

Abstract
The increase in the cytosolic Ca2+ concentration ([Ca2+]i) following repetitive stimulation with ATP or sphingosylphosphorylcholine (SPC) in single porcine aortic smooth muscle cells was investigated using the Ca2+ indicator, fura-2.
The ATP-induced [Ca2+]i increase resulted from both Ca2+ release and Ca2+ influx. The former was stimulated by phospholipase C activation, while the latter occurred predominantly via the receptor-operated Ca2+ channels (ROC), rather than the store-operated Ca2+ channels (SOC) or the voltage-operated Ca2+ channel (VOC). Furthermore, the P2X5 receptor was shown to be responsible for the ATP-induced Ca2+ influx.
A reproducible [Ca2+]i increase was induced by repetitive ATP stimulation, but was abolished by removal of extracellular Ca2+ or inhibition of intracellular Ca2+ release using U-73122 or thapsigargin, and was restored by Ca2+ readdition in the former case.
SPC only caused Ca2+ release, and the amplitude of the repetitive SPC-induced [Ca2+]i increases declined gradually. However, a reproducible [Ca2+]i increase was seen in cells in which protein kinase C being inhibited, which increased the SPC-induced Ca2+ influx, rather than IP3 generation.
In conclusion, although the amplitude of the ATP-induced Ca2+ release, measured when Ca2+ influx was blocked, or of the Ca2+ influx when Ca2+ release was blocked, progressively decreased following repetitive stimulation, the overall [Ca2+]i increase for each stimulation under physiological conditions remained the same, suggesting that the Ca2+ stores were replenished by an influx of Ca2+ during stimulation. The SPC-induced [Ca2+]i increase resulted solely from Ca2+ release and decreased gradually following repetitive stimulation, but the decrease could be prevented by stimulating Ca2+ influx, further supporting involvement of the intracellular Ca2+ stores in Ca2+ signalling.
Keywords: Purinoceptor, sphingosylphosphorylcholine, cytosolic Ca2+ concentration, aortic smooth muscle cells, desensitization, receptor-operated Ca2+ channel, store-operated Ca2+ channel, voltage-operated Ca2+ channel
Introduction
Intracellular Ca2+ levels ([Ca2+]i) regulate many cellular functions, including secretion, contraction, neurotransmission, proliferation and apoptosis, and cells have therefore evolved distinct mechanisms to rapidly and precisely control the Ca2+ signalling process (Berridge, 1997). In the response to a receptor agonist, Ca2+ release from intracellular Ca2+ stores (via IP3 generation) and Ca2+ influx via receptor-operated Ca2+ channels (ROC) are two distinct mechanisms responsible for the increased [Ca2+]i (Marks, 1997; Furuichi & Mikoshiba, 1995; Barnard, 1996), while Ca2+ influx via voltage-operated Ca2+ channels (VOC) is predominantly responsible for the increase in the [Ca2+]i seen on membrane depolarization (Hess, 1990). In addition to the IP3-mediated Ca2+ release, intracellular Ca2+ release can be triggered by many other messengers, including GTP (Chueh & Gill, 1986), cyclic ADP ribose (Lee et al., 1989), arachidonic acid (Wu et al., 1994), sphingolipid metabolites (Ghosh et al., 1990), and nicotinic acid adenine dinucleotide phosphate (Dousa et al., 1996). It has also been shown that the permeability of the plasma membrane to Ca2+ increases after depletion of the intracellular Ca2+ stores by second messengers (Birnbaumer et al., 1996). The Ca2+ influx pathway linked to a decreased Ca2+ content of the stores has been termed capacitative Ca2+ entry via so-called store-operated Ca2+ channels (SOC) (Putney, 1990); however, the mechanism by which this occurs is unknown.
In many tissues, extracellular ATP levels act as a signal for the induction of various cellular responses via activation of a distinct receptor (Harden et al., 1995). In neuronal and muscle cells, ATP activates a nonselective cation channel (P2x receptor) that is permeable to Ca2+ and Na+ (Surprenant et al., 1995; Lewis et al., 1995). In many tissues, activation of P2y or P2u receptors is directly coupled to G proteins, with subsequent modulation of the activities of two effector systems, phospholipase and adenylyl cyclase. Phospholipases A2, C and D have all been reported to be stimulated by extracellular ATP in different cell types (Wilkinson et al., 1993; Winitz et al., 1994), while the generation of cytosolic cyclic AMP is either inhibited or increased by the same stimulation (Sipma et al., 1994; Sato et al., 1992). In platelets, ADP, but not ATP, is the active agonist of P2t receptors, activation of which stimulates Ca2+ release from intracellular Ca2+ stores via phospholipase C activation (Hourani & Hall, 1994). In the inflammatory and immune systems, activation of P2z receptors opens nonselective pores which allows molecules with molecular masses of less than 1000 daltons to pass freely (Surprenant et al., 1996). Of the above mentioned ATP signalling cascades, activation of nonselective cation channels (P2x receptor), nonselective pores (P2z receptor), or phospholipase C (via activation of P2y, P2u or P2t receptors) results in an increase in the [Ca2+]i.
