

Abstract
Extracellular purine and pyrimidine nucleotides have been implicated in the regulation of several cellular functions including mitogenesis. In this study, experiments were conducted to characterize the P2Y receptor on C6 glioma cells responsible for stimulating cell proliferation associated with mitogen-activated protein kinase (MAPK) activation.
UTP and ATP produced a similar effect on [3H]-thymidine incorporation in a time- and concentration-dependent manner, suggesting the involvement of P2Y2 receptor in mediating proliferation of C6 glioma cells.
In response to UTP, both p42 and p44 MAPK were activated in a time- and concentration-dependent manner using Western blot analysis with an anti-phospho-p42/p44 MAPK antibody. The phosphorylation reached maximal levels after 5 min and declining by 30 min.
Pretreatment with pertussis toxin (PTX) did not change these responses to UTP. Both DNA synthesis and phosphorylation of MAPK in response to UTP were attenuated by tyrosine kinase inhibitors, genistein and herbimycin A, protein kinase C (PKC) inhibitors, staurosporine and GF109203X, and removal of Ca2+ by addition of BAPTA/AM plus EGTA.
UTP-induced [3H]-thymidine incorporation and p42/p44 MAPK phosphorylation was completely inhibited by PD98059 (an inhibitor of MEK1/2). Furthermore, we showed that overexpression of dominant negative mutants of Ras (RasN17) and Raf (Raf-301) completely suppressed MEK1/2 and p42/p44 MAPK activation induced by ATP and UTP.
These results conclude that the mitogenic effect of UTP is mediated through a P2Y2 receptor that involves the activation of Ras/Raf/MEK/MAPK pathway. UTP-mediated MAPK activation was modulated by Ca2+, PKC, and tyrosine kinase associated with cell proliferation in cultured C6 glioma cells.
Keywords: Purinergic receptors, proliferation, mitogen-activated protein kinase, glioma cells, tyrosine kinase, protein kinase C
Introduction
ATP is released from cells and acts in a well-established physiological role as an extracellular signalling molecule (Burnstock, 1997). Receptors for extracellular ATP were subdivided into P2X and P2Y receptors on the basis of pharmacological studies using isolated preparations from several species (Burnstock & Kennedy, 1985). These receptor subtypes differed in their transduction mechanisms; P2X receptors are transmitter-gated ion channels whereas P2Y receptors are members of the G protein-coupled receptor superfamily (Fredholm et al., 1997). So far, P2X receptor subtypes have been subclassified mainly as P2X1 to P2X7 (Surprenant et al., 1996). The stable analogue of ATP, α,β-MeATP, is a potent agonist for the P2X1 receptor (Burnstock & Kennedy, 1985). P2Y receptors have been subclassified as P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors (Hourani & Hall, 1994; Communi et al., 1997). The P2Y1 receptor is activated by adenine nucleotides and not by uridine nucleotides. ADP is the most potent natural agonist for this receptor, and whether or not ATP is an agonist for this receptor remains uncertain (Leon et al., 1997). The P2Y2 receptor is activated equipotently by ATP and UTP but is activated, at most only poorly, by diphosphate nucleotides (Nicholas et al., 1996). The P2Y4 receptor is activated by UTP and weakly, if at all, by ATP, UDP, or ADP (Nicholas et al., 1996). The P2Y6 receptor is UDP-sensitive and is activated weakly, if at all, by UTP, ATP, or ADP (Nicholas et al., 1996). The P2Y11 receptor has been shown to be activated by ATP and ADP, but not by UTP or UDP (Communi et al., 1997). In several cell types, P2Y receptors are G protein coupled receptors which are associated with phospholipase C (PLC) activation with subsequent increases in inositol 1,4,5-trisphosphate (IP3) formation, intracellular Ca2+ release, diacylglycerol production, and activation of PKC (Boarder et al., 1995; Harden et al., 1995; Yang et al., 1997).
ATP has been shown to increase [3H]-thymidine incorporation in Swiss 3T3 (Huang et al., 1989), A431 (Huang et al., 1989), and renal mesangial cells (Huwilder & Pfeilschifter, 1994). However, ATP-induced cell proliferation in C6 glioma cells needs to be investigated. It is conceivable that phosphorylation of cellular proteins by protein kinases plays an important role in regulation of cellular functions including cell growth, migration, differentiation, and response to extracellular stimuli. Cell proliferation induced by growth factors can be divided into at least two distinct classes that act through different signal transduction pathways. One is activated by receptors that contain an intrinsic protein tyrosine kinase such as those activated by PDGF and EGF (Pouyssegur & Seuwen, 1992). Tyrosine phosphorylation of cytosolic protein has been shown to be important in relaying extracellular messages to the nucleus, thus promoting cell division (Malarkey et al., 1995). The activation of MAPKs, in particular, the p42 and p44 MAPK isoforms, seems to be a key component in growth signal transduction through tyrosine kinase receptors such as the PDGF-β receptor (Blenis, 1993). The other pathway is activated by G protein-coupled receptors (Davis, 1993; Lange-Carter et al., 1993). Several studies reveal that the cellular growth mediated by the intracellular signal pathway for the G protein-linked receptor also involves MAPKs (Pouyssegur & Seuwen, 1992; Davis, 1993). These MAPKs are activated during proliferation and cell cycle transition triggered by various stimuli (Blenis, 1993). Thus, MAPKs are indeed important integrators of G protein-coupled receptor- and tyrosine kinase receptor-mediated signals for cell growth. Moreover, many growth factor receptors have been shown to activate a signal transduction pathway that includes MAPK kinase (MEK) and MAPK (Davis, 1993; Lange-Carter et al., 1993; Marshall, 1995). The requirement for the activation of the Ras, Raf, MEK, and MAPK, associated with cell proliferation, had been demonstrated for receptor tyrosine kinases such as PDGF in several cell types (Blenis, 1993; Post & Brown, 1996). Utilizing selective expression of dominant negative mutants, Ras, Raf, or MEK has further been shown to play a key step toward MAPK activation (Stacey et al., 1991; Schap et al., 1993; Abdellatif et al., 1998).
