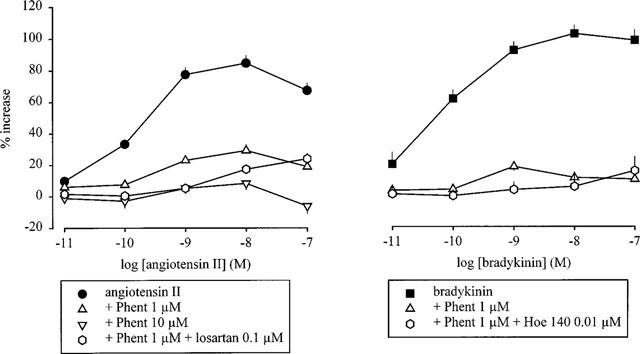
Effect of angiotensin II (left panel) and bradykinin (right panel) on the overflow of tritium, evoked by stimulation trains leading to marked autoinhibition, in the absence and presence of phentolamine. The preparations were stimulated for 6 periods (S1–S6) by 120 p at 3 Hz. Angiotensin II and bradykinin were introduced into the PSS either alone or in the presence of phentolamine (Phent, 1 or 10 μM), which was present throughout superfusion. In some experiments, losartan (0.1 μM) or Hoe 140 (0.01 μM) was also present throughout superfusion. Angiotensin II and bradykinin were introduced in increasing concentrations, 12 min before S2–S6. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of angiotensin II and bradykinin. The vertical lines represent the s.e.mean from 4–30 preparations. The curves showing the effect in the absence of phentolamine are from Figure 1.
Effect of angiotensin II and bradykinin with reduced pulse number and reduced calcium concentration
The overflow of tritium from preparations stimulated with 120 p at 3 Hz was markedly enhanced by the α-adrenoceptor antagonists (see above). The possibility was considered that the increase in overflow per se rather than the blockade of α2-adrenoceptors might minimise the effect of angiotensin II and bradykinin, or that the release of noradrenaline might be maximally enhanced by the α2-adrenoceptor antagonists, as previously suggested (Starke & Schümann, 1972), and thus no further increase by angiotensin II or bradykinin be possible. Two approaches were employed in order to reduce the evoked overflow, in the presence of phentolamine, to levels obtained with 120 p at 3 Hz in the absence of phentolamine. Firstly, the number of electrical pulses was reduced from 120 to 60 and 36. Even when S1 overflow values were reduced from 3.40% (120 p, phentolamine 1 μM) to 1.58 and 0.98% (60 and 36 p, respectively, phentolamine 1 μM; Table 1), the facilitatory effect of angiotensin II was still greatly attenuated by phentolamine (Figure 3). In the absence of phentolamine, angiotensin II enhanced tritium overflow even to a greater extent, by about 180% maximally (Figure 3), than in experiments with 120 p at 3 Hz (80%; Figure 1).
Figure 3.
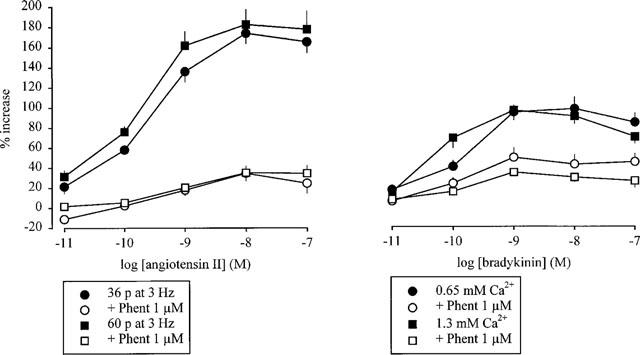
Effect of angiotensin II (left panel) and bradykinin (right panel) on the overflow of tritium, evoked by stimulation trains leading to marked autoinhibition, in the absence and presence of phentolamine: influence of a reduction of S1 overflow values. The preparations were stimulated for six periods (S1–S6). Angiotensin II and bradykinin were introduced into the PSS either alone or in the presence of phentolamine (Phent, 1 μM), which was present throughout superfusion. Angiotensin II and bradykinin were introduced into the PSS in increasing concentrations, 12 min before S2–S6. Left panel: preparations were stimulated with either 36 or 60 p (instead of the usual 120 p) at 3 Hz. Right panel: preparations were stimulated with 120 p at 3 Hz; the PSS contained either 0.65 or 1.3 mM (instead of the usual 2.5 mM) Ca2+. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of angiotensin II and bradykinin. The vertical lines represent the s.e.mean from 4–14 preparations.
