

Abstract
The effects of endothelium-derived hyperpolarizing factor (EDHF: elicited using substance P or bradykinin) were compared with those of 11,12-EET in pig coronary artery. Smooth muscle cells were usually impaled with microelectrodes through the adventitial surface.
Substance P (100 nM) and 11,12-EET (11,12-epoxyeicosatrienoic acid; 3 μM) hyperpolarized endothelial cells in intact arteries. These actions were unaffected by 100 nM iberiotoxin but were abolished by charybdotoxin plus apamin (each 100 nM).
Substance P (100 nM) and bradykinin (30 nM) hyperpolarized intact artery smooth muscle; Substance P had no effect after endothelium removal.
11,12-EET hyperpolarized de-endothelialized vessels by 12.6±0.3 mV, an effect abolished by 100 nM iberiotoxin. 11,12-EET hyperpolarized intact arteries by 18.6±0.8 mV, an action reduced by iberiotoxin, which was ineffective against substance P. Hyperpolarizations to 11,12-EET and substance P were partially inhibited by 100 nM charybdotoxin and abolished by further addition of 100 nM apamin.
30 μM barium plus 500 nM ouabain depolarized intact artery smooth muscle but responses to substance P and bradykinin were unchanged. 500 μM gap 27 markedly reduced hyperpolarizations to substance P and bradykinin which were abolished in the additional presence of barium plus ouabain.
Substance P-induced hyperpolarizations of smooth muscle cells immediately below the internal elastic lamina were unaffected by gap 27, even in the presence of barium plus ouabain.
In pig coronary artery, 11,12-EET is not EDHF. Smooth muscle hyperpolarizations attributed to ‘EDHF' are initiated by endothelial cell hyperpolarization involving charybdotoxin- (but not iberiotoxin) and apamin-sensitive K+ channels. This may spread electrotonically via myoendothelial gap junctions but the involvement of an unknown endothelial factor cannot be excluded.
Keywords: Epoxyeicosatrienoic acid; EDHF; porcine coronary artery; hyperpolarization; substance P; bradykinin; gap junctions; gap 27; endothelium; 11,12-EET
Introduction
In the coronary circulation, the identity of endothelium-derived hyperpolarizing factor (EDHF) remains to be determined. Based on studies in rat hepatic and mesenteric arteries, Edwards et al. (1998) first showed the critical importance of endothelial cell hyperpolarization in the EDHF pathway. These workers concluded that ‘EDHF' was likely to be K+ released as a consequence of endothelial cell K+ channel opening and hyperpolarization. In other vessels such as rabbit aorta, it has been proposed that EDHF might not even be a ‘factor' per se. Instead, the EDHF phenomenon might result from the transfer of endothelial cell hyperpolarization to the smooth muscle through gap junctions (Chaytor et al., 1998). In bovine and porcine coronary arteries, an alternative current view is that EDHF may be a cytochrome P450 metabolite, such as an epoxyeicosatrienoic acid (EET) (Hecker et al., 1994; Popp et al., 1996; Li & Campbell, 1997; Fisslthaler et al., 1999). Indeed, EETs are produced from coronary artery endothelial cells (Rosolowsky et al., 1990; Campbell et al., 1996; Rosolowsky & Campbell, 1996) and are capable of hyperpolarizing vascular smooth muscle (Campbell et al., 1996; Eckman et al., 1998). In bovine coronary artery myocytes, EETs open the large-conductance calcium-sensitive K+ channel (BKCa: Gebremedhin et al., 1998), which is selectively blocked by iberiotoxin (Kaczorowski et al., 1996). Similarly, in segments of guinea-pig coronary artery 11,12-EET induces an iberiotoxin-sensitive hyperpolarization (Eckman et al., 1998). However, iberiotoxin has little, if any, effect on the electrical or mechanical responses to EDHF in vessels including the pig and guinea-pig coronary arteries (Zygmunt & Högestätt, 1996; Corriu et al., 1996; Chataigneau et al., 1998; Eckman et al., 1998; Quignard et al., 1999).
In the porcine coronary artery it seems unlikely that EDHF is simply K+ derived from endothelial cells since small increases in extracellular K+ do not hyperpolarize its smooth muscle (Quignard et al., 1999). Gap junctions may be involved in the EDHF response in this artery, but whether these structures play an important role remains to be established. Additionally, a role for EETs cannot be excluded. Thus the aim of the present study was to characterize the electrophysiological aspects of the EDHF response in the porcine coronary artery paying particular attention to the contribution of gap junctions and of EETs in this phenomenon.
Methods
Experiments using bolus application of agonists were performed on porcine left descending coronary arteries which were obtained from the local abattoir and transported to the laboratory in ice-cold Krebs solution. The delay in removal of the heart from the animal and dissection of the coronary artery was approximately 30 min.
For the experiments with continuous application of agonists, Large-White pigs (male or female, 25 kg) were anaesthetized with a mixture of tiletamine plus zolazepam, (i.m., each 10 mg kg−1). The heart was removed and rapidly placed in an ice-cold oxygenated Krebs solution. The left anterior descending, left circumflex and right coronary arteries were then dissected free, cleaned of adherent fat and connective tissue, and maintained in oxygenated Krebs solution at room temperature.
Micro-electrode experiments
Intact vessels were pinned to the Sylgard base of a heated bath (volume 10 ml) and superfused (10 ml min−1), at 37°C, with Krebs solution containing 300 μM NG-nitro-L-arginine and 10 μM indomethacin and gassed with 95% O2/5% CO2. Smooth muscle cells were impaled from the adventitial side (unless otherwise indicated) and endothelial cells from the intimal side, using micro-electrodes filled with 3 M KCl (resistance 40–80 MΩ). Successful impalements were signalled by a sudden change in membrane potential which remained stable for at least 2 min before the experiment was commenced. In some experiments the endothelial layer was removed by gentle rubbing and the lack of endothelium in each vessel segment was confirmed by the absence of a response to substance P. Acetylcholine, bradykinin, 11,12-EET (11,12-epoxyeicosatrienoic acid), levcromakalim, NS1619 (1,3-dihydro-1-(2-hydroxy-5-(trifluoromethyl) phenyl) -5- (trifluoromethyl) - 2H - benzimidazol-2-one) and substance P were each added as bolus injections directly into the bath in quantities calculated to give (transiently) the final concentrations indicated. Barium, ouabain and the toxin K+-channel inhibitors were added to the reservoir of Krebs superfusing the bath. To investigate the effects of the gap junction inhibitor gap 27 (Chaytor et al., 1997; 1998), tissues were pre-incubated at 37°C with the peptide for 60 min prior to the recordings which were performed in the absence of the inhibitor (i.e. during its wash-out). In some experiments the coronary artery segments were continuously superfused with substance P so that responses were observed under equilibrium conditions. Recordings were made using a conventional high impedance amplifier (Intra 767; WPI Instruments or V 180, Biologic). The membrane potential was monitored simultaneously on a digital storage osciloscope (2211, Tektronix) and a pen-chart recorder (3400, Gould). Alternatively, signals were digitized and analysed using a MacLab system (AD Instruments) and 50 Hz interference at the amplifier output was selectively removed using an active processing circuit (Humbug; Digitimer).