The important role of glial cells in various pathological conditions in brain was first recognized by Del-Rio Hortega (1932). One of the characteristics of glial cells is their activation at a very early stage in response to injury (Gehrmann et al., 1995). Glia activation often precedes reactions of any other cell type in the brain. Furthermore, glial cells have receptors for signalling molecules such as ATP (Langosch et al., 1994), and can react ATP with changes in their extracellular ionic milieu (Langosch et al., 1994) and by activation of transcriptional mechanism (Priller et al., 1995). An understanding of intercellular signalling pathways for glia proliferation and activation could form a rational basis for targeted intervention on glial reactions to injuries in the brain. C6 glioma cells cloned from a rat glial tumour were widely used as a cell culture model to study physiological functions. However, whether the activation of MAPK by ATP associated with cell proliferation of C6 glioma cells has not been fully elucidated.
In this study, the experiments were undertaken to investigate the effect of ATP on activation of Ras/Raf/MEK/MAPK pathway, associated with cell proliferation of C6 glioma cells. In order to characterize the purinoceptor receptor subtype mediating mitogenic effects in C6 cells, we examined the responses to ATP and its analogues selective for different receptor subtypes.
Methods
Materials
Dulbecco's modified Eagle's medium (DMEM)/Ham's nutrient mixture F-12 (F-12) medium, OPTI-MEM I medium, Lipofectamine Plus reagent, and foetal bovine serum (FBS) were purchased from Gibco BRL (Gaithersburg, MD, U.S.A.). [3H]-Methyl thymidine, Hybond C membrane, and enhanced chemiluminescence (ECL) Western blotting detection system were from Amersham (Buckinghamshire, England). Fura-2/AM was from Molecular Probes (Eugene, OR, U.S.A.). PhosphoPlus p42/p44 MAPK and phosphoPlus MEK1/2 antibody kits were from New England Biolabs (Beverly, MA, U.S.A.). p42 MAPK antibody was from Santa Cruz (Santa Cruz, CA, U.S.A.). Genistein, herbimycin A, staurosporine, GF109203X, BAPTA/AM, PD98059, and SB203580 were from Calbiochem (San Diego, CA, U.S.A.). Bicinchoninic acid (BCA) protein assay reagent was from Pierce (Rockford, IL, U.S.A.). Enzymes and other chemicals were from Sigma (St Louis, MO, U.S.A.).
Cell culture
The C6 glioma cells were obtained from ATCC (CCL 107) and established in monolayer culture in DMEM/F-12 medium supplemented with 10% FBS and antibiotics (100 U ml−1 penicillin G, 100 μg ml−1 streptomycin, and 250 ng ml−1 fungizone). Cells were maintained in continuous culture at 37°C in a humidified atmosphere containing 5% CO2. At subconfluence, cultures were replated by enzymatic dissociation with trypsin/EDTA.
[3H]-Thymidine incorporation and cell proliferation
Cells were plated onto 24-well culture plates and grown to confluence. Cells were growth-arrested by incubation in serum-free DMEM/F-12 for 48 h. Confluent, growth-arrested cells were used because cells can be synchronized in G0/G1 phase of the cell cycle and at this baseline, minimally incorporate [3H]-thymidine. After 48 h in serum-free media, the cells were stimulated with ATP and its analogues. After 6 h of stimulation, cells were labelled with 1 μCi ml−1 of [3H]-thymidine for another 18 h in the presence of agonists. The experiments were terminated by washing the cells with cold PBS, precipitation of acid-insoluble materials with 10% TCA, and extraction of the DNA with 0.1 N NaOH. The precipitants were filtered through Whatman GF/B filters and washed three times with cold PBS using a cell harvester. The radioactivity was counted using a scintillation counter (Beckman LS5000TA, Fullerton, CA, U.S.A.).
Plasmids and transfection
The plasmids encoding RasN17 and Raf-301 (dominant negative mutants of Ras and Raf-1), cloned into pJ3H and pkRSPA, respectively, were kindly provided by Dr H.-F. Yang Yen (Institute of Molecular Biology, Academia Sinica). The dominant negative mutant of MEK1, MEK K97R, was a gift from Dr K.-L. Guan at the University of Michigan and subcloned into pAS-CYH. All plasmids were prepared using QIAGEN plasmid DNA preparation kits.
C6 glioma cells were plated at 3×105 cells ml−1 in 12-well culture plates for 24 h, reaching about 80% confluence. Cells were washed once with PBS and once with serum-free DMEM, and 0.5 ml of serum-free OPTI-MEM I medium was added to each well. The DNA PLUS-Lipofectamine reagent complex was prepared according to the instructions of the manufacturer. The amount of transfected plasmid was kept constant (2 μg of RasN17 or Raf-301 for each well). The DNA PLUS-Lipofectamine reagent complex was added to each well, then incubated at 37°C for 5 h. At that time, 0.5 ml of OPTI-MEM I medium containing 20% FBS was added and incubated for 19 h. After 24 h of transfection, the cells were washed twice with PBS and maintained in DMEM/F-12 containing 10% FBS for 48 h. Cells were then washed once with PBS and incubated with serum-free DMEM/F-12 for 24 h before treatment with either ATP or UTP.
Preparation of cell extracts and Western blot analysis of MAPK isoforms
For experiments, cells were plated in 100-mm dishes and made quiescent at confluence by incubation in fresh DMEM/F-12 for 48 h. Growth-arrested C6 glioma cells were incubated with or without UTP at 37°C for various times. When inhibitors were used, they were applied 1 h prior to the addition of UTP. After incubation, the cells were rapidly washed with ice-cold PBS, scraped and collected by centrifugation at 1000×g for 10 min. The collected cells were lysed with ice-cold lysis buffer containing (mM): Tris-HCl 25, pH 7.4, NaCl 25, NaF 25, sodium pyrophosphate 25, sodium vanadate 1, EDTA 2.5, EGTA 2.5, 0.05% Triton X-100, 0.5% SDS, 0.5% deoxycholate, 0.5% NP-40, 5 μg ml−1 leupeptin, 5 μg ml−1 aprotinin, and PMSF 1. The lysates were centrifuged at 45,000×g for 1 h at 4°C to yield the whole cell extract. The protein concentration was determined by the BCA reagents according to the manufacturer's instructions. Samples (100 μg protein) were denatured and subjected to SDS–PAGE using a 10% running gel. Proteins were transferred to nitrocellulose membrane and the membrane was successively incubated at room temperature with 5% BSA in TTBS (Tris-HCl 50 mM, NaCl 150 mM, 0.05% Tween 20, pH 7.4) for 1 h. The phosphorylation of MEK1/2 and p42/p44 MAPK isoforms were identified and quantified by Western blot analysis using anti-phospho-MEK1/2 or anti-phospho-p42/p44 MAPK polyclonal antibody kit according to the manufacturer's instructions. Briefly, membranes were incubated overnight at 4°C with the anti-phospho-MEK1/2 or anti-phospho-p42/p44 MAPK polyclonal antibody used at a dilution of 1 : 1000 in TTBS. Membranes were washed with TTBS four times for 5 min each, incubated with a 1 : 1500 dilution of anti-rabbit horseradish peroxidase antibody for 1 h. Following each incubation, the membrane was washed extensively with TTBS. The immunoreactive bands detected by ECL reagents were developed by Hyperfilm-ECL (Amersham International).
