

Abstract
We investigated the role of bacterial lipopolysaccharide (LPS) in the reactivation of autoimmune disease by using collagen-induced arthritis (CIA) in mice in which autoimmunity to the joint cartilage component type II collagen (CII) was involved.
CIA was induced by immunization with CII emulsified with complete Freund's adjuvant at the base of the tail (day 0) followed by a booster injection on day 21. Varying doses of LPS from E. coli were i.p. injected on day 50.
Arthritis began to develop on day 25 after immunization with CII and reached a peak on day 35. Thereafter, arthritis subsided gradually but moderate joint inflammation was still observed on day 50. An i.p. injection of LPS on day 50 markedly reactivated arthritis on a dose-related fashion. Histologically, on day 55, there were marked oedema of synovium which had proliferated by the day of LPS injection, new formation of fibrin, and intense infiltration of neutrophils accompanied with a large number of mononuclear cells. The reactivation of CIA by LPS was associated with increases in anti-CII IgG and IgG2a antibodies as well as various cytokines including IL-12, IFN-γ, IL-1β, and TNF-α. LPS from S. enteritidis, S. typhimurium, and K. neumoniae and its component, lipid A from E. coli also reactivated the disease. Polymyxin B sulphate suppressed LPS- or lipid A-induced reactivation of CIA.
These results suggest that LPS may play an important role in the reactivation of autoimmune joint inflammatory diseases such as rheumatoid arthritis in humans.
Keywords: Lipopolysaccharide, collagen-induced arthritis, cytokines, autoimmune disease, rheumatoid arthritis
Introduction
Lipopolysaccharide (LPS) is a biologically unique substance produced by Gram-negative bacteria. LPS activates B cells non-specifically, resulting in marked production of polyclonal antibodies (Dziarski, 1982). LPS also plays a role in the secretion of various mediators including IL-12 and IFN-γ involved in cellular immune responses (Fong & Mosmann, 1989; Panina-Bordignon et al., 1997). Therefore, some studies demonstrated the role of LPS in diseases in which autoimmune responses were involved. For instance, injection of LPS was followed by augmentation of autoimmune nephritis in BXSB, MRL/n, and NZW mice which was associated with increased deposition of pathogenic immune complexes in the microcirculation (Granholm & Cavallo, 1991; Hang et al., 1983). The endotoxin also enhances adoptive transfer of experimental allergic encephalomyelitis in rats by lymphoid cells sensitized with myelin basic protein (Hanada et al., 1989). However, few studies clearly demonstrated the role of LPS in autoimmune arthritis.
Collagen-induced arthritis (CIA) in mice is an experimental model of autoimmune diseases induced by immunization with type II collagen (CII) (Courtenay et al., 1980). Some clinical and histological features of CIA resemble those of rheumatoid arthritis (RA) in humans (Trentham, 1982; Stuart et al., 1982b). It has been shown that both cellular and humoral immune responses to CII are involved in the pathogenesis of CIA. For instance, the disease can be passively transferred to naive recipients by IgG antibodies specific for CII and their isotype IgG2a (Stuart et al., 1982b; Hirofuji et al., 1985). Lymphoid cells from animals immunized with CII (Trentham et al., 1978) and CII-specific T cell lines and clones (Holmdahl et al., 1985) also transmit the disease.
In the present study, we show that an i.p. injection of LPS from E. coli, S. enteritidis, S. typhimurium and K. neumoniae, and the LPS active site lipid A from E. coli reactivated CIA. We also show that the reactivated arthritis was associated with increased production of anti-CII IgG and IgG2a antibodies as well as varying kinds of cytokines including IL-12, IFN-γ, IL-1β, and TNF-α, suggesting that LPS plays a role in the exacerbation of the autoimmune joint inflammation.
Methods
Animals
Male DBA/1J mice, 8–9 weeks of age, were used in all experiments. The mice were bred in the animal breeding unit of Saga Medical School, Saga, Japan. They were maintained in a temperature- and light-controlled environmental with free access to standard rodent chow and water.
Induction of collagen-induced arthritis (CIA)
To induce CIA, 1 mg of type II collagen (CII) extracted from native calf articular cartilage (Funakoshi Co., Tokyo, Japan) was dissolved in 1 m of 0.1 N acetic acid and emulsified with an equal volume of complete Freund's adjuvant (CFA) (Difco Laboratories, Detroit, MI, U.S.A.) (Yoshino, 1998a). One hundred microliters of the emulsion containing 50 μg of CII was injected s.c. into the base of the tail (day 0). Twenty-one days later, the animals were given a booster injection of the same amount of the emulsion at the same site. In some experiments, on day 50, 100 μg of CII dissolved in 100 μl of 0.005 N acetic acid was i.p. injected to further stimulate CII-specific immune response. To evaluate the severity of arthritis, the lesions of the four paws were each graded from 0–3 according to the increasing extent of erythema and oedema of the periarticular tissue as described elsewhere (Yoshino & Cleland, 1992). The maximum possible score is 12.