Drugs and solutions
Krebs solution comprised (mM) Na+ 143, K+ 4.6, Ca2+ 2.5, Mg2+ 1.2, Cl− 126.4, H2PO4− 1.2, SO42− 1.2, HCO3− 25, glucose 11.1 and was bubbled with 95% O2 and 5% CO2.
The following substances were used: acetylcholine chloride, bradykinin, synthetic apamin and charybdotoxin (Latoxan, France), (±)11,12-epoxyeicosa-5Z,8Z14Z-trienoic acid (11,12-EET, Affiniti Research Products Ltd), gap 27 (amino acid sequence SRPTEKTIFII, purity >99%, synthesized by Severn Biotech, U.K.), synthetic iberiotoxin (Latoxan, France), indomethacin, levcromakalim (SmithKline Beecham), NS1619 (1,3-dihydro-1-(2-hydroxy -5- (trifluoromethyl) phenyl) -5- (trifluoromethyl)-2H-benzimidazol-2-one; RBI), ouabain, and substance P (RBI). Unless otherwise stated, all compounds were obtained from Sigma.
Statistics
Student's t-test (paired or unpaired as appropriate) was used to assess the probability that differences between mean values had arisen by chance; P<0.05 was considered to be significant.
Results
Effects of substance P and 11,12-EET
Smooth muscle impalements
The resting membrane potential of the porcine coronary artery smooth muscle was slightly but significantly depolarized by removal of the endothelial layer (+ endothelium: −51.6±0.2 mV, n=15; de-endothelialized: −50.0±0.3 mV, n=5). Substance P (100 nM) had no effect on the smooth muscle cells in the absence of the endothelium but hyperpolarized those in intact vessel segments by 28.4±0.8 mV (n=8; Figures 123). Iberiotoxin (100 nM) alone had no effect on responses to substance P or 10 μM levcromakalim in intact segments but abolished hyperpolarizations to 33 μM NS 1619. However, the combination of 100 nM iberiotoxin+100 nM apamin produced a small but significant inhibition of substance P-induced hyperpolarizations. Charybdotoxin (100 nM) also produced a small but significant inhibition of substance P, responses to which were abolished in the additional presence of 100 nM apamin (Figures 1 and 3).
Figure 1.
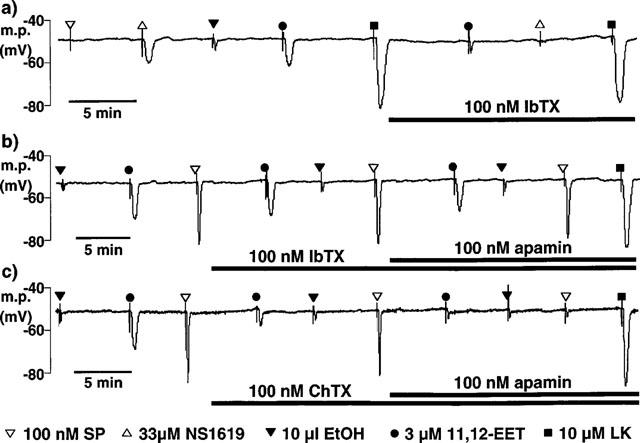
Typical traces comparing the smooth muscle hyperpolarizations in pig coronary artery induced by bolus application of 11,12-EET and its vehicle, ethanol (EtOH), with those induced by bolus application of substance P (SP), NS1619 and levcromakalim (LK). Microelectrode impalements were made through the adventitial surface. (a) In de-endothelialized vessels, substance P was without effect. Iberiotoxin (IbTX) abolished the response to NS1619 and reduced the magnitude of the hyperpolarization to 11,12-EET to that of its vehicle. (b,c) In intact vessels, iberiotoxin had little effect on the response to 11,12-EET or to substance P in the absence or presence of apamin. In contrast, charybdotoxin (ChTX) markedly reduced the hyperpolarization to 11,12-EET and slightly inhibited the response to substance P. Responses to 11,12-EET and substance P were abolished in the presence of both charybdotoxin and apamin. (a–c) Hyperpolarizations induced by levcromakalim were similar in the presence or absence of the endothelium and were not modified by the presence of the toxin K+ channel inhibitors.
Figure 2.
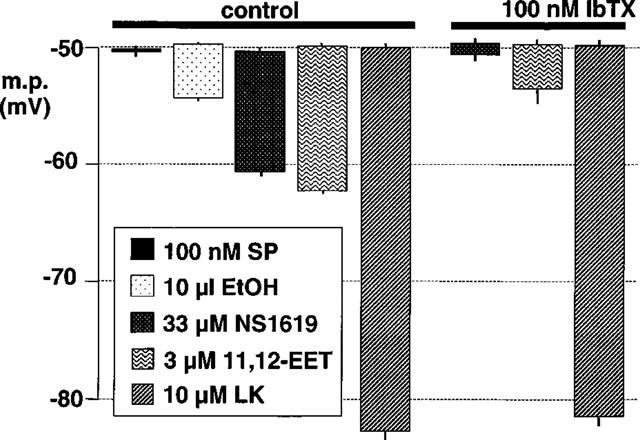
Effect of iberiotoxin on smooth muscle hyperpolarizations induced by bolus exposure to 11,12-EET, its vehicle (ethanol, EtOH), NS1619 and levcromakalim (LK) in de-endothelialized segments of porcine coronary artery. The results are the mean of four experiments typified by the trace in Figure 1(a). In the absence of the endothelium, substance P (SP) was without effect. Each column indicates the membrane potential before and after exposure to the indicated substance, +or−s.e.mean.
Figure 3.