Analysis of data
Concentration-effect curves were fitted and EC50 values were estimated using the Prizm Program (GraphPad, San Diego, CA, U.S.A.). Data were expressed as the means±s.e.mean and analysed with Student's t-test at a 0.01 level of significance. Error bars were omitted when they fell within the symbol representing values.
Results
Extracellular ATP- and UTP-stimulated DNA synthesis
To determine the mitogenic effect of ATP-induced DNA synthesis, we tested the ability of ATP and its analogues to stimulate [3H]-thymidine uptake in C6 glioma cells. ATP and UTP stimulated [3H]-thymidine incorporation in a time-dependent manner (Figure 1A). Onset of the effects of ATP and UTP on [3H]-thymidine incorporation was slow, a significant increase in this response being observed by 4 h after addition of [3H]-thymidine in the presence of these agonists at a concentration of 1 mM, and a maximal effect occurring by 24 h. Furthermore, ATP, UTP, ADP, and UDP produced a concentration-dependent increase in [3H]-thymidine incorporation into serum-deprived, quiescent C6 glioma cells (Figure 1B). The concentrations of ATP, UTP, ADP, and UDP that produced a half maximal increase (EC50) in [3H]-thymidine incorporation were 77±15, 21±12, 110±25, and 34±11 μM, n=3, respectively. ATP (1 mM) and UTP (1 mM) stimulated [3H]-thymidine incorporation to approximately 9.7±1.2 and 9.6±0.9 folds, over the basal level (2850±220 d.p.m. per well), respectively. Whereas P1 receptor agonists AMP and N6-cyclopentyl adenosine (CPA), P2X1 receptor agonists α,β-methylene ATP (α,β-MeATP) and β,γ-methylene ATP (β,γ-MeATP) as well as the P2Y1 receptor agonist 2-methylthio ATP (2MeSATP) did not cause any significant DNA synthesis (data not shown). These results suggest that the mitogenic effects of ATP and UTP were triggered by activation of P2Y2 receptors.
Figure 1.

[3H]-Thymidine incorporation induced by ATP and UTP in C6 glioma cells. (A) For time course, after 48 h in serum-free medium, the cells were stimulated with vehicle (basal), ATP or UTP at a concentration of 100 μM. The cells were labelled with 1 μCi ml−1 [3H]-thymidine for the times indicated in the continuous presence of agonists. (B) For concentration dependence, the cells were stimulated with various concentrations of ATP, UTP, ADP or UDP. After stimulation for 6 h, cells were labelled with 1 μCi ml−1 [3H]-thymidine for another 18 h in the presence of agonists. The incorporation of [3H]-thymidine was determined as described in Methods. Data are expressed as the means±s.e.mean of three separate experiments determined in triplicate.
Extracellular UTP stimulated MAPK isoforms activation
MAPKs, another group of components in the signal transduction pathway, have shown to be activated during stimulation of cell proliferation. Therefore, we determined whether UTP activated MAPKs, in C6 glioma cells. Tyrosine phosphorylation of p42 and p44 MAPK isoforms was monitored by Western blot with polyclonal antisera reactive for the tyrosine phosphorylated state of these two forms of MAPK. As shown in Figure 2, UTP (100 μM) stimulated a marked increase in the levels of p42 and p44 kDa phosphorylation within 2 min. Densitometric analysis of the blot revealed that at 5 min, UTP induced a 3.0±0.4 and 3.6±1.5 fold increase in p42 and p44 MAPK isoforms, respectively, above the basal level. After 30 min, phosphorylation declined below the basal level. Parallel blots run as controls that used antibody against the total p42 MAPK did not show any change (Figure 2). Moreover, as demonstrated in Figure 3, the UTP-induced phosphorylation of p42 and p44 MAPK isoforms was concentration-dependent. Densitometric analysis of the blot indicated that the maximal effect was achieved with 100 μM UTP. Similar results were obtained with ATP (data not shown). The following experiments were performed using UTP.
Figure 2.

Time course of UTP-stimulated activation of p42 and p44 MAPK isoforms in C6 glioma cells. The cells were grown to confluence, made quiescent by serum-deprivation for 48 h and incubated with 100 μM UTP for 2–60 min. The cell lysates were subjected to 10% SDS–PAGE and transferred to nitrocellulose membrane. Western blot analysis was performed using an antiserum reactive with anti-phospho-p42/p44 MAPK or total p42 MAPK (as a control) polyclonal antibody. Bands were visualized by an ECL method and quantified by a densitometer. Similar results were obtained in three independent experiments.
Figure 3.

Concentration-dependence of UTP-stimulated activation of p42 and p44 MAPK isoforms in C6 glioma cells. The cells were grown to confluence, made quiescent by serum-deprivation for 48 h and incubated with various concentrations of UTP for 5 min. The cell lysates were subjected to 10% SDS–PAGE and transferred to nitrocellulose membrane. Western blot analysis was performed using an antiserum reactive with anti-phospho-p42/p44 MAPK or total p42 MAPK (as a control) polyclonal antibody. Bands were visualized by an ECL method. Similar results were obtained in three independent experiments.
Effects of pertussis toxin and tyrosine kinase inhibitors on UTP-induced DNA synthesis and MAPK phosphorylation
To examine whether the effect of UTP on DNA synthesis and p42/p44 MAPK activation was mediated through the activation of a receptor coupled to a pertussis toxin (PTX)-sensitive G protein, C6 glioma cells were pretreated with 100 ng/ml PTX for 24 h and then stimulated with UTP. As shown in Figure 4, pretreatment of these cells with PTX did not significantly attenuate the UTP-induced [3H]-thymidine incorporation (P>0.01, n=3) and p42/p44 MAPK activation (P>0.01, n=3), indicating that the effect of UTP on these responses was mediated through a PTX-insensitive G protein.
Figure 4.