The second procedure to reduce S1 overflow was to keep the number of pulses constant (120 p at 3 Hz) but to decrease the extracellular calcium concentration to 1.3 or 0.65 mM. This approach was used for experiments with bradykinin. Even when S1 overflow values were reduced from 3.40% (2.5 mM calcium, phentolamine 1 μM) to 1.96 and 1.33% (1.3 and 0.65 mM calcium respectively, phentolamine 1 μM; Table 1), the facilitatory effect of bradykinin was still greatly attenuated by phentolamine (Figure 3). In the absence of phentolamine, bradykinin enhanced tritium overflow to the same extent, about 100% (Figure 3), as it did at 2.5 mM calcium (Figure 1).
It should be noted that marked α2-autoinhibition prevailed in trains of 60 and 36 p, as well as in trains of 120 p applied in low calcium media: phentolamine (1 μM) in each case greatly increased the evoked overflow of tritium (Table 1).
Effect of angiotensin II and bradykinin in the presence of phentolamine plus DSLET or NPY
The possibility was considered that the Gq/11- coupled receptors for angiotensin and bradykinin require the activation of a Gi/o-coupled receptor such as the α2-autoreceptor in order to facilitate noradrenaline release. To investigate this possibility, prejunctional non-α2, Gi/o-coupled receptors were activated under conditions where the α2-autoreceptor was blocked: namely the δ-opioid and the Y2 NPY receptor. The experiments consisted of three periods of stimulation with 120 p at 3 Hz. The δ-opioid receptor agonist DSLET and the Y2 NPY receptor agonist NPY (both 0.1 μM), when added together with phentolamine (1 μM) from t=30 min of superfusion onwards, reduced the evoked overflow of tritium as compared to phentolamine alone (S1; Table 1). In the presence of phentolamine alone, the effect of angiotensin II and bradykinin was greatly attenuated (Figure 4). Addition of either DSLET or NPY together with phentolamine restored the effect of angiotensin II and bradykinin (Figure 4).
Figure 4.
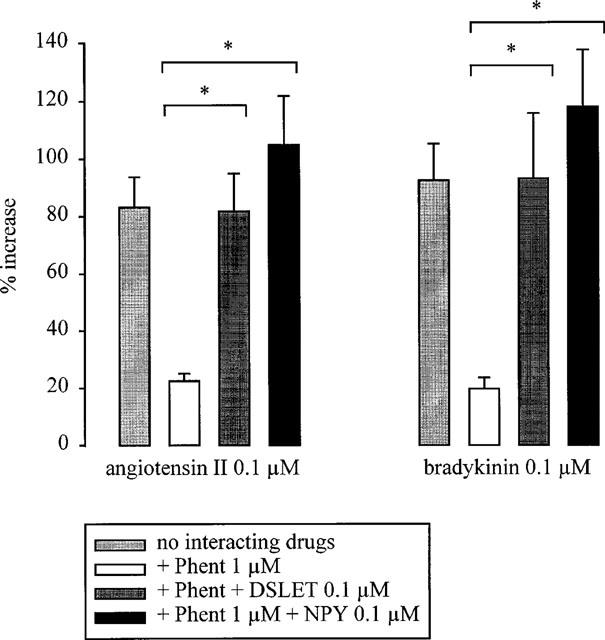
Effect of angiotensin II and bradykinin on the overflow of tritium, evoked by stimulation trains leading to marked autoinhibition, in the absence and presence of phentolamine: interaction with DSLET and NPY. The preparations were stimulated for three periods (S1–S3) by 120 p at 3 Hz. Angiotensin II and bradykinin were introduced into the PSS either alone, or in the presence of phentolamine (Phent, 1 μM), or in the presence of Phent plus DSLET (0.1 μM) or NPY (0.1 μM). Phent, when given alone, was present from the beginning of superfusion. The combinations of Phent plus DSLET or NPY were present from t=30 min. Angiotensin II and bradykinin were introduced 12 min before S3. The columns represent the mean percentage increase, calculated from S3/S1 values and corrected for changes observed in control experiments in the absence of angiotensin II or bradykinin. The vertical lines represent the s.e.mean from 5–15 preparations. * Indicates a significant difference (P<0.05). All increases were significant (P<0.05) as compared to control experiments without angiotensin II and bradykinin (not shown).