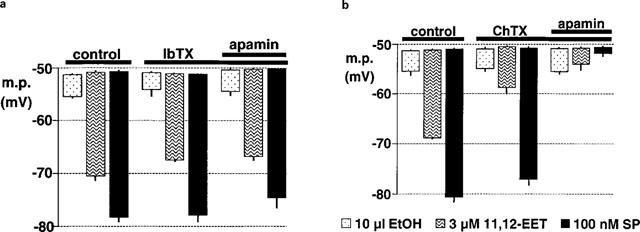
Effects of iberiotoxin (a) and charybdotoxin (b) on smooth muscle hyperpolarizations induced by bolus application of 11,12-EET, its vehicle (ethanol, EtOH) and substance P (SP) in intact segments of porcine coronary artery. The results in each graph are the mean of four experiments typified by the traces in Figure 1(b) and (c). Each column indicates the membrane potential before and after exposure to the indicated substance, +or−s.e.mean.
In contrast to substance P, 11,12-EET hyperpolarized the smooth muscle in both intact and de-endothelialized preparations but the magnitude of these effects and their toxin-sensitivities were very different. Thus, in the presence of the endothelium, 3 μM 11,12-EET hyperpolarized the smooth muscle by 18.6±0.8 mV (n=8). This value was significantly greater than in the absence of the endothelium (12.6±0.3 mV, n=4) and significantly more than the effect of ethanol, the vehicle used to dissolve 11,12-EET (smooth muscle hyperpolarization to ethanol: de-endothelialized vessel segments, 4.5±0.2 mV, n=4; intact vessel segments, 4.1±0.9 mV, n=8).
In the absence of the endothelium, the smooth muscle hyperpolarization induced by 3 μM 11,12-EET was abolished by 100 nM iberiotoxin (i.e. the hyperpolarization was not significantly different from that produced by vehicle alone) (Figures 1 and 2). However, iberiotoxin only slightly inhibited the response to 3 μM 11,12-EET in intact vessels and subsequent exposure to apamin produced no further inhibition (Figures 1 and 3). In contrast charybdotoxin markedly inhibited the response to 11,12-EET, with abolition of the residual hyperpolarization by apamin (Figures 123).
Endothelial cell impalements
The resting membrane potential of endothelial cells in intact segments of porcine coronary artery was −47.9±0.3 mV (n=8). As shown in Figure 4, 11,12-EET (3 μM) and substance P (100 nM) hyperpolarized these cells by 22.3±0.4 mV (n=8) and 31.0±0.6 mV (n=8), respectively, values which were significantly greater than for smooth muscle cells in both intact and de-endothelialized preparations (Figures 2 and 3). In intact vessel segments, iberiotoxin (100 nM) did not modify the endothelial cell hyperpolarization induced either by 11,12-EET or by substance P (Figure 4a,c). Charybdotoxin (100 nM) markedly inhibited the 11,12-EET response and slightly (but significantly; P<0.05) inhibited the substance P-induced hyperpolarization (Figure 4b,d). In the additional presence of apamin the responses to 11,12-EET and substance P were abolished (Figure 4b,d).
Figure 4.
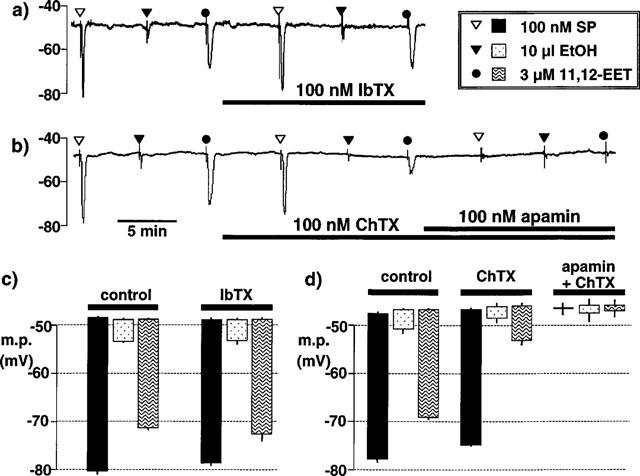
Typical traces comparing the endothelial cell hyperpolarizations in pig coronary artery induced by bolus application of 11,12-EET and its vehicle, ethanol (EtOH), with those induced by bolus application of substance P (SP). (a) Iberiotoxin (IbTX) was without effect on the hyperpolarizations to substance P, to 11,12-EET or its vehicle, ethanol (EtOH). (b) Charybdotoxin (ChTX) markedly reduced the hyperpolarization to 11,12-EET but had little effect on the response to substance P. Responses to 11,12-EET and substance P were abolished in the presence of both charybdotoxin and apamin. (c and d) Graphical representation of data from four separate experiments of the types shown in (a) and (b), respectively in which each column represents the endothelial cell membrane potential (m.p.), +or−s.e.mean, before and after exposure to substance P, EtOH or 11,12-EET. Responses are shown before (control) or after exposure to (c) 100 nM iberiotoxin or (d) 100 nM charybdotoxin in the absence and presence of 100 nM apamin.
Effects of barium+ouabain and gap junction inhibition
In this series of experiments the membrane potential of the smooth muscle cells of the porcine coronary artery was recorded with microelectrodes inserted either from the adventitial side (i.e. membrane potential of the outer layer of smooth muscle cells) or from the intimal side (i.e. membrane potential of the inner layer of smooth muscle cells).
Adventitial side
In the present study, we confirmed that acetylcholine does not hyperpolarize porcine coronary artery smooth muscle in the presence of the endothelium (Figure 5, representative of n=4 experiments), consistent with the reported lack of relaxation to acetylcholine in this preparation (Harasawa et al., 1989). Exposure to 30 μM barium and 500 nM ouabain (concentrations which inhibit inward-rectifier K+ channels and Na+, K+-ATPase and abolish the response to EDHF in the rat hepatic artery: Edwards et al., 1998; 1999b) rapidly depolarized the smooth muscle of the coronary artery and induced a series of rapid, transient depolarizations to 0 mV (mean number of transient depolarizations 4.7±0.7, n=6). Although in the presence of barium and ouabain the membrane did not hyperpolarize to the same potential on exposure to substance P, bradykinin or levcromakalim (Figure 6a), the magnitude of the hyperpolarizations was similar before and after exposure to the pump and channel inhibitors (Figure 6b). After 60 min pre-exposure to 500 μM gap 27, the resting membrane potential of the coronary artery smooth muscle (−51.5±0.4 mV, n=6) was similar to that of untreated segments from the same animals (−51.9±0.4 mV, n=6). However, after exposure to the gap 27 peptide the response to substance P was markedly reduced and was abolished in the additional presence of barium and ouabain (Figures 5 and 6). The hyperpolarization induced by bradykinin was also greatly inhibited by gap 27 and further reduced by exposure to barium+ouabain (Figure 6). Exposure to the gap 27 also reduced the number of transient depolarizations on exposure to barium+ouabain (0.8±0.4, n=6; Figure 5).