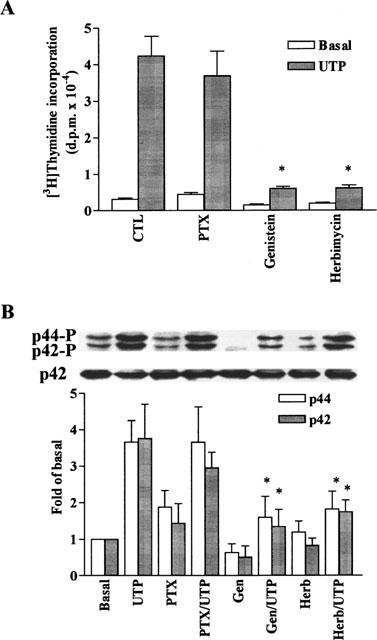
Involvement of G protein and tyrosine kinase in DNA synthesis and MAPK phosphorylation induced by UTP in C6 glioma cells. The cells were preincubated with pertussis toxin (PTX, 100 ng ml−1, 24 h), genistein (Gen, 10 μM, 1 h), or herbimycin A (Herb, 10 μM), 1 h), and then stimulated with vehicle or 100 μM UTP. (A) For DNA synthesis, after 6 h incubation, cells were labelled with 1 μCi ml−1 [3H]-thymidine for another 18 h in the continuous presence of agonists. The incorporation of [3H]-thymidine was determined as described in Methods. Data are expressed as the means±s.e.mean of three separate experiments determined in triplicate. (B) For MAPK experiment, after treatment with these agents, the cells were stimulated with vehicle or 100 μM UTP for 5 min. The phosphorylation of p42/p44 MAPK was determined as described in Figure 2. Similar results were obtained in three independent experiments. *P<0.01, as compared with the control cells exposed to UTP.
To determine whether the effect of UTP on DNA synthesis and p42/p44 MAPK activation was mediated through activation of tyrosine kinase, C6 glioma cells were treated with 10 μM genistein or herbimycin A for 1 h and then stimulated with 100 μM UTP. As shown in Figure 4, pretreatment of these cells with genistein and herbimycin A also inhibited the UTP-induced [3H]-thymidine incorporation (P<0.01, n=3) and p42/p44 MAPK activation (P<0.01, n=3), suggesting the implication of tyrosine kinase in these responses.
Effect of PKC on UTP-induced DNA synthesis and MAPK phosphorylation
In order to determine whether PKC activation is involved in DNA synthesis and p42/p44 MAPK activation in response to UTP, C6 glioma cells were pretreated with PKC inhibitors staurosporine (1 μM) or GF109203X (1 μM) for 1 h and then stimulated with UTP. As shown in Figure 5, pretreatment of the cells with these PKC inhibitors attenuated [3H]-thymidine incorporation (P<0.01, n=3) and p42/p44 MAPK activation (P<0.01, n=3) in response to UTP. These results suggest that UTP-stimulated [3H]-thymidine incorporation and p42/p44 MAPK activation was mediated through the activation of PKC in C6 glioma cells.
Figure 5.
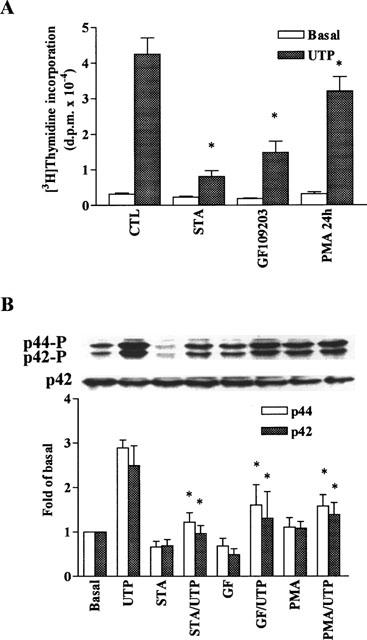
Effects of PKC on DNA synthesis and MAPK phosphorylation induced by UTP in C6 glioma cells. The cells were preincubated with staurosporine (STA, 1 μM, 1 h) or GF109203X (GF, 1 μM, 1 h) and then stimulated with vehicle or 100 μM UTP. (A) For DNA synthesis, after 6 h incubation, cells were labelled with 1 μCi ml−1 [3H]-thymidine for another 18 h in the presence of agonists. The incorporation of [3H]-thymidine was determined as described in Methods. Data are expressed as the means±s.e.mean of three separate experiments determined in triplicate. (B) For MAPK experiment, after treatment with these agents, the cells were stimulated with vehicle or 100 μM UTP for 5 min. The phosphorylation of p42/p44 MAPK was determined as described in Figure 2. Similar results were obtained in three independent experiments. *P<0.01, as compared with the control cells exposed to UTP.
Effect of Ca2+ on UTP-induced DNA synthesis and MAPK phosphorylation
To elucidate whether the UTP-induced increase in [Ca2+]i is implicated in the [3H]-thymidine incorporation and p42/p44 MAPK activation, C6 glioma cells were preincubated with 30 μM BAPTA/AM (a potent intracellular Ca2+ chelator) and 5 mM EGTA and then stimulated with UTP. Results in Figure 6 demonstrate that pretreatment of these cells with BAPTA/AM reduced both [3H]-thymidine incorporation (P<0.01, n=3) and p42/p44 MAPK activation (P<0.01, n=3), indicating that Ca2+ is required for the UTP-mediated responses.
Figure 6.

Effects of Ca2+ and MAPK kinase inhibitors on DNA synthesis and MAPK phosphorylation induced by UTP in C6 glioma cells. The cells were preincubated with BAPTA/AM (10 μM) plus EGTA (5 mM) for 1 h, PD98059 (PD, 10 μM, 1 h), or SB203580 (SB, 10 μM, 1 h) and then stimulated with vehicle or 100 μM UTP. (A) For DNA synthesis, after 6 h incubation, cells were labelled with 1 μCi ml−1 [3H]-thymidine for another 18 h in the continuous presence of agonists. The incorporation of [3H]-thymidine was determined as described in Methods. Data are expressed as the means±s.e.mean of three separate experiments determined in triplicate. (B) For MAPK experiment, after treatment with these agents, the cells were stimulated with vehicle or 100 μM UTP for 5 min. The phosphorylation of p42/p44 MAPK was determined as described in Figure 2. Similar results were obtained in three independent experiments. *P<0.01, as compared with the control cells exposed to UTP.