The combination of DSLET and phentolamine slightly increased basal tritium efflux. The combination of NPY and phentolamine was without significant effect on basal tritium efflux (data not shown).
Effect of terbutaline
The results thus far suggested an interaction between Gi/o- and Gq/11-coupled receptors. To investigate whether this was a specific phenomenon, we investigated the interaction between α2-autoreceptors and a GS-coupled release-enhancing receptor, namely the prejunctional β2-adrenoceptor. The β2-adrenoceptor agonist terbutaline enhanced tritium overflow in the same, concentration-dependent manner, irrespective of whether pulse trains leading to marked (120 p at 3 Hz) or little (20 p at 50 Hz) autoinhibition were used or whether, in the former case, the autoinhibition was blocked by phentolamine (1 μM) (Figure 5). The maximal increase by terbutaline was about 50% in each case (Figure 5). Terbutaline did not alter basal tritium efflux (data not shown).
Figure 5.
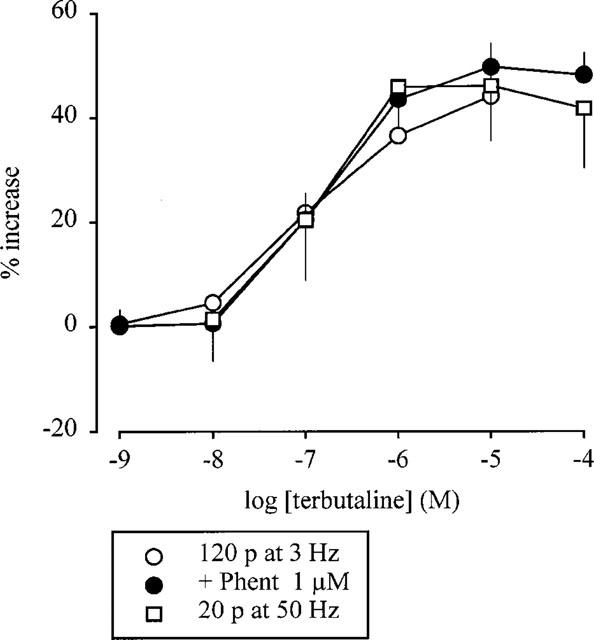
Effect of terbutaline on the evoked overflow of tritium. The preparations were stimulated for six periods (S1–S6) by either 120 p at 3 Hz or 20 p at 50 Hz. Terbutaline was introduced into the PSS either alone or in the presence of phentolamine (Phent, 1 μM), which was present throughout superfusion. Terbutaline was introduced in increasing concentrations, 12 min before S2–S6. The symbols represent the mean percentage increase, calculated from Sn/S1 values and corrected for changes observed in control experiments in the absence of terbutaline. The vertical lines represent the s.e.mean from 4–10 preparations.
Discussion
Under appropriate conditions, angiotensin II, angiotensin III and bradykinin all greatly enhanced, by up to 80–180%, the electrically evoked overflow of tritium, or, in other words (see Starke, 1977), the action potential-evoked release of [3H]-noradrenaline. These effects have previously been documented in mouse atria (Musgrave & Majewski, 1989; Rajanayagam et al., 1989; Chulak et al., 1998; Cox et al., 1999). They were blocked respectively by losartan and Hoe 140, which supports the general consensus that they were mediated via AT1- and B2-receptors, respectively (Chulak et al., 1998; Cox et al., 1999).