Figure 5.
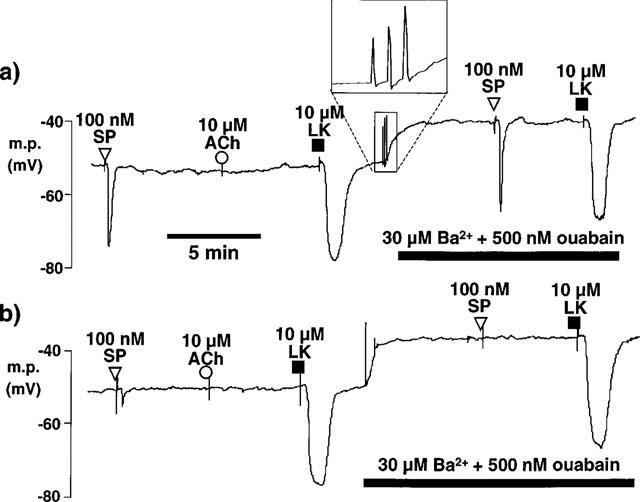
Effects of Ba2+ and ouabain and of pre-treatment with gap 27 on hyperpolarizations induced by bolus exposure to substance P (SP), acetylcholine (ACh) and levcromakalim (LK) in smooth muscle cells of pig intact coronary arteries. Microelectrode impalements were made through the adventitial surface. (a) The upper trace shows that substance P and levcromakalim each produced smooth muscle hyperpolarization whereas acetylcholine was without effect. Exposure to 30 μM Ba2++500 nM ouabain produced a depolarization usually accompanied by rapid membrane potential oscillations (inset panel) and responses to substance P and levcromakalim were essentially unchanged. (b) After 60 min incubation of a different vessel in Krebs solution containing 500 μM gap 27, substance P generated only a small hyperpolarization whereas a substantial effect of levcromakalin was always observed. Exposure to Ba2++ouabain produced a depolarization which was accompanied by fewer membrane potential oscillations. The effects of substance P were essentially absent while the effects of levcromakalim were unchanged or enhanced. Note that the rapid oscillations usually depolarized the membrane to approximately zero mV and that their indicated magnitude was limited by the speed of the A-D converter.
Figure 6.
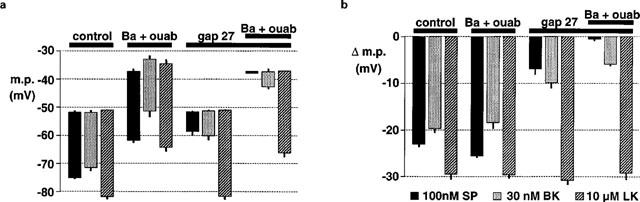
Graphical representation of data from four separate experiments of the types shown in Figure 5 in which the columns represent the membrane potential (m.p.)+or−s.e.mean in vessels exposed to 30 μM Ba2++500 nM ouabain (Ba+ouab) and 500 μM gap 27. (a) Shows absolute values of membrane potential (m.p.) before and after exposure to the agonists to highlight the depolarizing effect of Ba2++ouabain. (b) Hyperpolarizations (Δ m.p.) produced by substance P (SP), bradykinin (BK) and levcromakalim (LK) to emphasise the inhibitory effects of gap 27 and Ba2++ouabain on responses to substance P and bradykinin while responses to levcromakalim were unaffected.
Intimal side
When the smooth muscle was impaled from the intimal surface (resting membrane potential, −51.0±0.3 mV; Figure 6c) the hyperpolarizations to substance P (29.0±0.7 mV, n=5) and to levcromakalim (33.6±0.4 mV, n=5) were similar to those obtained when the muscle was impaled from the adventitial side (26.6±0.9 mV, n=12 and 31.4±0.8 mV, respectively, n=14) (compare Figure 6b with Figure 7d). As shown in Figure 7a, exposure to barium+ouabain produced an almost instantaneous depolarization of the smooth muscle which was associated with a single transient depolarization to approximately 0 mV. Again, in the presence of barium and ouabain, neither substance P nor levcromakalim hyperpolarized the membrane to the same potential as before exposure to the inhibitors but the absolute magnitude of the hyperpolarizations was increased (Figure 7a). After 60 min exposure to gap 27 (500 μM), the smooth muscle resting membrane potential (−50.9±0.5 mV, n=5; Figure 7c) was not significantly different from that of control segments from the same animals (−51.0±0.3 mV, n=5; Figure 7c). However, after pre-exposure to gap junction inhibitor the transient, rapid depolarizations to barium and ouabain were not recorded when the smooth muscle was impaled from the intimal side and the sustained depolarization developed more slowly (Figure 7b). Under these conditions there was no reduction in the magnitude of the substance P- and levcromakalim-induced hyperpolarizations compared to those in the absence of the gap peptide inhibitor (Figure 7d).
Figure 7.