Effects of MAPK kinase inhibitors on UTP-induced DNA synthesis and MAPK phosphorylation
To ensure that the mitogenic effect of UTP is mediated through the activation of MAPK pathway, the effect of UTP on the p42/p44 MAPK activation and DNA synthesis was examined after treatment of C6 glioma cells with 10 μM PD98059 (a synthetic MEK1/2 inhibitor) or 10 μM SB203580 (a p38 MEK inhibitor) for 1 h. As shown in Figure 6B, the effect of PD98059 on MAPK phosphorylation was assessed by Western blot analysis. PD98059 completely inhibited the phosphorylation of p42/p44 MAPK isoforms induced by UTP, conforming that MEK1/2 is required for MAPK activation in these cells. This hypothesis was further supported by the results that UTP-induced MEK1/2 activation in a time- and concentration-dependent manner (Figure 7). However, the phosphorylation of these MAPK isoforms was not inhibited by SB203580. In contrast, treatment of C6 glioma cells with these inhibitors caused a significant inhibition of the UTP-induced [3H]-thymidine incorporation (Figure 6A). The inhibitory effect of SB203580 on UTP-stimulated [3H]-thymidine incorporation implied that an alternative pathway may be implicated in this motogenic effect, namely the p38 MAPK pathway.
Figure 7.
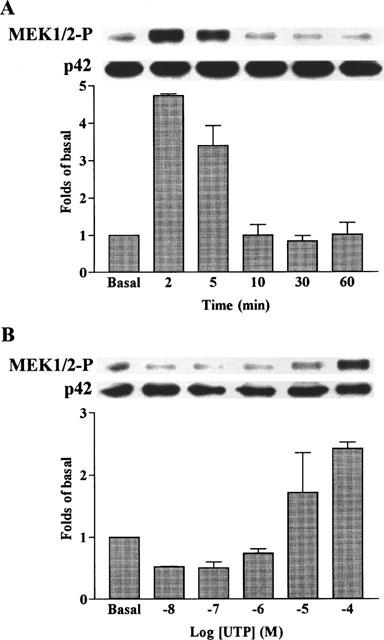
Time course and concentration-dependence of UTP-stimulated activation of MEK1/2 in C6 glioma cells. The cells were grown to confluence, made quiescent by serum-deprivation for 48 h and incubated with 100 μM UTP for 2–60 min (A) or various concentrations of UTP for 5 min (B). The cell lysates were subjected to 10% SDS–PAGE and transferred to nitrocellulose membrane. Western blot analysis was performed using an antiserum reactive with anti-phospho-MEK1/2 or total p42 MAPK (as a control) polyclonal antibody. Bands were visualized by an ECL method and quantified by a densitometer. Similar results were obtained in three independent experiments.
Activation of MAPK requires Ras and Raf-1
Several lines of evidence have suggested that Ras plays an important role in a variety of cell functions mediated through sequential activation of Raf-1, MEK1/2, and MAPK (Blenis, 1993; Post & Brown, 1996). To elucidate whether the activation of Ras/Raf is required for MAPK phosphorylation induced by ATP and UTP, C6 glioma cells were transfected with or without a dominant negative Ras (RasN127), Raf (Raf-301), or MEK (MEK K97R), and then treated with ATP or UTP. As shown in Figure 8, both ATP and UTP induced phosphorylation of p42/p44 MAPK in C6 glioma cells. In addition, transfection with RasN17, Raf-301, or MEK K97R almost completely abolished p42/p44 MAPK phosphorylation induced by these two agents, suggesting that ATP and UTP might activate MAPK through a Ras/Raf/MEK-dependent pathway in C6 glioma cells. Moreover, both ATP and UTP also activated MEK1/2 that is an upstream component of p42/p44 MAPK pathway, these stimulatory effects were almost suppressed by transfection with RasN17, Raf-301, or MEK K97R (Figure 8). These results demonstrated that ATP and UTP, at least in part, activated Ras/Raf/MEK/MAPK pathway in C6 glioma cells.
Figure 8.

Requirement of Ras, Raf, and MEK1/2 for UTP- and ATP-induced activation of MEK1/2 and p42/p44 MAPK in VSMCs. Cells were transfected with plasmids encoding RasN17, Raf-301, or MEK K97R, and then stimulated with 100 μM UTP or ATP for 5 min. The cell lysates were subjected to 10% SDS–PAGE and transferred to nitrocellulose membrane. Western blot analysis was performed using an antiserum reactive with the anti-phospho-MEK1/2 or total p42 MAPK (as a control) polyclonal antibody (a) and anti-phospho-p42/p44 MAPK or total p42 MAPK polyclonal antibody (b). Bands were visualized by an ECL method. Data are expressed as the means±s.e.mean of three independent experiments of anti-phospho-p42/p44 MAPK in the bar graph. *P<0.01, as compared with the control cells exposed to UTP; †P<0.01, as compared with the control cells exposed to ATP.
Discussion
In the present study, we examined the relative capacity of ATP and its analogues to stimulate proliferation of C6 glioma cells as measured by the incorporation of [3H]-thymidine. The mitogenic effects of ATP and UTP were concentration-dependent, related to the duration of exposure of cells to stimulus, and presumably mediated by a P2Y2 receptor. In an attempt to define the subtype of P2Y receptor mediating this effect, we compared the ability of a range of ATP analogues to stimulate mitogenesis. ATP and related analogues act on several cell types via receptors subclassified into P1 receptors for adenosine and P2 receptors for ATP and ADP (Burnstock & Kennedy, 1985). The P2 receptors have been further classified into P2X and P2Y (Burnstock & Kennedy, 1985; Fredholm et al., 1997). In this study, α,β-MeATP, β,γ-MeATP, and 2MeSATP had little effect on [3H]-thymidine incorporation, suggesting that the mitogenic effect was not mediated by the P2X1 or P2Y1 receptor. Moreover, P1 agonists, AMP and CPA, did not show any mitogenic effect. ATP and UTP possessed similar potency in the mitogenic effect. Therefore, these results suggest that ATP and UTP mediate their mitogenic effects by activating P2Y2 receptors, which are consistent with the results obtained from several cell types (Neary et al., 1994; Huwilder & Pfeilschiefter, 1994; Albert et al., 1997; Soltoff et al., 1998).