The ‘appropriate conditions' for a pronounced effect of the peptides were: stimulation by trains of 120, 60 or 36 p at 3 Hz in 2.5 mM calcium medium and stimulation by 120 p at 3 Hz in 1.3 or 0.65 mM calcium medium. In contrast, the facilitatory effects of the angiotensins and bradykinin were much smaller or even absent when atria were stimulated by trains of 20 p at 50 Hz, or by trains of 120, 60 or 36 p at 3 Hz in 2.5, 1.3 or 0.65 mM calcium medium in the presence of phentolamine or rauwolscine. This difference was not due to a difference in the pre-angiotensin or pre-bradykinin release (S1), or to an upper limit of release when release was very high (120 p at 3 Hz in the presence of phentolamine or rauwolscine). In fact, the pre-angiotensin and pre-bradykinin release values overlapped in the two groups: the ‘appropriate conditions' produced release values (S1) between 0.24% (120 p at 3 Hz, 0.65 mM calcium) and 0.98% (120 p at 3 Hz, 2.5 mM calcium), whereas the conditions permitting at best a minor effect of the peptides produced values between 0.46% (20 p at 50 Hz) and 3.7% (120 p at 3 Hz, phentolamine 10 μM; Table 1). Rather, what distinguished conditions that permitted a marked effect of angiotensin or bradykinin from those that did not, was the operation of α2-autoinhibition: the angiotensins and bradykinin were very effective when α2-autoinhibition was pronounced, as shown by the large release-enhancing effects of phentolamine (or rauwolscine); the effects of the peptides were weak when either pulse trains were too brief for autoinhibition (20 p at 50 Hz), or autoinhibition was interrupted by α2-antagonists.
Our findings in mouse atria confirm and extend previous observations with angiotensin II in the rabbit heart and in guinea-pig atria (Starke & Schümann, 1972; Brasch et al., 1995). They support the view of the latter authors that it is the degree of α2-autoinhibition that determines the effectiveness of angiotensin II. Bradykinin, it seems, requires the same conditions for its prejunctional effect.
Interestingly, some electrophysiological observations may also be explained by dependence of the effect of angiotensin on α2-autoinhibition. In three guinea-pig tissues, the uterine artery and vas deferens (Bell, 1972; Ziogas & Cunnane, 1991) and the mesenteric artery (Onaka et al., 1997), angiotensin II did not increase the amplitude of the first excitatory junction potential (e.j.p.) in a train of pulses but increased the amplitudes of subsequent e.j.p.s. Transmitter release (in this case cotransmitter ATP release) by the first pulse, after a period of rest, is free of α2-autoinhibition; α2-autoinhibition develops from the second pulse onwards (Illes & Starke, 1983; Brock et al., 1990). In the pulmonary artery of the rabbit, phentolamine actually increased the release-facilitating effect of angiotensin II (Costa & Majewski, 1988), the opposite of what one would expect from the results discussed so far; the reason for the difference is not known.
Our experiments provide some evidence for the view that the interaction between prejunctional α2-autoreceptors, prejunctional AT1-receptors and prejunctional B2-receptors takes place at the level of the cellular transduction pathways (see Brasch et al., 1995; see also Limberger et al., 1988; Hertting et al., 1990; Schlicker & Göthert, 1998). In support of this idea, angiotensin II and bradykinin shared both the dependence on α2-autoinhibition and a common transduction step, as shown by the lack of additivity of their releasing-enhancing effects. The common transduction step might for example be activation of one and the same Gq/11 protein. Also in support of an interaction during signal transduction, α2-autoinhibition did not modify the facilitation of noradrenaline release by terbutaline, which acts through prejunctional β2-adrenoceptors and hence, the GS pathway. We suspected that the two Gq/11-coupled receptors might require an ongoing Gi/o-mediated inhibition for a marked effect. For this reason, we activated two Gi/o-coupled receptors, namely the δ-opioid (Werling et al., 1989) and the Y2 NPY (Rump et al., 1997b) receptor, while the α2-autoreceptors were blocked by phentolamine. In fact, the δ-opioid agonist DSLET as well as NPY restored the effects of angiotensin II and bradykinin after they had been minimised by phentolamine.