Sphingolipid metabolites, including sphingosine 1-phosphate (S1P) and sphingosylphosphorylcholine (SPC), have been shown to act as extracellular signals (Meyer zu Heringdorf et al., 1997). Although the gene product of edg-1 has been identified as a S1P receptor (Lee et al., 1998), no SPC receptor has yet been cloned. Nevertheless, SPC transduces its signal via receptor-G protein-effector system coupling, since all SPC-stimulated signalling events are inhibited by pertussis toxin (Okajima & Kondo, 1995; Seufferlein & Rozengurt, 1995; van Koppen et al., 1996). Recently, we have shown that, in porcine aortic smooth muscle cells, the [Ca2+]i can be increased by G protein-coupled receptor agonists, including ATP, bradykinin, SPC, and lysophosphatidic acid (LPA), but only SPC and LPA are mitogenic and their effects sensitive to pertussis toxin. We further demonstrated that SPC-induced mitogen-activated protein kinase activation depends on SPC-induced intracellular Ca2+ release via IP3 generation and is not affected by the removal of extracellular Ca2+. Platelet-derived growth factor also increases the [Ca2+]i, but its mitogenic effect is insensitive to [Ca2+]i change (Chin & Chueh, 1998).
In the current study, again using porcine aortic smooth muscle cells, we further characterized the desensitization mechanisms involved in the [Ca2+]i increase induced by repetitive ATP or SPC stimulation. The Ca2+ signals remained at the same level throughout repetitive ATP stimulation, while those induced by SPC gradually declined with repetitive stimulation. Our data indicate that replenishment of the intracellular Ca2+ stores by influxed Ca2+ plays a pivotal role in maintaining the reproducible Ca2+ signalling.
Methods
Materials
Dulbecco's modified Eagle's medium, foetal bovine serum and trypsin/EDTA were purchased from Life Technologies (Grand Island, NY, U.S.A.). Fura-2 acetoxymethyl ester was obtained from Molecular Probes (Eugene, OR, U.S.A.). ATP, DIDS, BzATP, ATPγS, SPC, verapamil, ω-conotoxin, and nifedipine were purchased from Sigma (St. Louis, MO, U.S.A.). Thapsigargin, Rp-cAMPS, PPADS, AMPCPP, 2-MeSATP and U-73122 were obtained from Research Biochemicals International (Natick, MA, U.S.A.). Arachidonyl trifluoromethyl ketone (AACOCF3), KN-62, Gö 6983 and KT5823 were purchased from Calbiochem (San Diego, CA, U.S.A.). SK&F96365 was purchased from Biomol (Plymouth Meeting, PA, U.S.A.). The [3H]-IP3 assay system was obtained from the Amersham Corp (Buckinghamshire, U.K.). All other chemicals were analytical grade and obtained from Merck (Darmstadt, Germany).
Culture of porcine aortic smooth muscle cells
Porcine aortic smooth muscle cells were prepared using the explant method of Ross (1971), as previously described (Song & Chueh, 1996; Chin & Chueh, 1998). Explants of porcine aorta, obtained from a local slaughterhouse, were cultured in Dulbecco's modified Eagle's medium, supplemented with 10% foetal bovine serum, 100 units ml−1 of penicillin, and 100 μg ml−1 of streptomycin, and maintained at 37°C in an atmosphere of 95% air and 5% CO2. Over the next 10 days, the smooth muscle cells slowly migrated over the dish surface. After reaching confluency, they were subcultured by incubation with Ca2+- and Mg2+-free 0.05% trypsin/EDTA solution and maintained for five passages in culture. For [Ca2+]i measurement and Mn2+ influx experiments, cells were plated onto glass coverslips 48 h before use. In some experiments, cells were preincubated with pertussis toxin (100 ng ml−1) or cholera toxin (1 μg ml−1) for 16 h.
Measurement of [Ca2+]i
The [Ca2+]i change in a single cell was measured using the fluorescent Ca2+ indicator, fura-2, as described previously (Chin & Chueh, 1998). The [Ca2+]i was calculated from the ratio of the fluorescence at 340 nm and 380 nm according to the equation derived by Grynkiewicz et al. (1985) using parameters obtained on our instrument for fura-2 in aortic smooth muscle cells: Rmin=1.18; Rmax=3.5; Sf2/Sb2=1.22; Kd=135 nM. In some experiments performed in the absence of extracellular Ca2+, extracellular Ca2+ was omitted from the loading buffer and 0.5 mM EGTA was added after fura-2 loading. The loading buffer consisted of (mM): NaCl 150, KCl 5, glucose 5, MgCl2 1, CaCl2 2.2 and HEPES 10, pH 7.4. The results of one representative experiment are illustrated in the figures, and the mean±s.d. values for the [Ca2+]i changes, calculated for n experiments using different batches of cells, are given in the text.
Measurement of IP3 production
After near-confluency was reached in the six-well plates, the cells were pretreated with either buffer or 0.1 μM staurosporine for 5 min, then washed and incubated with the indicated agonists at 37°C for the indicated times (15 s to 4 min) in a volume of 1 ml of loading buffer per well. The amount of IP3 generated in each well was determined by radioreceptor assay using the D-myo[3H]-IP3 assay system (Amersham) according to the manufacturer's instructions. The data presented are the means±s.d. for six independent experiments using different batches of cells.
Determination of fluorescence quenching by Mn2+ influx
Fura-2-loaded cells were bathed in loading buffer containing 1 mM CaCl2. Where indicated, the cells were pretreated with (μM): SK&F96365 30, staurosporine 0.1, Rp-cAMPS 30, Gö 6983 0.3, KN-62 10 or KT5823 1 at room temperature for 5 min. At the indicated times, 1 mM Mn2+, either alone or together with thapsigargin, ATP, or SPC, was added. The Mn2+ influx into the cells was monitored by quenching of fura-2 fluorescence at the isosbestic wavelength of 360 nm. All experiments were repeated at least nine times using different batches of cells with similar results. The results for one representative experiment are shown in the figures.