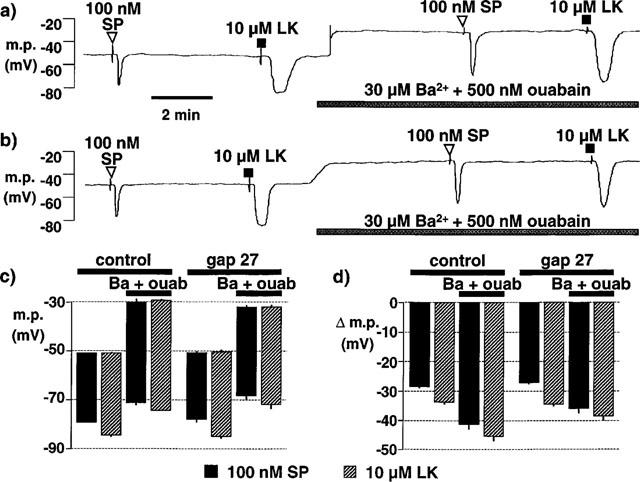
(a,b) Typical traces showing effects of Ba2+ and ouabain and of pre-treatment with gap 27 on hyperpolarizations induced by bolus application of substance P (SP) and levcromakalim (LK) in smooth muscle cells of pig intact coronary arteries impaled via the intimal surface. (a) Substance P and levcromakalim each produced smooth muscle hyperpolarization. Exposure to 30 μM Ba2++500 nM ouabain produced an almost instantaneous sustained depolarization usually accompanied by a single, rapid, transient depolarization to 0 mV. The responses to substance P and levcromakalim were not reduced by the presence of barium and ouabain. (b) After 60 min incubation of a different vessel in Krebs solution containing 500 μM gap 27, responses to substance P and levcromakalim were similar to those before exposure to the gap junction inhibitor (see panel (a)). Subsequent exposure to barium+ouabain produced a sustained depolarization which developed more slowly than in the absence of gap 27 and which was never associated with a rapid, transient depolarization. The effects of substance P and levcromakalim were not reduced in the presence of barium and ouabain after exposure to gap 27. (c,d) Graphical representation of data from 4–5 separate experiments in which the columns represent the membrane potential (m.p.), +or−s.e.mean, before and after exposure to substance P or levcromakalim under control conditions and after exposure to 30 μM Ba2++500 nM ouabain (Ba+ouab) and 500 μM gap 27 as indicated. (c) Representation showing absolute values of membrane potential (m.p.) to highlight the depolarizing effect of Ba2++ouabain. (d) Hyperpolarizations (Δ m.p.) produced by 100 nM substance P and levcromakalim to emphasise the lack of inhibitory effects of gap 27 and Ba2++ouabain.
When applied as a continuous superfusion, substance P (100 nM) induced hyperpolarizations of 19.6±1.1 mV (n=14) and 14.0±1.0 mV (n=7) when the smooth muscle cells were impaled through the intima or the adventitia, respectively. Furthermore, the rate of onset of the hyperpolarization was greater when impalements were made from the intimal side (compare Figure 8a(i) and (ii)). The resting membrane potential was unchanged after 60 min exposure to gap 27 (500 μM), irrespective of whether cells were impaled from the intimal or adventitial sides. Depolarization was induced throughout the smooth muscle by subsequent exposure to 30 μM barium plus 500 nM ouabain (Figure 8c). Under these conditions the hyperpolarization to substance P recorded just below the internal elastic lamina was not reduced (28.0±1.3 mV; n=8) (Figures 8b(i) and c–d), whereas near the adventitia it was markedly inhibited (4.0±1.5 mV, n=6; Figures 8b(ii) and c–d).
Figure 8.
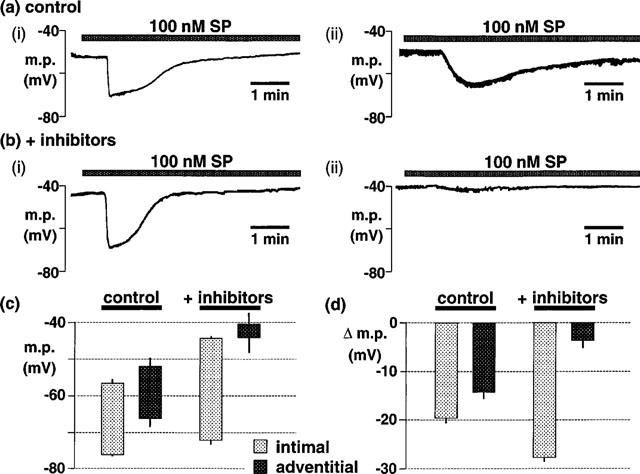
(a,b) Typical traces showing effects of gap 27 plus Ba2+ and ouabain on smooth muscle hyperpolarization induced by continuous superfusion of substance P (SP) in pig intact coronary arteries impaled via the intimal (i) or adventitial (ii) surfaces. (a) The onset of the response to substance P was more rapid when myocytes were impaled from the intimal side. (b) Following 60 min exposure to gap 27, tissues were exposed to 30 μM barium (Ba2+)+500 nM ouabain (+inhibitors). Under these conditions, the magnitude of the hyperpolarization induced by substance P was not reduced in recordings made from the intimal side but was substantially diminished in recordings from the adventitial side. (c,d) Graphical representation of data from 7–14 separate experiments in which the columns represent the membrane potential (m.p.), +or−s.e.mean, when myocytes were impaled from either the intimal or adventitial sides under control conditions or after exposure to 30 μM Ba2++500 nM ouabain and 500 μM gap 27 (+inhibitors). (c) Absolute values of membrane potential (m.p.) before and after exposure to 100 nM substance P to highlight the depolarizing effect of Ba2++ouabain. (d) Hyperpolarizations (Δ m.p.) produced by substance P to emphasise the effects of the inhibitors.
Discussion
EDHF responses in the pig coronary artery
In the present study, exposure of endothelium-intact pig coronary arteries to substance P produced smooth muscle hyperpolarizations which showed the axiomatic features of ‘EDHF' (Mombouli & Vanhoutte, 1997; Edwards & Weston, 1998). Thus the substance P-induced increase in membrane potential was produced in spite of inhibition of both cyclo-oxygenase and nitric oxide synthase. Furthermore, it was not inhibited by iberiotoxin+apamin but was abolished by charybdotoxin+apamin. These findings are similar to the results of studies (using acetylcholine) in the rat hepatic and mesenteric arteries and in the guinea-pig internal carotid artery (Corriu et al., 1996; Edwards et al., 1998; 1999b; Chataigneau et al., 1998) and confirm previous observations in the porcine coronary artery (Quignard et al., 1999).
Substance P hyperpolarized the endothelial cells in pig intact coronary vessels, an action which was unaffected by iberiotoxin but which was abolished by charybdotoxin+apamin. These findings, together with those of earlier studies in other vessels (Marchenko & Sage, 1996; Edwards et al., 1998; Doughty et al., 1999; Ohashi et al., 1999) combine to confirm that the charybdotoxin- and apamin-sensitive K+ channels involved in the EDHF pathway in the pig coronary vasculature are also on the endothelial cells in this tissue. The inability of iberiotoxin, at a concentration which abolished de-endothelialized smooth muscle responses to 11,12-EET, to reduce the endothelial cell hyperpolarizations to this eicosanoid indicates that the recording electrodes were in fact inserted into endothelial cells.
Are EETs involved in the EDHF response?