To assess the possible mechanisms that might mediate the mitogenic action of ATP, we attempted to pharmacologically analyse some of the potential pathways. Several lines of evidence demonstrate that P2Y2 receptors couple to a Gq protein in most cell types (Neary et al., 1994; Huwilder & Pfeilschiefter, 1994; Albert et al., 1997; Soltoff et al., 1998). This is strengthened by our observation that the UTP-stimulated MAPK activation and [3H]-thymidine incorporation in C6 glioma cells was not affected by pretreatment with PTX. These findings suggest that the mitogenic effect and activation of p42/p44 MAPK induced by UTP were mediated through a receptor coupled to a PTX-insensitive G protein. The activation of the P2Y2 receptor is linked to the stimulation of phosphoinositide hydrolysis, which produces two second messengers, diacylglycerol and IP3 (Boarder et al., 1995), which activates PKC and release Ca2+ from the intracellular stores, respectively. The activation of PKC and increase of [Ca2+]i seem to account for most of the early proliferative growth-promoting agents coupled to G proteins, such as ATP (Albert et al., 1997) and endothelin (Simonson et al., 1989). Elevation of [Ca2+]i is an important intracellular signal stimulating many cellular responses including mitogenesis. ATP and its analogues have been demonstrated to induce inositol phosphate accumulation and elevation of [Ca2+]i in several cell types (Harden et al., 1995) with striking similarities to our findings in the mitogenesis assay. The mitogenic effects of UTP in our experiments required the presence of extracellular Ca2+. This result indicates a role for Ca2+ in mediating the mitogenic effect of UTP. This hypothesis was further supported by the results that removal of Ca2+ by BAPTA/EGTA significantly attenuated the p42/p44 MAPK activation and [3H]-thymidine incorporation in C6 glioma cells.
Activation of p42/p44 MAPK is known to require both tyrosine and threonine phosphorylations by the dual specificity MEK1/2. Several lines of evidence indicate that complexity in the mechanisms of agonist stimulation of MAPK in cells including the possible involvement of tyrosine kinase upstream of MAPK kinase (Davis, 1993; Blenis, 1993). MAPK isoforms are activated by various growth factors including PDGF (Blenis, 1993; Post & Brown, 1996), EGF (Pouyssegur & Seuwen, 1992), and ATP (Albert et al., 1997; Soltoff et al., 1998). In this study, we have shown that stimulation with UTP resulted in activation of p42/p44 MAPK in a time- and concentration-dependent manner. These results are consistent with the findings that MAPK isoforms are activated by agonists in several cell types (Pouyssegur & Seuwen, 1992; Albert et al., 1997; Soltoff et al., 1998). Furthermore, we also investigated the implication of a tyrosine kinase in the MAPK cascade of C6 glioma cells stimulated by UTP, using the tyrosine kinase inhibitors genistein and herbimycin A. The results with tyrosine kinase inhibitors showed that the UTP-induced DNA synthesis and activation of p42/p44 MAPK was significantly attenuated by these inhibitors. These results may imply that the mitogenic response to UTP was dependent on tyrosine kinase.
ATP and UTP have been shown to activate PLC to generate diacylglycerol and elevate PKC activity in several cell types (Harden et al., 1995); this time course of PKC activation appears to be essential for late responses such as proliferation and differentiation (Nishizuka, 1992). PKC is a predominant component in the kinase cascade initiating by ligand attachment to both G protein-coupled receptors and receptors containing intrinsic tyrosine kinase activity. In this study, we further investigated the regulatory mechanisms which are involved in UTP-stimulated [3H]-thymidine incorporation and activation of p42/p44 MAPK by PKC. Pretreatment with the PKC inhibitors, staurosporine and GF109203X, attenuated the UTP-stimulated [3H]-thymidine incorporation and activation of p42/p44 MAPK, indicating that a component of the MAPK signal involves PKC-mediated activation of an intermediate kinase. This is likely to be either Raf-1 which has been shown to be phosphorylated by PKC (Kolch et al., 1993) or possibly MEK1/2 which is also activated in a PKC-dependent manner (Lange-Carter et al., 1993). These results are consistent with studies which examine MAPK activation in response to other G-protein coupled agonists including angiotensin II, vasopressin, and endothelin-1 (Molloy et al., 1993; Malarkey et al., 1995).
Although mitogenic signals from the activation of specific tyrosine kinase-coupled growth factors have been well characterized, the mechanism by which the G protein-coupled receptors activate the components of MAPK pathway is not completely understood. PD98059, a synthetic and highly specific MEK1/2 inhibitor, has been shown to inhibit the activation of p42/p44 MAPK by several stimuli (Alessi et al., 1995; Dudley et al., 1995). Because activation of components in the MAPK cascade originates from stimulation of cells by growth factors, it has been proposed that transmission of the signal along this pathway is required for the induction of mitogenesis. In support of this hypothesis, inhibition of MEK1/2 by PD98059 has been associated with a decrease not only in PDGF-stimulated [3H]-thymidine incorporation in 3T3 cells (Dudley et al., 1995), but also with an attenuation in nerve growth factor-induced differentiation in PC12 cells (Pang et al., 1995). In the current study, pretreatment with PD98059 attenuated the UTP-induced activation of p42/p44 MAPK and DNA synthesis in C6 glioma cells, revealing that stimulation of MEK1/2 is required for UTP-induced responses in these cells. In addition, SB203580 showed an inhibitory effect on [3H]-thymidine incorporation but no inhibitory effect on p42/p44 MAPK activation, indicating that an alternate pathway such as p38 MAPK was also involved in the mitogenic effect of UTP in C6 glioma cells.
It has been well established that growth factors activate phosphorylation of cascade of protein kinases including tyrosine kinases, Ras, Raf-1, MEK and MAPK (Blenis, 1993; Post & Brown, 1996). Several studies have depicted a picture that MAPK could be a convergence point for growth signals originating from tyrosine kinase receptors, G protein-coupled receptors, and cytokines (Blenis, 1993; Post & Brown, 1996). In the proposed signalling pathway, several lines of evidence have suggested that Ras is activated by various stimuli for growth and differentiation and that the activated Ras evoked the phosphorylation cascade of protein kinases including Raf-1, MEK1/2, and MAPK (Kerkhoff & Rapp, 1998; Satoh et al., 1992; Blenis, 1993; Post & Brown, 1996). It has been demonstrated that PDGF-BB-induced cell proliferation may be mediated through the activation of Ras (Arvidsson et al., 1994; Weber et al., 1997). In this study, to elucidate whether Ras is required for UTP-induced activation of MAPKs, C6 glioma cells were transfected with a dominant negative mutant of Ras (RasN17) that preferentially interacts with guanine nucleotide exchange factors and inhibits Ras functions (Thorburn et al., 1993; Hara et al., 1993). We found that UTP- and ATP-induced p42/p44 MAPK activation were completely suppressed by transfection with RasN17 in C6 glioma cells, as previously reported in other cell types (Stacey et al., 1991; Abdellatif et al., 1998). Several studies have also shown that activated Ras binds and activates Raf-1, resulting in the activation of MEK1/2 and MAPK (Blenis, 1993; Satoh et al., 1992). Consistent with these reports, we demonstrated that in C6 glioma cells, UTP- and ATP-induced MAPK activation are completely suppressed by transfection with dominant negative mutants of Raf-1 (Raf-301), suggesting that Raf-1 plays a key role in UTP- and ATP-induced activation of MEK/MAPK cascade in C6 glioma cells.