Results
In this study, we first characterized the Ca2+ signalling mechanism induced by the purinoceptor agonist, ATP, in single porcine aortic smooth muscle cells. As shown in Figure 1A (trace a), the [Ca2+]i increased rapidly from the basal level of 50±17 nM (n=76) to a peak level of 729±85 nM (n=76) within 5 s of the addition of 30 μM ATP in the presence of extracellular Ca2+, then fell to the basal level over the next 180 s. When the extracellular Ca2+ was removed and 0.5 mM EGTA added to the bathing solution, this increase was reduced by approximately 38% (Figure 1A, trace b), the basal and peak levels of [Ca2+]i being 42±19 nM and 475±30 nM (n=68), respectively, and the [Ca2+]i fell to the basal level within 90 s. Figure 1B shows the dependence of the [Ca2+]i changes, either at the peak (panel a) or 60 s after ATP addition (panel b), on the concentration of ATP. The difference between the ATP-induced [Ca2+]i increase in the presence and absence of extracellular Ca2+ (open and filled circles, respectively) reflects the ATP-induced Ca2+ influx. These ATP-induced [Ca2+]i increases were mediated by a purinoceptor, since they were inhibited by approximately 46–68% by pretreatment of the cells with 30 μM PPADS or 30 μM DIDS (data not shown).
Figure 1.
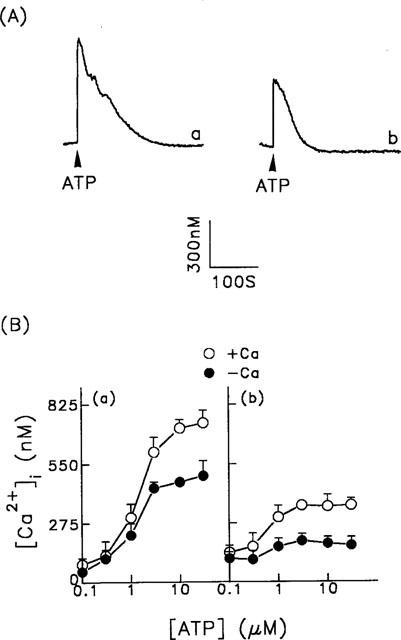
ATP-induced changes in [Ca2+]i in porcine aortic smooth muscle cells. (A) ATP (30 μM) was added as indicated by the arrowheads to fura-2-loaded cell in the presence (trace a) or absence (trace b) of extracellular Ca2+. (B) Various concentrations of ATP were added to cells in the presence or absence of extracellular Ca2+. The [Ca2+]i peak height (panel a) or the [Ca2+]i increase 60 s after ATP addition (panel b) are plotted versus the ATP concentration. The data are the mean±s.d. for 68–76 cells.
In the presence of extracellular Ca2+, the amplitude of the [Ca2+]i increases induced by repetitive ATP challenge was not significantly reduced (Figure 2A, trace a). However, in the absence of extracellular Ca2+, desensitization occurred, as the amplitude of [Ca2+]i increase progressively decreased (Figure 2A, trace b). The data for the reduction in the [Ca2+]i increase induced by repetitive ATP stimulation in the presence or absence of extracellular Ca2+ are summarized in Figure 2B, in which the responses are expressed as the ratio of the response to the nth stimulus over that to the initial stimulus.
Figure 2.
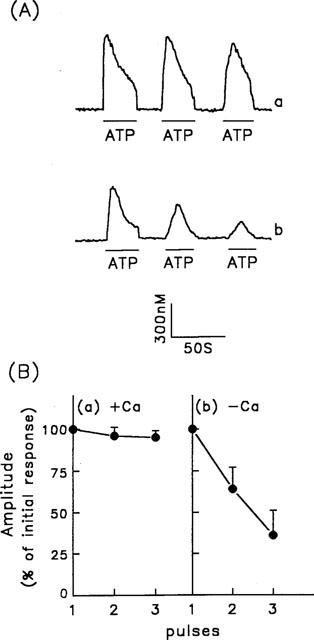
[Ca2+]i increases induced by repetitive ATP stimulation in porcine aortic smooth muscle cells. (A) [Ca2+]i changes in fura-2-loaded cells were measured in response to repetitive stimulation with 30 μM ATP as indicated by the horizontal bars in the presence (trace a) or absence (trace b) of extracellular Ca2+. (B) Summarized data for the [Ca2+]i changes induced by repetitive ATP stimulation in the presence (panel a) or absence (panel b) of extracellular Ca2+ under the conditions described above. The amplitudes of the [Ca2+]i changes are expressed as the peak height for response n relative to that for the initial response. The values are the mean±s.d. for 86–96 cells.