Administration of LPS
LPS from E. coli 011:B4 (Difco) was used in all experiments. Varying doses of LPS were dissolved in 100 μl of sterile, pyrogen-free saline and injected i.p. on day 50. As a control, 100 μl of saline alone was given on the same day. In some experiments, LPS from S. enteritidis, S. typhimurium (Difco), and K. neumoniae (Sigma Chemical Co., St. Louis, MO, U.S.A.) and lipid A from E. coli K12D31m4 (Funakoshi Co., Tokyo, Japan) were also i.p. administered.
Histology
Mice were killed on days 50 (immediately before administration of LPS) and 55. Hindpaws were amputated, fixed in 4% formalin, and decalcified (Yoshino et al., 1991). The tissues were embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin.
Measurement of antibodies to CII
Mice were killed on day 65 and sera collected were heat inactivated at 56°C for 30 min. Anti-CII IgG and IgG2a antibodies were measured by using an ELISA (Yoshino & Ohsawa, 1997). In brief, 96-well flat-bottomed microtiter plates were incubated with 100 μl well−1 of CII (100 μg ml−1) at 37°C for 1 h and washed three times with PBS containing 0.05% Tween 20. The wells were then blocked by incubation with 100 μl of PBS containing 1% ovalbumin (Sigma) at 37°C for 1 h. After washing, the plates were incubated with 100 μl of a 1 : 600 dilution of each serum sample at 37°C for 30 min. The plates were washed, and 100 μl well−1 of a 1 : 1000 dilution of rat anti-mouse IgG or IgG2a labelled with alkaline phosphatase (Pharmigen, San Diego, CA, U.S.A.) was added and incubated at 37°C for 1 h. After washing, 100 μl of 3 mM of p-nitrophenylphosphate (Bio-Rad Laboratories, Hercules, CA, U.S.A.) was added per well and the plates were incubated in the dark at room temperature for 15 min. The absorbance was then measured at 405 nm in a Titertec Multiscan spectrophotometer (EFLAB, Helsinki, Finland). The results were expressed as absorbance units at OD405±s.e.mean.
Measurement of cytokines
Spleens were removed on day 53 and cell suspensions were prepared. Erythrocytes in the cells were lysed with Tris-NH4Cl. A total 5×106 cells in 1 ml of RPMI 1640 (Flow Laboratories, Inc., McLean, VA, U.S.A.) containing 1 mM glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 5×10−5 M 2-mercaptoethanol, and 1% heat inactivated autologous mouse serum were cultured in 24-well tissue culture plates with 50 μg ml−1 CII (Yoshino, 1998b). Forty-eight hours later, supernatants were harvested and stored at −70°C until assayed. Cytokine production was quantified by ELISA. The ELISA kits for IL-12, IFN-γ, IL-1β, and TNF-α were purchased from Funakoshi Co. (Tokyo, Japan).
Administration of polymyxin B sulphate (PMB)
Fifty micrograms of PBM (Sigma) dissolved in 100 μl of sterile, pyrogen-free saline was mixed with 5 μg of LPS or 2 μg of lipid A and i.p. injected on day 50.
Treatment with anti-IFN-γ monoclonal antibody (mAb)
Anti-IFN-γ-mAb produced by the hybridoma R4-6A2 (American Type Culture Collection, Rockville, MD, U.S.A.) was precipitated by ammonium sulphate from ascitic fluid of SCID mice inoculated with the cells and purified with a protein G Sepharole 4 FF column (Pharmacia Biotec, Tokyo, Japan) (Yoshino, 1998a). The protein content was quantified by absorbance measurement at 280 nm. Purified anti-IFN-γ mAb (500 μg) dissolved in 0.1 ml saline was injected i.p. on day 50, immediately before injection of LPS, and then daily from days 51–55. The same amount of anti-IFN-γ mAb alone was also i.p. injected on the same days.
Statistics
To analyse data statistically, the Mann-Whitney U-test was used as one of non-parametric statistical methods since sample sizes in our experiments were small; therefore, the normality obtained was poor.
Results
The role of LPS in the reactivation of CIA
Mice immunized with CII had signs of arthritis on day 25 and the joint inflammation reached a peak on day 35 (Figure 1). Thereafter, the joint inflammation decreased gradually by day 50. Injection of LPS on day 50 resulted in marked reactivation of CIA. The reactivation of the disease by LPS was already observed on day 51 and reached a peak on day 55, followed by a relatively rapid decline by day 70. Not only already affected but also unaffected joints flared up following injection of the endotoxin. Injection of saline on day 50 failed to reactivate the joint inflammation. Administration of varying doses of LPS showed that the reactivation of CIA was dose-dependent (Figure 2).