If indeed Gq/11-mediated enhancement of noradrenaline release requires, or is at least much amplified by, an ongoing Gi/o-mediated inhibition, what might be the mechanism? A likely possibility is that protein kinase C (PKC), activated by the Gq/11 pathway, interferes with the ongoing Gi/o pathway. The prejunctional Gi/o pathway is thought to involve inter alia the inhibition of N-type calcium channels (see Starke et al., 1989; Lipscombe et al., 1989), and PKC has been reported to attenuate the Gi/o-mediated inhibition, although not of prejunctional, at least of somato-dendritic N-type calcium channels (see Hamid et al., 1999). PKC might disrupt the Gi/o pathway by phosphorylating either the Gi/o-coupled receptor (for example the α2A-adrenoceptor; Liang et al., 1998), Gi/o itself (Katada et al., 1985), or the N-type calcium channel (see Hamid et al., 1999). PKC activation increases transmitter release in atrial preparations (Musgrave et al., 1991; Brasch, 1993; Chulak et al., 1995). Interestingly, a PKC activating phorbol ester did not increase noradrenaline release in guinea-pig atria after α2-adrenoceptor blockade on the one hand, and under autoinhibition-free stimulation conditions on the other hand (Brasch, 1993), like angiotensin II and bradykinin in our experiments. Taken together, these findings and those of our study are compatible with the idea that prejunctional AT1- and B2-receptors, once activated, (i) stimulate Gq/11 and thereby PKC; that PKC then (ii) phosphorylates and inactivates a protein involved in the Gi/o-mediated prejunctional α2-autoinhibition; and that (iii) ongoing autoinhibition is thus disrupted and transmitter release disinhibited.
In conclusion, apart from confirming the requirement of α2-autoinhibition for a marked facilitatory effect of angiotensin II, we extend these observations to angiotensin III and also provide evidence for the same requirement in the case of another Gq/11-coupled receptor, the bradykinin B2-receptor. When α2-autoinhibition is disrupted, activation of other prejunctional Gi/o-coupled receptors, namely opioid and NPY receptors, restores a marked effect of angiotensin II and bradykinin. No such interaction is obtained between α2-autoinhibition and the GS-coupled prejunctional β2-adrenoceptor. Thus, we provide further evidence in support of a special type of prejunctional receptor cross-talk in mouse atria. The mechanism of this cross-talk and whether it occurs in other tissues and species is of particular interest.
Acknowledgments
S.L. Cox is a recipient of an Alexander von Humboldt Fellowship.
Abbreviations
- DSLET
[D-Ser2]-leucine enkephalin-Thr
- Hoe 140
D-Arg[Hyp3,Thi5,D-Tic7,Oic8]bradykinin
- NPY
neuropeptide Y
- Phent
phentolamine
- PSS
physiological salt solution
References
- BELL C. Mechanism of enhancement by angiotensin II of sympathetic adrenergic transmission in the guinea pig. Circ. Res. 1972;31:348–355. doi: 10.1161/01.res.31.3.348. [DOI] [PubMed] [Google Scholar]
- BRASCH H. Field stimulation-induced noradrenaline release from guinea-pig atria is modulated by prejunctional α2-adrenoceptors and protein kinase C. Basic Res. Cardiol. 1993;88:545–556. doi: 10.1007/BF00788873. [DOI] [PubMed] [Google Scholar]
- BRASCH H., SIEROSLAWSKI L., BERGMANN N., DOMINIAK P.In field-stimulated guinea-pig atria an AT1-receptor mediated increase of noradrenaline release by angiotensin II is seen only in the presence of prejunctional autoinhibition Tissue renin-angiotensin systems 1995Plenum Press: New York; 293–298.eds. Mukhopadhyay, A.K. & Raizada, M.K. pp [DOI] [PubMed] [Google Scholar]
- BROCK J.A., CUNNANE T.C., STARKE K., WARDELL C.F. α2-Adrenoceptor-mediated autoinhibition of sympathetic transmitter release in guinea-pig vas deferens studied by intracellular and focal extracellular recording of junction potentials and currents. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:45–52. doi: 10.1007/BF00178971. [DOI] [PubMed] [Google Scholar]