Our data indicate that both intracellular Ca2+ release and extracellular Ca2+ influx contribute to the Ca2+ signal induced by ATP in porcine aortic smooth muscle cells. The reduction in the [Ca2+]i increase seen on repetitive stimulation in the absence of extracellular Ca2+ further indicates either that releasable Ca2+ was not available due to depletion of the intracellular Ca2+ stores after repetitive stimulation or that the phospholipase C signalling cascade was desensitized. We therefore examined the amount of available Ca2+ remaining within the intracellular Ca2+ stores after repetitive exposure to ATP, using the amplitude of the [Ca2+]i transient induced by ionomycin in the absence of extracellular Ca2+ as an index of the size of the intracellular Ca2+ stores (Montero et al., 1990). As shown in Figure 3A, after repetitive ATP stimulation, the [Ca2+]i transient induced by ionomycin was significantly less in the cells that had been subjected to ATP stimulation in Ca2+-free buffer (trace a) than in those stimulated in Ca2+-containing buffer (trace b), the peak levels of the [Ca2+]i increases being 170±18 (n=46) and 704±25 (n=46), respectively. The available Ca2+ remaining within the intracellular Ca2+ stores was not reduced following repetitive ATP stimulation in the presence of extracellular Ca2+, since the EGTA and ionomycin-induced [Ca2+]i increase was identical to that for control cells not previously subjected to ATP challenge (trace c). Our results therefore suggest that the decreased response to ATP seen in Ca2+-free medium is at least partially due to the filling state of the intracellular Ca2+ stores. Indeed, when Ca2+ was subsequently added back to the bathing solution after repetitive ATP stimulation in the absence of extracellular Ca2+, reproducible ATP-induced [Ca2+]i increases were fully restored, regardless of pretreatment with staurosporine (Figure 3B, traces a and b).
Figure 3.
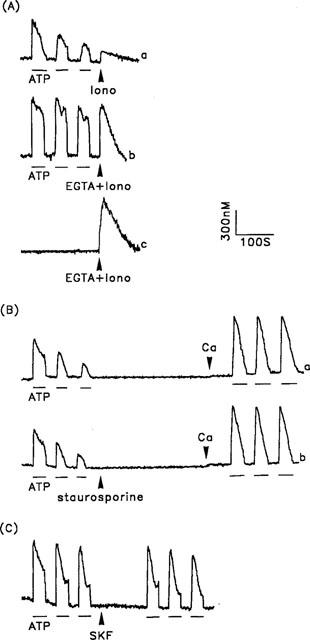
The effect of the Ca2+ content of the intracellular Ca2+ stores on repetitive ATP-induced [Ca2+]i changes in porcine aortic smooth muscle cells. (A) The extent of filling of the intracellular Ca2+ stores in cells after repeated exposure to ATP in the absence (trace a) or presence (trace b) of extracellular Ca2+ and in control cells without ATP stimulation (trace c) was measured by the addition of 10 μM ionomycin only (Iono) (trace a), or 5 mM EGTA and 10 μM ionomycin (EGTA+Iono) (traces b and c), as shown by the arrowheads. (B) [Ca2+]i changes induced by repetitive ATP stimulations were initially measured in the absence of extracellular Ca2+. The cells shown in (a) were left untreated while those in (b) were treated with 0.1 μM staurosporine, then 2.2 mM CaCl2 were reintroduced into the bathing solution in both cases, as indicated by the arrowheads, and the [Ca2+]i changes induced by repetitive ATP stimulation again measured. (C) [Ca2+]i changes induced by repetitive ATP stimulation in the presence of extracellular Ca2+ were measured before, and after, the introduction of 30 μM SK&F96365 (SKF), as shown by the arrowhead. ATP (30 μM) was applied as indicated by the horizontal bars. The experiments were performed 46–58 times with similar results; one representative example is shown.
Our data indicate that the ATP-induced [Ca2+]i increase is attributable to both intracellular Ca2+ release and extracellular Ca2+ influx. Since ROC, VOC, and SOC are three pathways responsible for ATP-induced Ca2+ influx, we next analysed the role of each of these in ATP-induced Ca2+ signalling. First, we examined the contribution of Ca2+ influx via SOC to the ATP-induced Ca2+ signal. When the effect of SK&F96365, a SOC inhibitor, on the repetitive ATP stimulation-induced [Ca2+]i increase was tested, identical results were seen before and after SK&F96365 exposure (Figure 3C). To further characterize the contribution of SOC to the Ca2+ influx, we examined the effect of thapsigargin on the [Ca2+]i increase. Thapsigargin blocks Ca2+ loading of the intracellular Ca2+ stores by inhibiting the endoplasmic reticulum Ca2+ pump and depleting intracellular Ca2+ stores in the absence of receptor activation; this depletion of Ca2+ stores then activates SOC. As shown in Figure 4A, the thapsigargin-induced [Ca2+]i increase in cells in Ca2+-free buffer (trace b) was as great as that in cells in Ca2+-containing buffer (trace a), the mean values for the net thapsigargin-induced [Ca2+]i changes being 444±60 nM (n=36) and 476±63 nM (n=36), respectively. These results indicate that depletion of Ca2+ stores alone appears insufficient to alter the Ca2+ permeability of the plasma membrane, and this is further supported by the results shown in Figures 4B and 5.
Figure 4.