Figure 1.
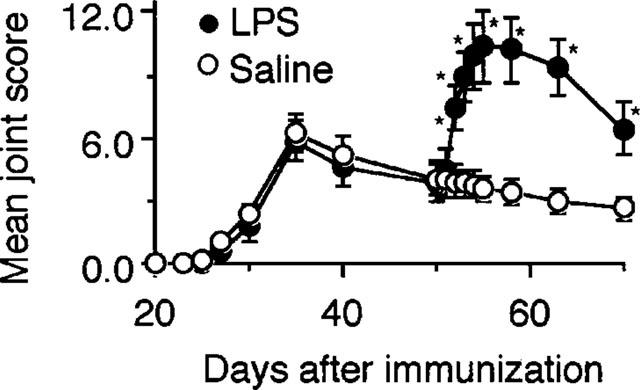
Reactivation of CIA by LPS. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. The severity of arthritis was determined on days 20, 23, 25, 27, 30 and 35, 40, 50, 51, 52, 53, 54, 55, 58, 63 and 70. Five micrograms of LPS was i.p. injected on day 50. Saline was administered as a control on the same days. Bars show the mean±s.e.mean of 12 mice. *P<0.05 versus saline, Mann-Whitney U-test. Data are representative of three experiments.
Figure 2.
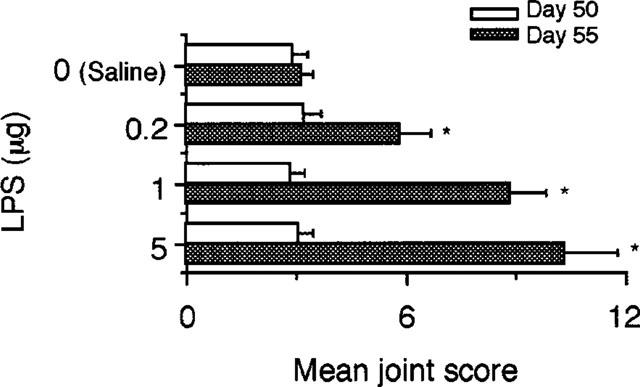
Dose-related reactivation of CIA by LPS. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. On day 50, saline and the indicated doses of LPS were i.p. injected. The severity of arthritis was determined on day 50, i.e. immediately before administration of LPS and on day 55. Bars show the mean±s.e.mean of ten mice. *P<0.05 versus saline, Mann-Whitney U-test. Data are representative of three experiments.
Histologically, on day 50 before injection of LPS, there was moderate proliferation of synovium with cell infiltrate in which mononuclear cells predominated (Figure 3A). Administration of LPS on day 50 was followed by marked oedema of the synovium, new formation of fibrin, and intense infiltration of neutrophils accompanied with a large number of mononuclear cells on day 55 (Figure 3B), while mice administered with saline showed histologic changes similar to those shown in Figure 3A (not shown).
Figure 3.
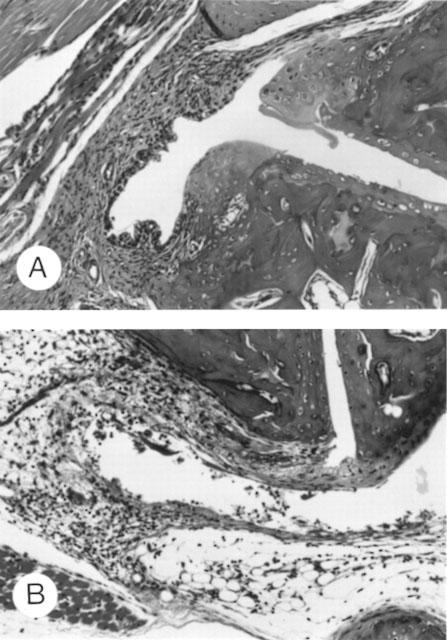
Histologic changes in tarsal joints of mice with CIA following administration of LPS. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. Saline or 5 μg of LPS was i.p. injected on day 50. Tarsal joints were histologically examined on day 50, i.e. immediately before injection of LPS (A) and on day 55 after injection of LPS (B). Haematoxylin and eosin stained, ×200 (original magnification).
The effect of LPS on anti-CII IgG and IgG2a antibody production
Since IgG and its isotype, IgG2a antibodies to CII play an important role in CIA (Stuart et al., 1982a; Hirofuji et al., 1985), production of these antibodies was examined after administration of LPS. As shown in Figure 4, injection of the endotoxin was followed by increases in anti-CII IgG and IgG2a antibodies, although the increase in anti-CII IgG2a appeared to be greater than that in anti-CII IgG.
Figure 4.