Metabolism of arachidonic acid by cytochrome P450 epoxygenases results in the formation of four regioisomeric epoxyeicosatrienoic acids, 5,6-, 8,9-, 11,12- and 14,15-EET (Fitzpatrick & Murphy, 1988). Endothelial cells are capable of producing EETs (Moore et al., 1988; Graier et al., 1995; Rosolowsky & Campbell, 1996; Hoebel et al., 1998) and, when applied exogenously, these are able to hyperpolarize, and thereby relax, some vascular smooth muscle preparations (Rosolowsky et al., 1990; Gebremedhim et al., 1992; 1998; Harder et al., 1994; Campbell et al., 1996; Leffler & Fedinec, 1997; Eckman et al., 1998; Fisslthaler et al., 1999). In some preparations such as the rabbit mesenteric artery (Hutcheson et al., 1999), the rat hepatic artery (Zygmunt et al., 1996) and the guinea-pig internal carotid artery (Chataigneau et al., 1998), exogenous EETs appear to have no relaxant or hyperpolarizing effects and are thus unlikely to represent an ‘EDHF'. However, the current view is that EETs represent an ‘EDHF' in porcine coronary arteries (Hecker et al., 1994; Popp et al., 1996; Li & Campbell, 1997; Gebremedhin et al., 1998; Fisslthaler et al., 1999). In the present study, 11,12-EET was selected for investigation since this is the isomer most likely to be involved in the EDHF response in the porcine coronary artery (Fisslthaler et al., 1999).
In de-endothelialized preparations of pig coronary artery, 11,12-EET did hyperpolarize the smooth muscle. This effect was essentially abolished by iberiotoxin at a concentration which also abolished hyperpolarizations to the BKCa opener, NS1619. However, 11,12-EET hyperpolarized the muscle to a greater extent in intact vessels and iberiotoxin (a selective inhibitor of BKCa; Kaczorowski et al., 1996) produced only a small inhibition of this hyperpolarization even in the presence of apamin. In contrast, the response to 11,12-EET in the intact vessel was abolished by apamin+charybdotoxin.
11,12-EET hyperpolarized the endothelial cells of pig coronary arteries, an action which was unaffected by iberiotoxin but was abolished by charybdotoxin plus apamin. Since iberiotoxin abolished the hyperpolarizing action of 11,12-EET on the pig coronary smooth muscle, the observed endothelial cell hyperpolarization produced by this agent must have been exerted directly on the endothelium and not transmitted to the endothelium from the muscle layer (possibly via gap junctions). Collectively, these results suggest that 11,12-EET can cause some smooth muscle hyperpolarization by opening BKCa channels on the myocytes. However, these channels have only a minor role in the EET-induced hyperpolarization in intact vessels in which the opening of charybdotoxin- (but not iberiotoxin-) sensitive and apamin-sensitive K+ channels (presumably IKCa and SKCa; Edwards et al., 1999b) on the endothelium is the prime cause of the observed response.
As already discussed, the smooth muscle hyperpolarization to both 11,12-EET and to substance P could be totally inhibited by the combination charybdotoxin+apamin. However, the EET-induced hyperpolarization was more sensitive to charybdotoxin alone than was the effect of substance P, a finding also made when endothelial cells were impaled. EETs are believed to stimulate endothelial cell calcium influx via calcium release-activated channels (CRAC) (Hoebel et al., 1997; Mombouli et al., 1999). Speculatively, the primary effect of 11,12-EET may thus be to stimulate calcium influx via endothelial CRAC at a site closely associated with IKCa. In the absence of the endothelial cell layer, the effect of 11,12-EET may result from the stimulation of calcium influx via smooth muscle CRAC channels which results in the opening of BKCa channels and a smaller hyperpolarization.
In the many studies on the modulation of smooth muscle K+ channels by exogenous EETs, the non-selective BKCa channel inhibitor charybdotoxin has surprisingly been used in preference to iberiotoxin. In vascular preparations, iberiotoxin selectively inhibits smooth muscle BKCa channels but does not inhibit endothelial cell IKCa channels whereas charybdotoxin inhibits both channel types (Edwards et al., 1999b; Kaczorowski & Garcia, 1999). In studies using isolated coronary artery smooth muscle cells, the K+ channel opened by exogenous EETs, including 11,12-EET, has a large unitary conductance. Since it is inhibited by iberiotoxin (Gebremedhin et al., 1992; 1998; Li & Campbell, 1997; Hayabuchi et al., 1998), it is likely to be BKCa (Kaczorowski et al., 1996) and this conclusion is consistent with the results of the present study. However, since the EDHF response in many preparations is inhibited by apamin+charybdotoxin and not by apamin +iberiotoxin (Zygmunt & Högestätt, 1996; Petersson et al., 1997; Eckman et al., 1998; Yamanaka et al., 1998), it seems unlikely that an EET per se is ‘EDHF'.
Gap junction involvement in the EDHF response: impalements through the adventitia
In a previous study (Edwards et al., 1999a), the gap junction inhibitors gap 27 and carbenoxolone (a water-soluble salt of β-glycyrrhetinic acid) were equi-effective inhibitors of ‘EDHF' in the guinea-pig carotid artery. However, gap 27, unlike carbenoxolone, did not depolarize the vascular smooth muscle membrane. Since the carbenoxolone-induced depolarization probably results from inhibition of Na+/K+ ATPase (Terasawa et al., 1992), gap 27 was used in the present study in an attempt to minimise such unwanted effects. This peptide inhibitor comprises the amino acid sequence of one of the extracellular loops (of a connexin, Cx43, found in both vascular smooth muscle and endothelial cells: Christ et al., 1996) which is thought to be involved in the docking of connexons to form gap junctions (Chaytor et al., 1997). It is thought that gap 27 interferes not only with formation but also with the stability of gap junctions (Chaytor et al., 1998).
When pig coronary artery smooth muscle cells lying immediately below the adventitia were impaled, gap 27 had no effect on the resting membrane potential but markedly inhibited the endothelium-dependent hyperpolarizations to substance P and to bradykinin. After exposure to the gap junction inhibitor, barium+ouabain further inhibited the bradykinin-induced hyperpolarizations and abolished those to substance P, whereas barium+ouabain alone were without effect on either response. These results are consistent with a major role for gap junctions in the EDHF response in the pig coronary vasculature, with only a minor contribution from smooth muscle inwardly-rectifying K+ channels and/or Na+/K+-ATPase (Edwards et al., 1998).