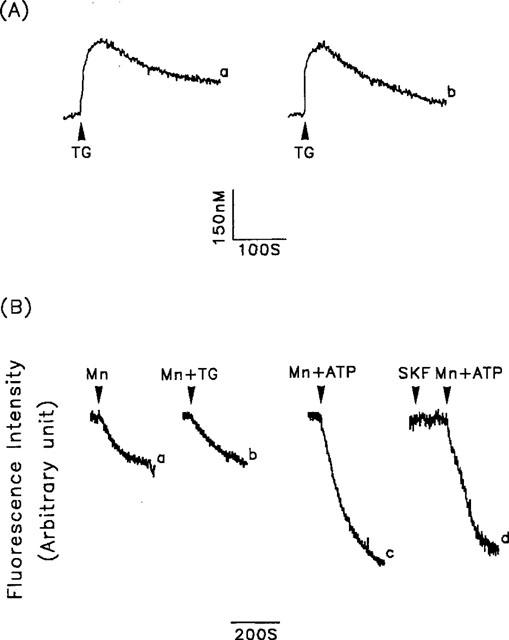
Contribution of the SOC to the thapsigargin-induced [Ca2+]i increase or the thapsigargin- or ATP-induced Mn2+ influx. (A) Thapsigargin (1 μM) was added as indicated (arrowheads) and [Ca2+]i changes were measured in fura-2-loaded cells in the presence (trace a) or absence (trace b) of extracellular Ca2+. (B) Fura-2-loaded cells were bathed in loading buffer containing 1 mM Ca2+, then 1 mM Mn2+ alone (Mn) (trace a), 1 mM Mn2+ plus 1 μM thapsigargin (Mn+TG) (trace b), or 1 mM Mn2+ plus 30 μM ATP (Mn+ATP (traces c and d) was added, and the fluorescence quenching caused by the Mn2+ influx measured. In trace d, the cells were treated with 30 μM SK&F96365 (SKF) for 2 min before the addition of Mn2+ plus ATP. Experiments were repeated 36–46 times with similar results; one representative example is shown.
Figure 5.
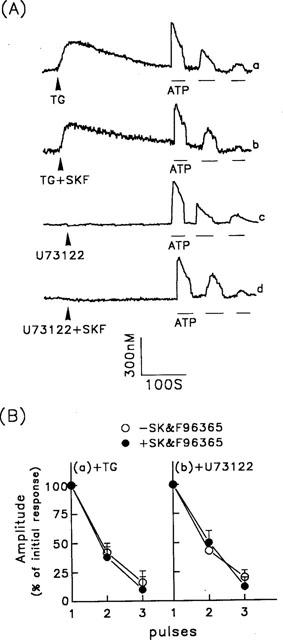
ATP-induced [Ca2+]i increase via Ca2+ influx. (A) Five min after the cells were treated with 1 μM thapsigargin (TG) (traces a and b) or 10 μM U-73122 (traces c and d), repetitive ATP-induced [Ca2+]i increases were measured. In some experiments, 30 μM SK&F96365 was added at the same time as the thapsigargin (TG+SKF) (trace b) or U-73122 (U73122+SKF) (trace d). ATP (30 μM) was applied as indicated by the horizontal bars. (B) Summarized data for the [Ca2+]i responses to repetitive ATP stimulation following pretreatment of cells with thapsigargin panel (a) or U-73122 panel (b) in the presence or absence of SK&F96365. The amplitudes of the [Ca2+]i changes are expressed as the peak height for response n relative to that for the initial response. The data are the mean±s.d. for 44–58 experiments.
In the experiment shown in Figure 4B, we used Mn2+, which presumably enters cells via the same pathways as Ca2+, as a substitute for Ca2+ in Ca2+ influx studies since it cannot be removed from the cytosol by Ca2+ transporters, including Ca2+ pumps and the Na+/Ca2+ exchanger, and the fluorescence quenching due to Mn2+ influx at the isosbestic wavelength can therefore be used as an index of Ca2+ influx (Mertz et al., 1990). In porcine aortic smooth muscle cells, no difference was seen in the fluorescence quenching induced by Mn2+ alone or Mn2+ plus thapsigargin (Figure 4B, traces a and b). However, when Mn2+ and ATP were added simultaneously, fluorescence quenching was significantly increased (Figure 4B, trace c), and this effect was not blocked by SK&F96365 (Figure 4B, trace d). Taken together, our results indicate a negligible contribution of SOC to the ATP-induced [Ca2+]i increase.
We then examined the contribution of other Ca2+ influx mechanisms to the ATP-induced Ca2+ signal. To block intracellular Ca2+ release and SOC activity, we used two different approaches. In addition to depleting the intracellular Ca2+ stores using thapsigargin, we also used the phospholipase C inhibitor, U-73122, to block IP3 generation. As shown in Figure 5A (trace a), following a thapsigargin-induced [Ca2+]i increase, the [Ca2+]i declined to a new steady state at which exposure of the cells to ATP still induced an increase in [Ca2+]i, but this was less marked than in control cells not pretreated with thapsigargin (Figure 2A, trace a). Furthermore, the [Ca2+]i increase was significantly reduced in response to a second exposure to ATP and was only marginal in response to a third exposure. Similar results were seen after treatment with 10 μM U-73122 (Figure 5A, trace c). We have previously shown that U-73122, at a concentration of 10 μM, completely blocks ATP- or SPC-induced IP3 generation in porcine aortic smooth muscle cells (Chin & Chueh, 1998). SK&F96365 had no effect on the ATP-induced Ca2+ influxes after treatment with thapsigargin or U-73122 (Figure 5A, traces b and d). The data for the [Ca2+]i changes induced by repetitive ATP stimulation under the above conditions are summarized in Figure 5B. Under identical experimental conditions, bradykinin did not induce any [Ca2+]i increase in cells pretreated with either thapsigargin or U-73122 (data not shown). These results indicate that following pretreatment of cells with concentrations of U-73122 or thapsigargin at which IP3 generation (U-73122) or Ca2+ refilling of, and Ca2+ release from, intracellular Ca2+ stores (thapsigargin) are efficiently inhibited, the ATP-induced Ca2+ influx desensitized on repeated stimulation.