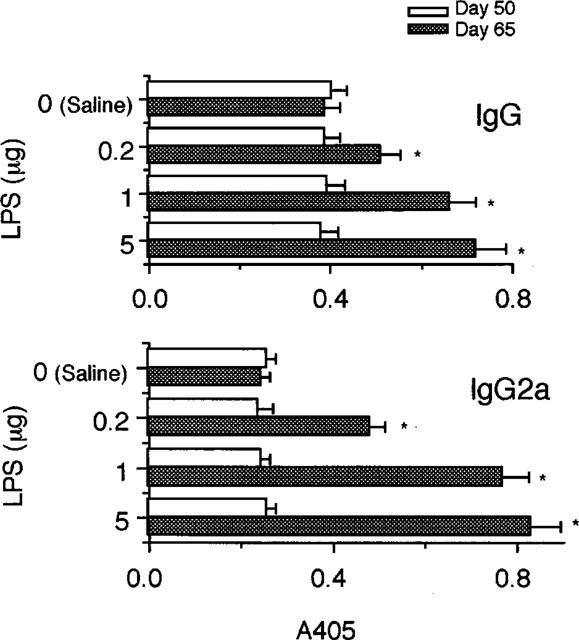
Enhancement of anti-CII antibody production by LPS. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. On day 50, saline and the indicated doses of LPS were i.p. injected and the serum levels of anti-CII IgG and IgG2a antibodies were determined on day 50, i.e. immediately before administration of LPS and on day 65. Bars show the mean±s.e.mean of six mice. *P<0.05 versus saline, Mann-Whitney U-test. Data are representative of three experiments.
The effect of LPS on cytokine secretion
To examine whether the reactivation of CIA by LPS was associated with secretion of cytokines, IL-12, IFN-γ, IL-1β, and TNF-α were measured on day 53. The results showed that administration of the endotoxin increased the levels of all these cytokines in a dose-related manner (Table 1). The increase rates of the secretion of IL-12, IFN-γ, IL-1β, and TNF-α by 5 μg of LPS were 1123, 746, 1198, and 1580%, respectively.
Table 1.
Secretion of cytokines in mice treated with LPS

The role of varying types of LPS and lipid A in the reactivation of CIA
LPS from other Gram-negative bacteria and lipid A from E. coli were also used to test their ability to reactivate CIA. As shown in Figure 5, administration of all types of LPS from S. enteritidis, S. typhimurium, and K. neumoniae resulted in the reactivation of joint inflammation and the extent of the reactivation was similar to that caused by the endotoxin from E. coli. Lipid A from E. coli was also active in exacerbating CIA significantly.
Figure 5.
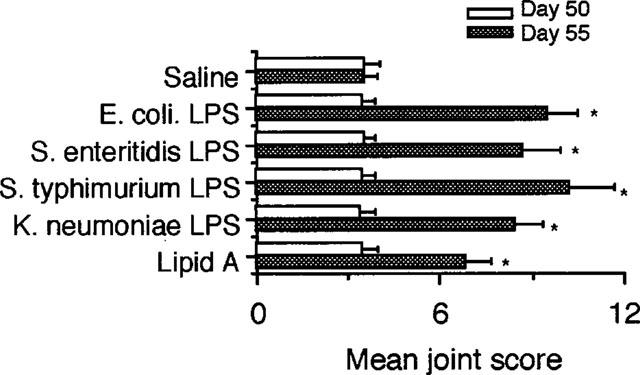
Reactivation of CIA by varying types of LPS and lipid A. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. On day 50, saline, 5 μg of LPS from E. coli, S. enteritidis, S. typhimurium, and K. peumoniae, and 2 μg of lipid A from E. coli were i.p. injected. The severity of arthritis was determined on day 50, i.e. immediately before administration of LPS and on day 55. Bars show the mean±s.e.mean of eight mice. *P<0.05 versus saline, Mann-Whitney U-test. Data are representative of two experiments.
The effect of PMB on LPS- or lipid A-induced reactivation of CIA
To investigate whether PMB which neutralizes LPS and lipid A (van Miert et al., 1997) can suppress the reactivation of CIA by these bacterial cell wall components, the antibiotic was mixed with LPS or lipid A before injection on day 50. The results showed that PMB markedly blocked the reactivation of joint inflammation by these substances (Figure 6).
Figure 6.