In conclusion, we reported here that ATP and UTP appeared to exert their mitogenic effects by binding to a receptor which was coupled to a PTX-insensitive G protein and signalled through Ras/Raf/MEK/MAPK to enhance DNA synthesis in C6 glioma cells. These results demonstrate that UTP stimulates activation of a MAPK pathway that is regulated by PKC, Ca2+, and tyrosine kinase. The mitogenic effects of these nucleotides were triggered by activation of P2Y2 receptors that subsequently lead to the activation of MAPK isoforms, DNA synthesis and cell proliferation.
Acknowledgments
We thank Professor Stephen J. Hill at the University of Nottingham for critical reading of manuscript. The authors also appreciate Drs H.-F. Yang Yen (Institute of Molecular Biology, Academia Sinica) K.-L. Guan (University zof Michigan) for providing RasN17 and Raf-301, as well as MEK K97R, respectively. This work was supported by grants CMRP680 from Chang Gung Medical Research Foundation and NSC86-2314-B182-114M10 from National Science Council, Taiwan.
Abbreviations
- BCA
bicinchoninic acid
- CPA
N6-cyclopentyl adenosine
- DMEM
Dulbecco's modified Eagle's medium
- EC50
concentration required for half-maximal stimulation
- ECL
enhanced chemiluminescence
- F-12
Ham's nutrient mixture F-12
- FBS
foetal bovine serum
- IP3
inositol 1,4,5-trisphosphate
- MAPK
mitogen-activated protein kinase
- αβ-MeATP
α,β-methylene ATP
- β,γ-MeATP
β,γ-methylene ATP
- MEK1/2
MAPK kinase
- 2MeSATP
2-methylthio ATP
- PKC
protein kinase C
- PLC
phospholipase C
- PTX
pertussis toxin
References
- ABDELLATIF M., PACKER S.E., MICHAEL L.H., ZHANG D., CHARNG M.J., SCHNEIDER M.D. A ras-dependent pathway regulates RNA polymerase II phosphorylation in cardiac myocytes: implications for cardiac hypertrophy. Mol. Cell Biol. 1998;18:6729–6736. doi: 10.1128/mcb.18.11.6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALBERT J.L., BOYLE J.P., ROBERTS J.A., CHALLIS R.A.J., GUBBY S.E., BOARDER M.R. Regulation of brain capillary endothelial cells by P2Y receptors coupled to Ca2+, phospholipase C and mitogen-activated protein kinase. Br. J. Pharmacol. 1997;122:935–941. doi: 10.1038/sj.bjp.0701453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALESSI D.R., CUENDA A., COHEN P., DUDLEY D.T., SATIEL A.R. PD098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- ARVIDSSON A.K., RUPP E., NANBERG E., DOWNWARD J., RONNSTRAND L., WENNSTROM S., SCHLESSINGER J., HELDIN C.H., CLAESSON-WELSH L. Tyr-716 in the plaelet-derived growth factor beta-receptor kinase insert is involved in GRB2 binding and Ras activation. Mol. Cell Biol. 1994;14:6715–6726. doi: 10.1128/mcb.14.10.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLENIS J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOARDER M.R., WEISMAN G.A., TURNER J.T., WILKINSON G.F. G protein-coupled P2 purinoceptors: from molecular biology to functional responses. Trends Pharmacol. Sci. 1995;16:133–138. doi: 10.1016/s0165-6147(00)89001-x. [DOI] [PubMed] [Google Scholar]
- BURNSTOCK G. The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology. 1997;36:1127–1139. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- BURNSTOCK G., KENNEDY C. Is there a basis for distinguishing two types of P2-purinoceptor. Gen. Pharmacol. 1985;16:433–440. doi: 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., GOVAERTS C., PARMENTIER M., BOEYNAEMS J.-M. Cloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J. Biol. Chem. 1997;272:31969–31973. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- DAVIS R.J. The mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- DEL-RIO HORTEGA P. Cytology and Cellular Pathology of the Nervous System 1932New York: W. Hoeber; 481–534.In: Penfield, W. (ed) [Google Scholar]
- DUDLEY D.T., PANG L., DECKER S.J., BRIDGES A.J., SATIEL A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREDHOLM B.B., ABBRACHIO M.P., BURNSTOCK G., DUBYAK G.R., HARDEN T.K., JACOBSON K.A., SCHWABE U., WILLIAMS M. Towards a revised nomenclature for P1 and P2 receptors. Trends Pharmacol. Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEHRMANN J., MATSUMOTI Y., KREUTZBERG G.W. Microglia: intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- HARA M., AKASAKA K., AKINAGA S., OKABE M., NAKANO H., GOMEZ R., WOOD D., UH M., TAMANOI F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. U.S.A. 1993;90:2281–2285. doi: 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDEN T.K., BOYER J.L., NICHOLAS R.A. P2-purinergic receptors: subtype-associated signaling responses and structure. Ann. Rev. Pharmacol. Toxicol. 1995;35:541–579. doi: 10.1146/annurev.pa.35.040195.002545. [DOI] [PubMed] [Google Scholar]
- HOURANI S.M.O., HALL D.A. Receptors for ADP on human blood platelets. Trends Pharmacol. Sci. 1994;15:103–108. doi: 10.1016/0165-6147(94)90045-0. [DOI] [PubMed] [Google Scholar]
- HUANG N.N., WANG D.J., HEPPEL L.A. Extracellular ATP is a mitogen for 3T3, 3T6, and A431 cells and acts synergistically with other growth factors. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7904–7908. doi: 10.1073/pnas.86.20.7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUWILDER A., PFEILSCHIFTER J. Stimulation by extracellular ATP and UTP of the mitogen-activated protein kinase cascade and proliferation of rat renal mesangial cells. Br. J. Phrmacol. 1994;113:1455–1463. doi: 10.1111/j.1476-5381.1994.tb17160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERKHOFF E., RAPP U.R. Cell cycle targets of Ras/Raf signalling. Oncogene. 1998;17:1457–1462. doi: 10.1038/sj.onc.1202185. [DOI] [PubMed] [Google Scholar]