We next determined the subtype of ATP-gated ion channels involved in the ATP-induced Ca2+ influx. As shown in Figure 6A, of the P2x agonists tested (2-MeSATP, AMPCPP, ATPγS, and BzATP), only 2-MeSATP was able to induce a [Ca2+]i increase; similar results were seen for induction of Mn2+ influx (data not shown). 2-MeSATP also acts as a P2y agonist in stimulating Ca2+ release via phospholipase C activation. To determine the contribution of the 2-MeSATP-mediated Ca2+ influx, the 2-MeSATP-induced [Ca2+]i increase was measured in thapsigargin-treated cells. As shown in Figure 6B, following thapsigargin pretreatment, the [Ca2+]i increase induced by a second and third exposure to 2-MeSATP was significantly reduced compared to that caused by the initial exposure. The 2-MeSATP-induced [Ca2+]i increase occurred via P2x receptor activation, since it was completely inhibited by pretreatment of cells with 30 μM PPADS for 30 min (Figure 6C).
Figure 6.
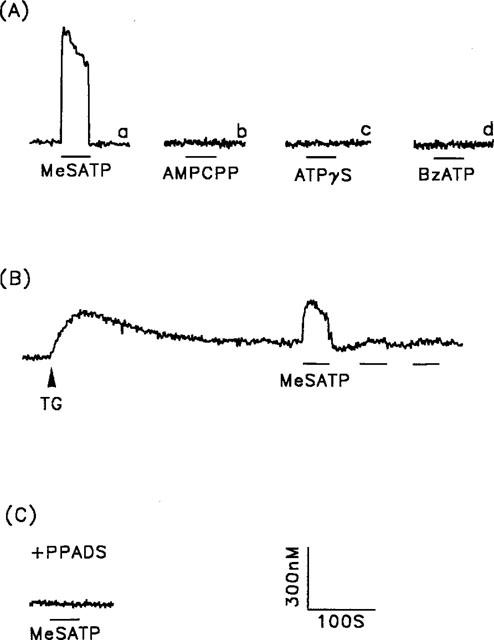
The [Ca2+]i increase induced by P2x purinoceptor agonists. (A) [Ca2+]i changes in response to 2-MeSATP (trace a), AMPCPP (trace b), ATPγS (trace c) and BzATP (trace d) (all 30 μM). (B) Five min after the cells were exposed to 1 μM thapsigargin (TG), responses to repetitive 2-MeSATP stimulation, indicated by the horizontal bars, were measured. (C) After 30 min pretreatment of the cells with 30 μM PPADS, 2-MeSATP-induced [Ca2+]i increases were measured. The concentration of 2-MeSATP was 30 μM in both B and C. Experiments were repeated 26–33 times with similar results; one representative example is shown.
It was possible that VOC might contribute to the ATP-induced Ca2+ signal, since Na+ influx via ATP-gated ion channels (2-MeSATP-sensitive P2x receptors, as described above) can depolarize cells. Furthermore, activation of the P2 purinoceptor stimulates phospholipase A2 to produce arachidonic acid (Winitz et al., 1994), which can then cause a Ca2+ influx, as seen in duckling nasal gland cells (Shuttleworth, 1996). We therefore examined the effects of inhibitors of VOC or phospholipase A2 on the effects of repetitive ATP stimulation. As shown in Figure 7A, neither a combination of VOC inhibitors (30 μM verapamil, 30 μM nifedipine, and 10 μM ω-conotoxin) (trace a) nor a phospholipase A2 inhibitor (3 μM arachidonyl trifluoromethyl ketone; AACOCF3) (trace b) had any effect on the repetitive ATP stimulation-induced [Ca2+]i increases. The data for these changes are summarized in Figure 7B.
Figure 7.
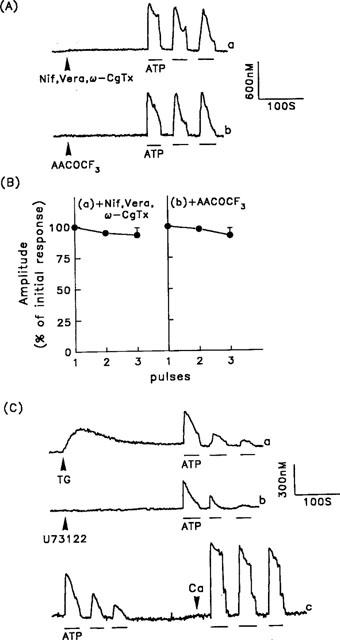
Effect of pertussis toxin, VOC blockers, and a phospholipase A2 inhibitor on the ATP-induced [Ca2+]i increases. (A) After treating the cells for 5 min with 30 μM nifedipine, 30 μM verapamil, and 10 μM ω-conotoxin (Nif, Vera, ω-CgTx) (trace a) or 3 μM arachidonyl trifluoromethyl ketone (AACOCF3) (trace b), repetitive ATP-induced [Ca2+]i increases, indicated by the horizontal bars, were measured in the presence of extracellular Ca2+. (B) Summarized data for the [Ca2+]i responses following pretreatment of cells with nifedipine, verapamil and ω-conotoxin (panel a) or AACOCF3 (panel b). The data are the mean±s.d. for 42–56 cells. (C) Cells were pretreated with 100 ng ml−1 of pertussis toxin for 16 h, then repetitive ATP-induced [Ca2+]i increases were measured 5 min after exposure of the cells to 1 μM thapsigargin (TG) (trace a) or 10 μM U-73122 (trace b). Repetitive ATP-induced [Ca2+]i increases were also measured in the absence of extracellular Ca2+ (trace c), and after extracellular Ca2+ (2.2 mM) was added back (indicated by the arrowhead). The experiments were repeated 22 times with similar results.