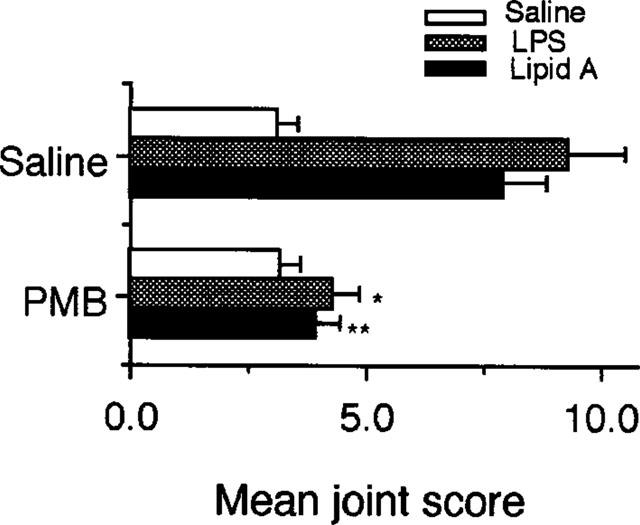
Suppression by polymyxin B of LPS-induced reactivation of CIA. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. On day 50, 100 μl of saline alone or 100 μl of saline containing 50 μg of polymyxin B sulphate (PMB) was mixed with saline, 5 μg of LPS, and 2 μg of lipid A and then i.p. injected on day 50. The severity of arthritis was determined on day 55. Bars show the mean±s.e.mean of ten mice. *P<0.05 versus saline- and LPS-treatment; **P<0.05 versus saline- and lipid A-treatment, Mann-Whitney U-test. Data are representative of two experiments.
The effect of anti-IFN-γ mAb on LPS-induced reactivation of CIA
The role of IFN-γ involved in TH1 responses (Fong & Mosmann, 1989) in LPS-induced reactivation of CIA was investigated by using anti-IFN-γ mAb. As shown in Figure 7, the administration of LPS plus anti-IFN-γ mAb significantly diminished the enhancement of the severity of arthritis by the injection of LPS alone. The rates of increase in the severity of joint inflammation in mice treated with LPS plus anti-IFN-γ mAb and LPS alone were 83 and 257%, respectively. The administration of anti-IFN-γ mAb alone failed to affect the ongoing disease. Soluble CII alone injected on day 50 significantly reactivated CIA with the increase rate 54% in the severity of arthritis.
Figure 7.
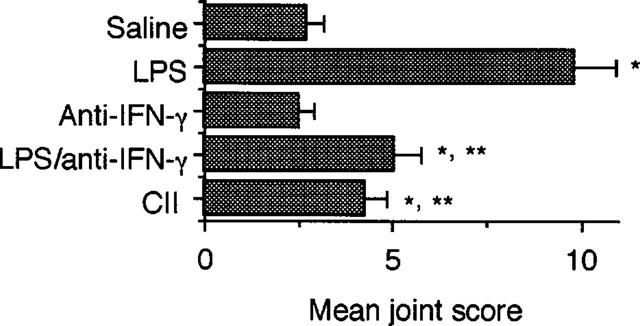
Effect of anti-IFN-γ mAb on reactivation of CIA by LPS. Mice were immunized with CII on day 0 followed by a booster injection on day 21 as described in Methods. On day 50, saline, 5 μg of LPS alone, 500 μg of anti-IFN-γ mAb alone, 5 μg of LPS plus 500 μg of anti-IFN-γ mAb, or 100 μg of CII was i.p. injected. Injection of anti-IFN-γ mAb was continued daily from days 51 to 55. The severity of arthritis was determined on day 60. Bars show the mean±s.e.mean of eight mice. *P<0.05 versus saline; **P<0.05 versus LPS alone, Mann-Whitney U-test. Data are representative of two experiments.
Discussion
In many autoimmune diseases including rheumatoid arthritis, remission and recurrence are often repeatedly observed. However, the cause of the recurrence as well as the mechanism of the remission are not well clarified at present. In the present study, we showed that bacterial LPS might play a role in the recurrence of autoimmune arthritis since systemic injection of the endotoxin from E. coli as well as from S. enteritidis, S. typhimurium, and K. neumonia markedly reactivated CIA in mice. The LPS active site lipid A was also effective in stimulating the existing joint inflammation. The reactivation of CIA by LPS and lipid A was blocked by PMB that neutralized these Gram negative bacterial cell wall components (van Miert et al., 1987), supporting the role of LPS and lipid A in the recurrence of joint inflammation. The reactivation of CIA by LPS was also confirmed by histologic changes in joints of LPS-treated mice showing marked oedema of synovium which had proliferated by the day of the endotoxin challenge, new formation of fibrin, and infiltration of many neutrophils accompanied with mononuclear cells, while control untreated animals failed to have such histologically acute inflammatory features.
There are a number of studies demonstrating a role for LPS in autoimmune diseases. For instance, it was previously shown that the injection of LPS enhanced autoimmune nephritis in BXSB, MRL/n, or NZW lupus-prone mice (Granholm & Cavallo, 1991; Hang et al., 1983) and adoptive transfer of experimental autoimmune encephalomyelitis (Hanada et al., 1989). It was also demonstrated that LPS acted as an adjuvant to induce autoimmune uveitis (Yokochi et al., 1993) and autoimmune myocarditis in rats (Kato et al., 1993). However, few studies clearly showed that LPS has ability to reactivate existing autoimmune arthritis.