- KOLCH W., HEIDECKER G., KOCHS G., HUMMEL R., VAHIDI H., MISCHAK H., FINKENZELLER G., MARME D., RAPP U.R. Protein kinase Cα activates RAF-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- LANGE-CARTER C.A., PLEIMAN C.M., GARDNER A.M., BLUMER K.J., JOHNSON G.L. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- LANGOSCH J.M., GEBICKE-HAERTER P.J., NORENBERG W., ILLES P. Characterization and transduction mechanisms of purinoceptors in activated rat microglia. Br. J. Pharmacol. 1994;113:29–34. doi: 10.1111/j.1476-5381.1994.tb16169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEON C., HECHLER B., VIAL C., LERAY C., CAZENAVE J.-P., GACHET T.C. The P2Y1 receptor is an ADP receptor antagonized by ATP and expressed in platelets and megakaryoblastic cells. FEBS Lett. 1997;403:26–30. doi: 10.1016/s0014-5793(97)00022-7. [DOI] [PubMed] [Google Scholar]
- MALARKEY K., BELHAM C.M., PAUL A., GRAHAM A., MCLEES A., SCOTT P.H., PLEVIN R. The regulation of tyrosine kinase signalling pathways in response to growth factors and G-protein coupled receptors. Biochem. J. 1995;309:361–375. doi: 10.1042/bj3090361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSHALL C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- MOLLOY C.J., TAYLOR D.S., WEBER H. Angiotensin II stimulation of protein tyrosine phosphorylation and protein kinase activation in rat aortic smooth muscle cells. J. Biol. Chem. 1993;263:7338–7345. [PubMed] [Google Scholar]
- NEARY J.T., ZHU Q., NORENBERG M.D. ATP signalling in astocytes: activation of MAP kinase. Drug Dev. Res. 1994;31:302. [Google Scholar]
- NICHOLAS R.A., WATT W.C., LAZAROWSKI E.R., LI Q., HARDEN T.K. Uridine nucleotide selectivity of three phospholipase C-activating P2 receptors: identification of a UDP-selective, a UTP-selective, and an ATP- and UTP-specific receptor. Mol. Pharmacol. 1996;50:224–229. [PubMed] [Google Scholar]
- NISHIZUKA Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- PANG L., SAWADA T., DECKER S.J., SALTIEL A.R. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J. Biol. Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- POST G.R., BROWN J.H. G protein-coupled receptors and signaling pathways regulating growth response. FASEB J. 1996;10:741–749. doi: 10.1096/fasebj.10.7.8635691. [DOI] [PubMed] [Google Scholar]
- POUYSSEGUR J., SEUWEN K. Transmembrane receptors and intracellular pathways that control cell proliferation. Annu. Rev. Physiol. 1992;54:195–210. doi: 10.1146/annurev.ph.54.030192.001211. [DOI] [PubMed] [Google Scholar]
- PRILLER J., HAAS C.A., REDDINGTON M., KREUTZBERG G.W. Calcitonin gene-related peptide and ATP induce early gene expression in cultured rat microglial cells. Glial. 1995;15:447–457. doi: 10.1002/glia.440150408. [DOI] [PubMed] [Google Scholar]
- SATOH T., NAKAFUKU M., KAZIRO Y. Function of Ras as a molecular switch in signal transduction. J. Biol. Chem. 1992;267:24149–24152. [PubMed] [Google Scholar]
- SCHAPP D., VAN DER WAL J., HOWES L.R., MARSHALL C.J., VAN BLITTERSWIJK W.J. A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J. Biol. Chem. 1993;268:20232–20236. [PubMed] [Google Scholar]
- SIMONSON M.S., WANN S., MENE P., DUBYAK G.R., KESTER M. Endothelin stimulates phospholipase C, Na+/H+ exchange, c-fos expression, and mitogenesis in rat mesangial cells. J. Clin. Invest. 1989;83:708–712. doi: 10.1172/JCI113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLTOFF S.P., AVRAHAM H., AVRAHAM S., CANTLEY L.C. Activation of P2Y2 receptors by UTP and ATP stimulates mitogen-activated kinase activity through a pathway that involves related adhesion focal tyrosine kinase and protein kinase C. J. Biol. Chem. 1998;273:2653–2660. doi: 10.1074/jbc.273.5.2653. [DOI] [PubMed] [Google Scholar]
- STACEY D.W., FEIG L.A., GIBBS J.B. Dominant inhibitory Ras mutants selectively inhibit the activity of either cellular or oncogenic Ras. Mol. Cell Biol. 1991;11:4053–4064. doi: 10.1128/mcb.11.8.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SURPRENANT A., RASESNDREN F., KAWASHIMA E., NORTH R.A., BUELL G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- THORBURN A., THORBURN J., CHEN S.Y., POWERS S., SHUBEITA H.E., FERAMISCO J.R., CHIEN K.R. Hras-dependent pathways can activate morphological and genetic markers of cardiac muscle hypertrophy. J. Biol. Chem. 1993;268:2244–2249. [PubMed] [Google Scholar]
- WEBER J.D., HU W., JEFCOAT S.C., JR, RABEN D.M., BALDASSARE J.J. Ras-stimulated extracellular signal-related kinase 1 and RhoA activities coordinate platelet-derived growth factor-induced G1 progression through the independent regulation of cyclin D1 and p27. J. Biol. Chem. 1997;272:32966–32971. doi: 10.1074/jbc.272.52.32966. [DOI] [PubMed] [Google Scholar]
- YANG C.M., TSAI Y.-J., PAN S.-L., TSAI C.-T., WU W.-B., CHIU C.-T., LUO S.-F., OU J.T. Purinoceptor-stimulated phosphoinositide hydrolysis in Madin-Darby canine kidney (MDCK) cells. Naunyn-Schmiedeberg's Arch Pharmacol. 1997;356:1–7. doi: 10.1007/pl00005015. [DOI] [PubMed] [Google Scholar]