We then characterized the toxin sensitivity of the G protein involved in the ATP-induced [Ca2+]i increase. As shown in Figure 7C (trace a), after 16 h pretreatment of cells with 100 ng ml−1 of pertussis toxin, neither the thapsigargin-induced [Ca2+]i increase nor the subsequent repetitive ATP stimulation-induced [Ca2+]i increases differed significantly from those in control cells (compare with Figure 5A, trace a). Similar results were seen when U-73122 was used instead of thapsigargin (Figure 7C, trace b). In the absence of extracellular Ca2+, the ATP-induced [Ca2+]i increase presumably occurs via the phospholipase C signalling cascade, which is pertussis toxin-insensitive, and when Ca2+ was reintroduced into the bathing solution, a reproducible [Ca2+]i increase profile was still seen in response to repetitive ATP stimulations (Figure 7C, trace c). A similar lack of sensitivity was seen when cells were treated with 1 μg ml−1 of cholera toxin for 16 h (data not shown).
SPC, the enzymic product of sphingomyelin deacylase, has been shown to act as an extracellular signal (Meyer zu Heringdorf et al., 1997). In porcine aortic smooth muscle cells, it induces an increase in [Ca2+]i predominantly via IP3-sensitive Ca2+ release (Chin & Chueh, 1998). We therefore measured its effect on the [Ca2+]i increases seen on repetitive stimulation. As shown in Figure 8, the [Ca2+]i increases gradually decreased and were hardly detectable on the third exposure both in the presence or absence of extracellular Ca2+ (Figure 8A and B, trace a). In contrast to the response to ATP (Figure 3B), the desensitized response to SPC, seen in the absence of extracellular Ca2+, was not restored by re-addition of Ca2+ to the bathing buffer (Figure 8B, trace a), but was restored by pretreatment of cells with staurosporine for 5 min (Figure 8A and B, trace b). Since staurosporine was a rather nonspecific kinase inhibitor inhibiting protein kinases A, C and G, we next examined the effect of Rp-cAMPS, Gö 6983, KT5823 and KN-62, the specific inhibitor for protein kinase A, C and G and CaM kinase II, respectively, on repetitive SPC stimulation-induced [Ca2+]i increase. Of the inhibitors tested, only Gö 6983 was able to restore the desensitized Ca2+ response to SPC (Figure 8A, traces c–f); similar results were seen in the absence of extracellular Ca2+ after Ca2+ was added back to the bathing buffer (Figure 8B, traces c–f).
Figure 8.
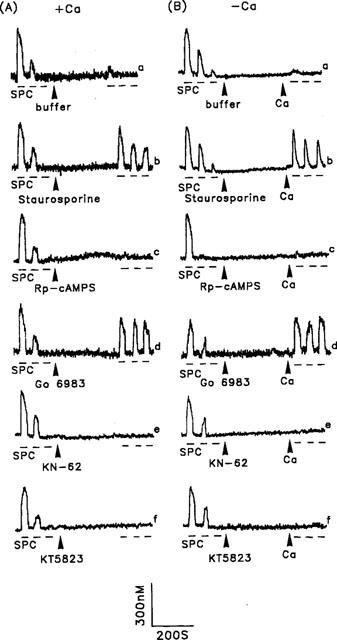
Effect of kinase inhibitors on SPC-induced [Ca2+]i increases in the presence or absence of extracellular Ca2+. Repetitive SPC-induced [Ca2+]i increases to 5 μM SPC were initially measured in the presence (A) or absence (B) of extracellular Ca2+, then after the cells had been treated with buffer (A and B, trace a), 0.1 μM staurosporine (A and B, trace b), 30 μM Rp-cAMPS (A and B, trace c), 0.3 μM Gö 6983 (A and B, trace d), 10 μM KN-62 (A and B, trace e) or 1 μM KT5823 (A and B, trace f), or after adding back 2.2 mM Ca2+ to the bathing solution (B, all traces), as indicated by the arrowheads. Experiments were repeated 32–51 times with similar results; one representative example is shown.
Our results indicate that, in addition to IP3 generation, ATP also stimulated Ca2+ influx via a 2-MeSATP-sensitive P2x receptor, leading to a [Ca2+]i increase. Thus, although both ATP and SPC activated phospholipase C, the action of SPC could be desensitized, while that of ATP could not. However, desensitization of the repetitive SPC-induced [Ca2+]i increase could be reversed by inhibition of protein kinase C-induced protein phosphorylation. To characterize the underlying mechanism, we next measured ATP- or SPC-induced IP3 generation, with or without prior treatment of the cells with staurosporine. As shown in Figure 9A, in control cells without staurosporine pretreatment, ATP-induced IP3 generation reached a peak of 20.3±1.9 (n=6) pmol mg−1 protein after 15 s, then fell to a plateau where it remained for at least 4 min, while the peak level after SPC treatment was significantly less, reaching a peak of 6.3±0.9 (n=6) pmol mg−1 protein in 15 s, and returning to the basal level within 60 s. Following pretreatment of cells with staurosporine, basal and ATP- or SPC-stimulated IP3 generation was similar to that seen in control cells (Figure 9B). Thus, ATP- or SPC-induced IP3 generation in control cells did not differ significantly from that in staurosporine-treated cells. In porcine aortic smooth muscle cells, Ca2+ release from intracellular Ca2+ stores was predominantly attributable to the action of IP3, since neither caffeine (20 mM) nor ryanodine (10 μM) induced [Ca2+]i increase (data not shown).