Although the precise mechanism of the reactivation of CIA by LPS is currently unclear, the increased production of in anti-CII IgG and the isotype IgG2a antibodies following treatment with the endotoxin may have in part contributed to the reactivation of the disease because it was previously demonstrated that IgG, especially the IgG2a antibodies to CII played a critical role in CIA (Stuart et al., 1982a; Hirofuji et al., 1985). The marked production of anti-CII IgG2a antibodies by LPS may be due to the augmented secretion of IFN-γ since this Th1 cytokine plays a role in production of this isotype of antibody (Snapper & Paul, 1987; Finkelman et al., 1988). Furthermore, since LPS does not normally activate T cells directly, but stimulates non-lymphocytes such as macrophages known to be a IL-12-producing cell (Verhasselt et al., 1997) and because this cytokine plays a role in the secretion of IFN-γ, the enhanced secretion of IFN-γ may be due to the increase in IL-12 in mice injected with the endotoxin.
The exacerbation of CIA by LPS may also be explained by the increased secretion by the endotoxin of other cytokines including IL-1β and TNF-α which are involved not only in immune responses but also in inflammation itself (Arend & Dayer, 1995; Brennan et al., 1992). Stimpson et al. (1987) demonstrated that arthritis induced by the toxic effect of peptidoglycan-polysaccharide polymers injected intra-articularly in rats was reactivated by a systemic injection of LPS. It is also likely that anti-CII antibodies and the above proinflammatory mediators acted synergistically to reactivate arthritis. Terato et al. (1995) showed that injection of LPS reduced the amount of anti-CII monoclonal antibodies required for the induction of joint inflammation, suggesting the synergistic effect of LPS on arthritis.
The involvement of proinflammatory cytokines as well as CII-specific immune responses in the reactivation of CIA by LPS was also supported by the experiments in which anti-IFN-γ mAb was used to neutralize the cytokine so that antigen-specific Th1 responses were expected to be suppressed (Fong & Mossmann, 1989). We previously demonstrated that CIA appeared to be a Th1-dominant disease (Yoshino, 1998a). It is of note that proinflammatory cytokines might have more contributed to the flare up than CII-specific Th1 immune responses since the treatment with LPS plus anti-IFN-γ-mAb was followed by an increase in the severity of joint inflammation up to 83%, while the injection of LPS alone resulted in a greater increase (257%). Furthermore, only 54% increase in the severity was seen in mice injected with soluble CII alone. The result showing no difference in the severity of CIA between mice treated with saline- and anti-IFN-γ alone on day 50 suggests that Th1 responses have not been critically involved in the ongoing disease at that time.
Aoki et al. (1996) reported that patients with RA had significantly increased titers of antibodies against E. coli in the sera and synovial fluids compared with control healthy subjects, indicating that the patients appeared to be more sensitized with the enteric bacteria. By using immunoblot analysis, they also found a ladder-like banding pattern equivalent to enterobacterial common antigen associated with LPS. Heumann et al. (1995) showed high levels of LPS binding protein in sera and in synovial fluids in RA patients. We recently found that LPS administered orally was detected in blood (unpublished data), suggesting the endotoxin from intestinal flora may cross the mucosa and be distributed systemically. In addition, CII is a major component of cartilage and anti-CII IgG antibodies are seen in sera from subjects with RA (Gay et al., 1980; Stuart et al., 1983). Furthermore, CII and anti-CII antibody complexes are present in sera from the patients (Claque & Moore 1984). Taken together, these findings suggest that LPS may play a role in the recurrence of RA, although the definite role of anti-CII antibodies in the disease has not been established.
In summary, the systemic injection of LPS resulted in the reactivation of CIA in mice that was associated with the increased production of anti-CII IgG and IgG2a antibodies as well as the enhanced secretion of cytokines including IL-12, IFN-γ, IL-1β, and TNF-α. Thus, LPS may play a role in the exacerbation of autoimmune diseases.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan.