Such results are thus similar to those obtained in studies using rabbit and guinea-pig arteries which have described a reduction in the EDHF response by inhibitors of gap junctions. From these it was concluded that such connections must play a role in endothelium-dependent hyperpolarization of the smooth muscle in these vessels. Furthermore, it was assumed that the inhibited gap junctions were those which formed a functional connection between the endothelial and smooth muscle layers (Kühberger et al., 1994; Chaytor et al., 1997; 1998; Yamamoto et al., 1998; 1999; Dora et al., 1999). However, in porcine (and other) coronary arteries, gap junctions link not only the endothelial cells to the smooth muscle layer but also adjacent endothelial and smooth muscle cells within each layer (Bény & Gribi, 1989; Bény & Connat, 1992; Bény & Pacicca, 1994). Thus, an equally tenable hypothesis is that inhibition of gap-junctional coupling within the endothelial or smooth muscle layers (and not between the different layers) could account for the observed inhibition of the smooth muscle hyperpolarization by agents such as carbenoxolone and gap 27.
Gap junction involvement in the EDHF response: impalements through the intima
In an attempt to clarify the site of action of gap 27, microelectrodes were carefully advanced through the endothelium and internal elastic lamina to impale the smooth muscle cells lying immediately below the endothelial layer. Under these conditions, gap 27 was without effect on hyperpolarizations produced by substance P. In fact, the only detectable action of gap 27 was a marked slowing of the rate of myocyte depolarization following exposure to barium+ouabain which, in combination with gap 27, also failed to inhibit hyperpolarizations to substance P. The observed major differences between the inhibitory effects of gap 27, barium and ouabain on ‘sub-adventitial' and ‘sub-intimal' smooth muscle cells cannot be attributed to the method of agonist exposure since identical results were obtained using either bolus or continuous application of agonists.
Collectively, these differences between the inhibitory effects of gap 27, which are dependent on the position of the microelectrode, suggest that the actions of this agent (in the pig coronary artery at least) are not solely exerted on myoendothelial gap junctions. Instead a major action at myocyte–myocyte junctions is strongly implicated, a possibility which could explain many of the previous findings reported using this peptide (Kühberger et al., 1994; Chaytor et al., 1997; 1998; Dora et al., 1999). The pig coronary vessels used contained a 15–20 myocyte-thick layer of cells (Burnham & Weston, unpublished). If 500 μM gap 27 effectively inhibited (say) 50% of both endothelial–myocyte and myocyte–myocyte gap junctions, the endothelial cell hyperpolarization would still be well-coupled to myocytes close to the endothelium. Additionally, and consistent with the findings of the present study, the electrical changes in the endothelium would be prevented from reaching the outermost layers of muscle cells immediately below the adventitia.
Conclusions
In pig coronary artery, 11,12-EET is not EDHF. Instead, the smooth muscle hyperpolarization attributed to ‘EDHF' is initiated by endothelial cell hyperpolarization involving charybdotoxin- (but not iberiotoxin-) and apamin-sensitive K+ channels. The results minimise the contribution to the porcine coronary EDHF pathway made by the coupling of endothelium-derived K+ to smooth muscle inward rectifiers and Na+/K+ ATPase. In contrast, the data appear to emphasise the role of gap-junctional coupling between the endothelium and smooth muscle in this vascular system. Nevertheless, the present results would also be consistent with the release of an endothelium-derived hyperpolarizing factor of unknown identity following endothelial cell hyperpolarization. This possibility is the subject of further study.
Acknowledgments
This study was supported by the British Heart Foundation, the Medical Research Council and by the Servier Research Institute.
Abbreviations
- EDHF
endothelium-derived hyperpolarizing factor
- EET
epoxyeicosatrienoic acid
References
- BÉNY J.-L., CONNAT J.L. An electron-microscope study of smooth muscle cell dye coupling in the pig coronary arteries. Role of gap junctions. Circ. Res. 1992;70:49–55. doi: 10.1161/01.res.70.1.49. [DOI] [PubMed] [Google Scholar]
- BÉNY J.-L., GRIBI F. Dye and electrical coupling of endothelial cells in situ. Tissue Cell. 1989;21:797–802. doi: 10.1016/0040-8166(89)90030-x. [DOI] [PubMed] [Google Scholar]
- BÉNY J.-L., PACICCA C. Bidirectional electrical communication between smooth muscle and endothelial cells in the pig coronary artery. Am. J. Physiol. 1994;266:H1465–H1472. doi: 10.1152/ajpheart.1994.266.4.H1465. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHATAIGNEAU T., FELETOU M., DUHAULT J., VANHOUTTE P.M. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Peptides homologous to extracellular loop motifs of connexin 43 reversibly abolish rhythmic contractile activity in rabbit arteries. J. Physiol. 1997;503:99–110. doi: 10.1111/j.1469-7793.1997.099bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRIST G.J., SPRAY D.C., EL-SABBAN M., MOORE L.K., BRINK P.R. Gap junctions in vascular tissues. Evaluating the role of intercellular communication in the modulation of vasomotor tone. Circ. Res. 1996;79:631–646. doi: 10.1161/01.res.79.4.631. [DOI] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarizations of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORA K.A., MARTIN P.E.M., CHAYTOR A.T., EVANS W.H., GARLAND C.J., GRIFFITH T.M. Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: inhibition by a connexin-mimetic peptide. Biochem. Biophys. Res. Commun. 1999;254:27–31. doi: 10.1006/bbrc.1998.9877. [DOI] [PubMed] [Google Scholar]
- DOUGHTY J.M., PLANE F., LANGTON P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999;276:H1107–H1112. doi: 10.1152/ajpheart.1999.276.3.H1107. [DOI] [PubMed] [Google Scholar]