- CHULAK C., COUTURE R., FOUCART S. Modulatory effect of bradykinin on the release of noradrenaline from rat isolated atria. Br. J. Pharmacol. 1995;115:330–334. doi: 10.1111/j.1476-5381.1995.tb15881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHULAK C., COUTURE R., FOUCART S. Modulatory effect of bradykinin on noradrenaline release in isolated atria from normal and B2 knockout transgenic mice. Eur. J. Pharmacol. 1998;346:167–174. doi: 10.1016/s0014-2999(98)00060-0. [DOI] [PubMed] [Google Scholar]
- COSTA M., MAJEWSKI H. Facilitation of noradrenaline release from sympathetic nerves through activation of ACTH receptors, β-adrenoceptors and angiotensin II receptors. Br. J. Pharmacol. 1988;95:993–1001. doi: 10.1111/j.1476-5381.1988.tb11730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COX S.L., TRENDELENBURG A.U., STARKE K. Prejunctional angiotensin receptors involved in the facilitation of noradrenaline release in mouse tissues. Br. J. Pharmacol. 1999;127:1256–1262. doi: 10.1038/sj.bjp.0702652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUDER H., MUSCHOLL E. Heteroreceptor-mediated modulation of noradrenaline and acetylcholine release from peripheral nerves. Rev. Physiol. Biochem. Pharmacol. 1995;126:265–412. doi: 10.1007/BFb0049778. [DOI] [PubMed] [Google Scholar]
- HAMID J., NELSON D., SPAETGENS R., DUBEL S.J., SNUTCH T.P., ZAMPONI G.W. Identification of an integration center for cross-talk between kinase C and G protein modulation of N-type calcium channels. J. Biol. Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- HERTTING G., WURSTER S., ALLGAIER C. Regulatory proteins in presynaptic function. Annals N.Y. Acad. Sci. 1990;604:289–304. doi: 10.1111/j.1749-6632.1990.tb32001.x. [DOI] [PubMed] [Google Scholar]
- ILLES P., STARKE K. An electrophysiological study of presynaptic α-adrenoceptors in the vas deferens of the mouse. Br. J. Pharmacol. 1983;78:365–373. doi: 10.1111/j.1476-5381.1983.tb09402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATADA T., GILMAN A.G., WATANABE Y., BAUER S., JAKOBS K.H. Protein kinase C phosphorylates the inhibitory guanine-nucleotide-binding regulatory component and apparently suppresses its function in hormonal inhibition of adenylate cyclase. Eur. J. Biochem. 1985;151:431–437. doi: 10.1111/j.1432-1033.1985.tb09120.x. [DOI] [PubMed] [Google Scholar]
- LIANG M., EASON M.G., JEWELL-MOTZ E.A., WILLIAMS M.A., THEISS C.T., DORN G.W., LIGGETT S.B. Phosphorylation and functional desensitization of the α2A-adrenergic receptor by protein kinase C. Mol. Pharmacol. 1998;54:44–49. doi: 10.1124/mol.54.1.44. [DOI] [PubMed] [Google Scholar]
- LIMBERGER N., SPÄTH L., STARKE K. Presynaptic α2-adrenoceptor, opioid κ-receptor and adenosine A1-receptor interactions on noradrenaline release in rabbit brain cortex. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;338:53–61. doi: 10.1007/BF00168812. [DOI] [PubMed] [Google Scholar]
- LIMBERGER N, TRENDELENBURG A.U., STARKE K. Pharmacological characterization of presynaptic α2-autoreceptors in rat submaxillary gland and heart atrium. Br. J. Pharmacol. 1992;107:246–255. doi: 10.1111/j.1476-5381.1992.tb14494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIPSCOMBE D., KONGSAMUT S., TSIEN R.W. α-Adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 1989;340:639–642. doi: 10.1038/340639a0. [DOI] [PubMed] [Google Scholar]
- MUSGRAVE I.F., FOUCART S., MAJEWSKI H. Evidence that angiotensin II enhances noradrenaline release from sympathetic nerves in mouse atria by activating protein kinase C. J. Auton. Pharmacol. 1991;11:211–220. doi: 10.1111/j.1474-8673.1991.tb00319.x. [DOI] [PubMed] [Google Scholar]