Abbreviations
- CIA
collagen-induced arthritis
- CII
type II collagen
- LPS
lipopolysaccharide
- PMB
polymyxin B sulphate
- RA
rheumatoid arthritis
References
- AOKI S., YOSHIKAWA K., YOKOYAMA T., NONOGAKI T., IWASAKI S., MITSUI T., NIWA S. Role of enteric bacteria in the pathogenesis of rheumatoid arthritis: evidence for antibodies to enterobacterial common antigens in rheumatoid sera and synovial fluids. Ann. Rheum. Dis. 1996;55:363–369. doi: 10.1136/ard.55.6.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AREND W.P., DAYER J.M. Inhibition of the production and effects of interleukin-1 and tumor necrosis factor alpha in rheumatoid arthritis. Arth. Rheum. 1995;38:151–160. doi: 10.1002/art.1780380202. [DOI] [PubMed] [Google Scholar]
- BRENNAN F.M., MAINI R.N., FELDMANN M. TNF-alpha: a pivotal role in rheumatoid arthritis. Br. J. Rheumatol. 1992;31:293–298. doi: 10.1093/rheumatology/31.5.293. [DOI] [PubMed] [Google Scholar]
- CLAQUE R.B., MOORE L.J. IgG and IgM antibody to native type II collagen in rheumatoid arthritis serum and synovial fluid: evidence for the presence of collagen-anticollagen immune complexes in synovial fluid. Arth. Rheum. 1984;27:1370–1377. doi: 10.1002/art.1780271207. [DOI] [PubMed] [Google Scholar]
- COURTENAY J.S., DALLMAN M.J., DAYAN A.D., MARTIN A., MOSEDALE B. Immunization against heterologous type II collagen induces arthritis in mice. Nature. 1980;283:666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- DZIARSKI R. Preferential induction of autoantibody secretion in polyclonal activation by peptidoglycan and lipopolysaccharide. I. In vitro studies. J. Immunol. 1982;128:1018–1025. [PubMed] [Google Scholar]
- FINKELMAN F.D., KATONA I.M., MOSMAN T.R., COFFMAN R.L. IFN-gamma regulates the isotypes of Ig secreted during in vivo humoral immune responses. J. Immunol. 1988;140:1022–1027. [PubMed] [Google Scholar]
- FONG T.A., MOSMANN T.R. The role of IFN-gamma in delayed type hypersensitivity mediated by Th1 clones. J. Immunol. 1989;143:2887–2893. [PubMed] [Google Scholar]
- GAY S., GAY R.E., MILLER E.J. The collagens of the joint. Arth. Rheum. 1980;23:937–941. doi: 10.1002/art.1780230810. [DOI] [PubMed] [Google Scholar]
- GRANHOLM N.A., CAVALLO T. Bacterial lipopolysaccharide enhances deposition of immune complexes and exacerbate nephritis in BXSB lupus-prone mice. Clin. Exp. Immunol. 1991;85:270–277. doi: 10.1111/j.1365-2249.1991.tb05717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANADA T., DRISCOLL B.F., KIES M.W., ALVORD E.C., Jr LPS augments adoptive transfer of experimental allergic encephalomyelitis in the Lewis rat. Autoimmun. 1989;2:275–284. doi: 10.3109/08916938908997153. [DOI] [PubMed] [Google Scholar]
- HANG L., SLACK J.H., AMUNDSON C., IZUI S., THEOFILOPOULOS A.N., DIXON F.J. Induction of murine autoimmune disease by chronic polyclonal B cell activation. J. Exp. Med. 1983;157:874–883. doi: 10.1084/jem.157.3.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEUMANN D., BAS S., GALLAY P., LE ROY D., BARRAS C., MENSI N., GLAUSER M.P., VISCHER T. Lipopolysaccharide binding protein as a marker of inflammation in synovial fluid of patients with arthritis: correlation with interleukin 6 and C-reactive protein. J. Rheumatol. 1995;22:1224–1229. [PubMed] [Google Scholar]
- HIROFUJI T., KAKIMOTO K., MORI H., NAGAI Y., SAISHO K., SUMIYASHI A., KOGA T. Characterization of monoclonal antibody specific for human type II collagen: possible implication in collagen-induced arthritis. Clin. Exp. Immunol. 1985;62:159–166. [PMC free article] [PubMed] [Google Scholar]
- HOLMDAHL R., KLARESKOG L., RUBIN K., LARSSON E., WIGZELL H. T lymphocytes in collagen II-induced arthritis in mice: characterization of arthritogenic collagen II-specific T-cell lines and clones. Scand. J. Immunol. 1985;22:295–306. doi: 10.1111/j.1365-3083.1985.tb01884.x. [DOI] [PubMed] [Google Scholar]
- KATO N., FUJII Y., AGATA N., KIDO N., OHTA M., NAITO S., YOKOCHI T.Experimental murine model for autoimmune myocarditis using Klebsiella pneumoniae O3 lipopolysaccharide as a potent immunological adjuvant Autoimmun. 199314231–236.1993 [DOI] [PubMed] [Google Scholar]