- ECKMAN D.M., HOPKINS N., MCBRIDE C., KEEF K.D. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998;124:181–189. doi: 10.1038/sj.bjp.0701778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Endothelium-derived hyperpolarizing factor: a critical appraisal. Prog. Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDNER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARDENER M.J., THOLLON C., VANHOUTTE P.M., WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999a;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., GARDENER M.J., FELETOU M., BRADY G., VANHOUTTE P.M. &, WESTON A.H. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br. J. Pharmacol. 1999b;128:1064–1070. doi: 10.1038/sj.bjp.0702916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FITZPATRICK F.A., MURPHY R.C. Cytochrome P-450 metabolism of arachidonic acid: formation and biological actions of ‘epoxygenase'-derived eicosanoids. Pharmacol. Rev. 1988;40:229–241. [PubMed] [Google Scholar]
- GEBREMEDHIM D., HARDER D.R., PRATT P.F., CAMPBELL W.B. Bioassay of an endothelium-derived hyperpolarizing factor from bovine coronary arteries: Role of a cytochrome P-450 metabolite. J. Vasc. Res. 1998;35:274–284. doi: 10.1159/000025594. [DOI] [PubMed] [Google Scholar]
- GEBREMEDHIN D., MA Y.H., FALCK J.R., ROMAN R.J., VANROLLINS M., HARDER D.R. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am. J. Physiol. 1992;263:H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., STUREK M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARASAWA Y., KIMURA M., OHNO Y., HAYASHI S. Coronary endothelium is responsive to bradykinin and arachidonate but not to acetylcholine. Arch. Int. Pharmacodyn. Ther. 1989;302:196–208. [PubMed] [Google Scholar]
- HARDER D.R., GEBREMEDHIN D., NARAYANAN J., JEFCOAT C., FALCK J.R., CAMPBELL W.B., ROMAN R. Formation and action of a P-450 4A metabolite of arachidonic acid in cat cerebral microvessels. Am. J. Physiol. 1994;266:H2098–H2107. doi: 10.1152/ajpheart.1994.266.5.H2098. [DOI] [PubMed] [Google Scholar]
- HAYABUCHI Y., NAKAYA Y., MATSUOKA S., KURODA Y. Endothelium-derived hyperpolarizing factor activates Ca2+-activated K+ channels in porcine coronary artery smooth muscle cells. J. Cardiovasc. Pharmacol. 1998;32:642–649. doi: 10.1097/00005344-199810000-00018. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOEBEL B.G., KOSTNER G.M., GRAIER W.F. Activation of microsomal cytochrome P450 mono-oxygenase by Ca2+ store depletion and its contribution to Ca2+ entry in porcine aortic endothelial cells. Br. J. Pharmacol. 1997;121:1579–1588. doi: 10.1038/sj.bjp.0701304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOEBEL B.G., STEYRER E., GRAIER W.F. Origin and function of epoxyeicosatrienoic acids in vascular endothelial cells: more than just endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 1998;10:826–830. doi: 10.1111/j.1440-1681.1998.tb02162.x. [DOI] [PubMed] [Google Scholar]
- HUTCHESON I.R., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Nitric oxide independent relaxations to acetylcholine and A23187 involve different routes of heterocellular communication – Role of gap junctions and phospholipase A2. Circ. Res. 1999;84:53–63. doi: 10.1161/01.res.84.1.53. [DOI] [PubMed] [Google Scholar]
- KACZOROWSKI G.J., GARCIA M. Pharmacology of voltage-gated and calcium-activated potassium channels. Curr. Opin. Chem. Biol. 1999;3:448–458. doi: 10.1016/S1367-5931(99)80066-0. [DOI] [PubMed] [Google Scholar]
- KACZOROWSKI G.J., KNAUS H.G., LEONARD R.J., MCMANUS O.B., GARCIA M.L. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J. Bioenerg. Biomembr. 1996;28:255–267. doi: 10.1007/BF02110699. [DOI] [PubMed] [Google Scholar]
- KÜHBERGER E., GROSCHNER K., KUKOVETZ W.R., BRUNNER F. The role of myoendothelial cell contact in non-nitric oxide-, non-prostanoid-mediated endothelium-dependent relaxation of porcine coronary artery. Br. J. Pharmacol. 1994;113:1289–1294. doi: 10.1111/j.1476-5381.1994.tb17138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFFLER C.W., FEDINEC A.L. Newborn piglet cerebral microvascular responses to epoxyeicosatrienoic acids. Am. J. Physiol. 1997;273:H333–H338. doi: 10.1152/ajpheart.1997.273.1.H333. [DOI] [PubMed] [Google Scholar]
- LI P.L., CAMPBELL W.B. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ. Res. 1997;80:877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOMBOULI J.V., HOLZMANN S., KOSTNER G.M., GRAIER W.F. Potentiation of Ca2+ signaling in endothelial cells by 11,12-epoxyeicosatrienoic acid. J. Cardiovasc. Pharmacol. 1999;33:779–784. doi: 10.1097/00005344-199905000-00015. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;17:252–256. [PubMed] [Google Scholar]
- MOORE S.A., SPECTOR A.A., HART M.N. Eicosanoid metabolism in cerebromicrovascular endothelium. Am. J. Physiol. 1988;254:C37–C44. doi: 10.1152/ajpcell.1988.254.1.C37. [DOI] [PubMed] [Google Scholar]
- OHASHI M., SATOH K., ITOH T. Acetylcholine-induced membrane potential changes in endothelial cells of rabbit aortic valve. Br. J. Pharmacol. 1999;126:19–26. doi: 10.1038/sj.bjp.0702262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HÖGESTÄTT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, beta-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIGNARD J.-F., FELETOU M., THOLLON C., VILAINE J.P., DUHAULT J., VANHOUTTE P.M. Potassium ions are not endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSOLOWSKY M., CAMPBELL W.B. Synthesis of hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Biochim. Biophys. Acta. 1996;1299:267–277. doi: 10.1016/0005-2760(95)00216-2. [DOI] [PubMed] [Google Scholar]
- ROSOLOWSKY M., FALCK J.R., WILLERSON J.T., CAMPBELL W.B. Synthesis of lipoxygenase and epoxygenase products of arachidonic acid by normal and stenosed canine coronary arteries. Circ. Res. 1990;66:608–621. doi: 10.1161/01.res.66.3.608. [DOI] [PubMed] [Google Scholar]
- TERASAWA T., OKADA T., HARA T., ITOH K. Glycyrrhetinic acid derivatives as potent inhibitors of Na+, K+-ATPase. Synthesis and structure-activity relationships. Eur. J. Med. Chem. 1992;27:345–351. [Google Scholar]
- YAMAMOTO Y., FUKUTIZ H., NAKAHIRA Y., SUZUKI H. Blockade by 18 beta-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. J. Physiol. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO Y., IMAEDA K., SUZUKI H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMANAKA A., ISHIKAWA T., GOTO K. Characterization of endothelium-dependent relaxation independent of NO and prostaglandins in guinea pig coronary artery. J. Exp. Pharmacol. Ther. 1998;285:480–489. [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D. Role of potassium channels in endothelium-dependent relaxation to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS S.C., HÖGESTÄTT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]