- MUSGRAVE I.F., MAJEWSKI H. Effect of phorbol ester and pertussis toxin on the enhancement of noradrenaline release by angiotensin II in mouse atria. Br. J. Pharmacol. 1989;96:609–616. doi: 10.1111/j.1476-5381.1989.tb11859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ONAKA U., FUJII K., ABE I., FUJISHIMA M. Enhancement by exogenous and locally generated angiotensin II of purinergic neurotransmission via angiotensin type 1 receptor in the guinea-pig isolated mesenteric artery. Br. J. Pharmacol. 1997;122:942–948. doi: 10.1038/sj.bjp.0701458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAJANAYAGAM M.A.S., MUSGRAVE I.F., RAND M.J., MAJEWSKI H. Facilitation of noradrenaline release by isoprenaline is not mediated by angiotensin II in mouse atria and rat tail artery. Arch. Int. Pharmacodyn. 1989;299:185–199. [PubMed] [Google Scholar]
- RUMP L.C., BERLIT T., SCHWERTFEGER E., BEYERSDORF F., SCHOLLMEYER P., BOHMANN C. Angiotensin converting enzyme inhibition unmasks the sympathofacilitatory effect of bradykinin in human right atrium. J. Hypertension. 1997a;15:1263–1270. doi: 10.1097/00004872-199715110-00010. [DOI] [PubMed] [Google Scholar]
- RUMP L.C., RIESS M., SCHWERTFEGER E., MICHEL M.C., BOHMANN C., SCHOLLMEYER P. Prejunctional neuropeptide Y receptors in human kidney and atrium. J. Cardiovasc. Pharmacol. 1997b;29:656–661. doi: 10.1097/00005344-199705000-00014. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., GÖTHERT M. Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res. Bull. 1998;47:129–132. doi: 10.1016/s0361-9230(98)00068-9. [DOI] [PubMed] [Google Scholar]
- SEYEDI N., WIN T., LANDER H.M., LEVI R. Bradykinin B2-receptor activation augments norepinephrine exocytosis from cardiac sympathetic nerve endings. Circ. Res. 1997;81:774–784. doi: 10.1161/01.res.81.5.774. [DOI] [PubMed] [Google Scholar]
- STARKE K. Regulation of noradrenaline release by presynaptic receptor systems. Rev. Physiol. Biochem. Pharmacol. 1977;77:1–124. doi: 10.1007/BFb0050157. [DOI] [PubMed] [Google Scholar]
- STARKE K., GÖTHERT M., KILBINGER H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- STARKE K., PESKAR B.A., SCHUMACHER K.A., TAUBE H.D. Bradykinin and postganglionic sympathetic transmission. Naunyn-Schmiedeberg's Arch. Pharmacol. 1977;299:23–32. doi: 10.1007/BF00508633. [DOI] [PubMed] [Google Scholar]
- STARKE K., SCHÜMANN H.J. Interactions of angiotensin, phenoxybenzamine and propranolol on noradrenaline release during sympathetic nerve stimulation. Eur. J. Pharmacol. 1972;18:27–30. doi: 10.1016/0014-2999(72)90127-6. [DOI] [PubMed] [Google Scholar]
- STARKE K., WERNER U., SCHÜMANN H.J. Wirkung von Angiotensin auf Funktion und Noradrenalinabgabe isolierter Kaninchenherzen in Ruhe und bei Sympathicusreizung. Naunyn-Schmiedeberg's Arch. Pharmakol. 1969;265:170–186. [PubMed] [Google Scholar]
- VAZ-DA-SILVA M., MAGINA S., DOMINGUES-COSTA A., MOURA D., GUIMARÃES S. The role of the endocardium in the facilitatory effect of bradykinin on electrically-induced release of noradrenaline in rat cardiac ventricle. Br. J. Pharmacol. 1996;118:364–368. doi: 10.1111/j.1476-5381.1996.tb15411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WERLING L.L., MCMAHON P.N., COX B.M. Effects of pertussis toxin on opioid regulation of catecholamine release from rat and guinea-pig brain slices. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989;339:509–513. doi: 10.1007/BF00167253. [DOI] [PubMed] [Google Scholar]
- ZIOGAS J., CUNNANE T.C. An electrophysiological study of the actions of angiotensin II at the sympathetic neuroeffector junction in the guinea-pig vas deferens. Br. J. Pharmacol. 1991;103:1196–1202. doi: 10.1111/j.1476-5381.1991.tb12323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]