- PANINA-BORDIGNON P., MAZZEO D., LUCIA P.D., D'AMBROSIO D., LANG R., FABBRI L., SELF C., SINIGAGLIA F. Beta 2-agonists prevent Th1 development by selective inhibition of interleukin 12. J. Clin. Invest. 1997;100:1513–1519. doi: 10.1172/JCI119674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNAPPER C.M., PAUL W.E. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–947. doi: 10.1126/science.3107127. [DOI] [PubMed] [Google Scholar]
- STIMPSON S.A., ESSER R.E., CARTER P.B., SARTOR R.B., CROMARTIE W.J., SCHWAB J.H. Lipopolysaccharide induces recurrence of arthritis in rat joints previously injured by peptidoglycan-polysaccharide. J. Exp. Med. 1987;165:1688–1702. doi: 10.1084/jem.165.6.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUART J.M., CREMER M.A., TOWNES A.S., KANG A.H. Type II collagen-induced arthritis in rats: passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. J. Exp. Med. 1982a;155:1–16. doi: 10.1084/jem.155.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STUART J.M., HUFFSTUTTER E.H., TOWNES A.S., KANG A.H. Incidence and specificity of antibodies to type I, II, III, IV, and V collagen in rheumatoid arthritis and other rheumatic diseases as measured by 125I-radioimmunoassay. Arth. Rheum. 1983;26:832–840. doi: 10.1002/art.1780260703. [DOI] [PubMed] [Google Scholar]
- STUART J.M., TOWNES A.S., KANG A.H. The role of collagen autoimmunity in animals and human diseases. J. Invest Dermatol. 1982b;79 suppl:121S–127S. doi: 10.1111/1523-1747.ep12545958. [DOI] [PubMed] [Google Scholar]
- TERATO K., HARPER D.S., GRIFFITHS M.M., HASTY D.L., YE X.J., CREMER M.A., SEYER J.M. Collagen-induced arthritis in mice: synergistic effect of E.Coli lipopolysaccharide bypasses epitope specificity in the induction of arthritis with monoclonal antibodies to type II collagen. Autoimmun. 1995;22:137–147. doi: 10.3109/08916939508995311. [DOI] [PubMed] [Google Scholar]
- TRENTHAM D.E. Collagen arthritis as a relevant model for rheumatoid arthritis: evidence pro and con. Arth. Rheum. 1982;25:911–916. doi: 10.1002/art.1780250801. [DOI] [PubMed] [Google Scholar]
- TRENTHAM D.E., DYNESIUS R.A., DAVID J.R. Passive transfer by cells by type II collagen-induced arthritis in rats. J. Clin. Invest. 1978;62:359–366. doi: 10.1172/JCI109136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN MIERT A.S., VAN DUIN C.T., WENSING T. Effects of pentoxifylline and polymyxin B on the acute-phase-response to Escherichia coli endotoxin in dwarf goats. J. Vet. Pharmacol. Ther. 1987;20:61–68. doi: 10.1046/j.1365-2885.1997.00041.x. [DOI] [PubMed] [Google Scholar]
- VERHASSELT V., BUELENS C., WILLEMS F., DE GROTTE D., HAEFFNER-CAVAILLON N., GOLDMAN M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: evidence for a soluble CD14-dependent pathway. J. Immunol. 1997;158:2919–2925. [PubMed] [Google Scholar]
- YOKOCHI T., FUJII Y., NAKASHIMA I., KIUCHI M., KOJIMA K., KATO N. A murine model of experimental autoimmune lens-induced uveitis using Klebsiella O3 lipopolysaccharide as a potent immunological adjuvant. Int. J. Exp. Pathol. 1993;74:573–582. [PMC free article] [PubMed] [Google Scholar]
- YOSHINO S. Effect of a monoclonal antibody against interleukin-4 on collagen-induced arthritis in mice. Br. J. Pharmacol. 1998a;123:237–242. doi: 10.1038/sj.bjp.0701607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHINO S. Treatment with an anti-IL-4 monoclonal antibody blocks suppression of collagen-induced arthritis in mice by oral administration of type II collagen. J. Immunol. 1998b;160:3067–3071. [PubMed] [Google Scholar]
- YOSHINO S., CLELAND G.L. Depletion of αβ T cells by a monoclonal antibody against the αβ T cell receptor suppresses established adjuvant arthritis, but not established collagen-induced arthritis in rats. J. Exp. Med. 1992;175:907–915. doi: 10.1084/jem.175.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHINO S., CLELAND L.G., MAYRHOFER G. Treatment of collagen-induced arthritis in rats with a monoclonal antibody against the α/β T cell antigen receptor. Arthritis Rheum. 1991;34:1039–1047. doi: 10.1002/art.1780340814. [DOI] [PubMed] [Google Scholar]
- YOSHINO S., OHSAWA M. Effect of a monoclonal antibody against interleukin-4 on the induction of oral tolerance in mice. Eur. J. Pharmacol. 1997;336:203–209. doi: 10.1016/s0014-2999(97)01244-2. [DOI] [PubMed] [Google Scholar]