Figure 9.
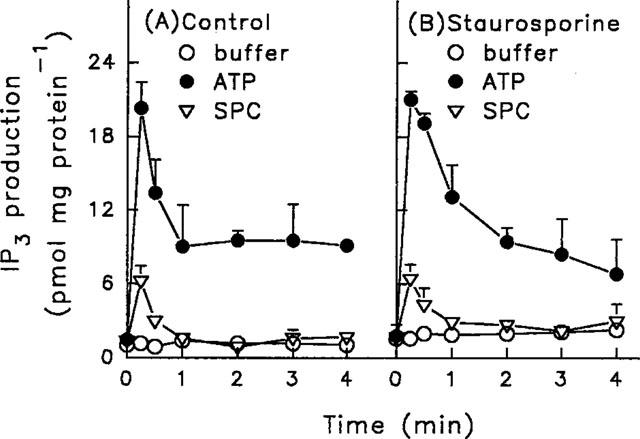
Effect of staurosporine on ATP- or SPC-induced IP3 generation in porcine aortic smooth muscle cells. Control cells (A) or cells pretreated with 0.1 μM staurosporine for 5 min (B) were stimulated with buffer, 30 μM ATP, or 5 μM SPC for the indicated time periods, then the IP3 was extracted and measured using a radioreceptor assay. The data are means±s.d. for six independent experiments.
We finally examined whether protein kinase C inhibitor increased the Ca2+ permeability in SPC-stimulated cells, using Mn2+ influx as an indicator of Ca2+ influx. As shown in Figure 10, SPC did not stimulate Mn2+ influx, since the fluorescence quenching was indistinguishable from that in control cells (traces a and b). The same was true after cells have been pretreated with Rp-cAMPS, KT5823 or KN-62 (data not shown). However, following pretreatment of cells with staurosporine or Gö 6983, the fluorescence quenching induced by SPC was significantly increased (traces c and d, and traces e and f, respectively)
Figure 10.
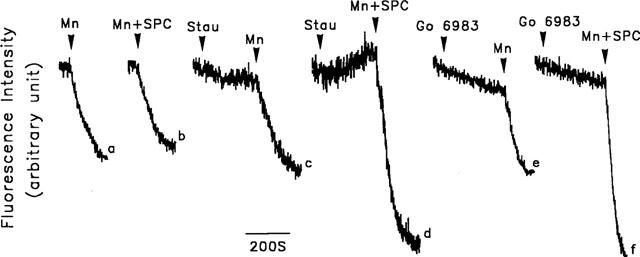
Effect of kinase inhibitors on the SPC-induced Mn2+ influx in porcine aortic smooth muscle cells. Fura-2-loaded cells were bathed in loading buffer containing 1 mM Ca2+. The cells were then treated for 5 min with buffer (traces a and b), 0.1 μM staurosporine (traces c and d) or 0.3 μM Gö 6983 (traces e and f). 1 mM Mn2+ (Mn) (traces a, c and e) or 1 mM Mn2+ plus 5 μM SPC (Mn+SPC) (traces b, d and f) was then added and the fluorescence quenching due to Mn2+ influx measured. The experiments were repeated 28–36 times with similar results; one representative result is shown.
Discussion
Our results show that, in porcine aortic smooth muscle cells, ATP induces both intracellular Ca2+ release and Ca2+ influx. Firstly, the ATP-induced [Ca2+]i increase was greater, and the time required for the [Ca2+]i to fall to the basal level longer, in Ca2+-containing than in Ca2+-free buffer (Figure 1). Secondly, the ATP-induced [Ca2+]i increase was still seen after cells were pretreated with U-73122 or thapsigargin (Figure 5), indicating that Ca2+ influx is activated by ATP, since U-73122 inhibits phospholipase C (Smallridge et al., 1992), while thapsigargin depletes the intracellular Ca2+ stores (Thastrup et al., 1990). Finally, the Mn2+ influx induced by ATP is further support for an ATP-induced Ca2+ influx. In general, Ca2+ influxes are mediated by activation of ROC, VOC and SOC. We further demonstrated that the P2X5 purinoceptor (ROC) was responsible for the ATP-induced Ca2+ influx, and that VOC and SOC were not involved (see below).
In many nonexcitable cells, capacitative Ca2+ entry via SOC can be stimulated by store depletion (Luckhoff & Clapham, 1992; Hoth & Penner, 1992). The results of this study showed SOC activity (trp channels) to be negligible in porcine aortic smooth cells. Firstly, not only was the Mn2+ influx unchanged from the basal level in the presence of thapsigargin (Figure 4B), but the thapsigargin-induced [Ca2+]i increases were identical in the presence or absence of extracellular Ca2+ (Figure 4A). If depletion of the Ca2+ stores altered the permeability of the plasma membrane to Ca2+ via activation of SOC, a higher [Ca2+]i increase in the presence of extracellular Ca2+ or a greater Mn2+ influx in response to thapsigargin would be expected. The ineffectiveness of thapsigargin in inducing Mn2+ or Ca2+ influx in the current study is consistent with a recent study in human embryonic kidney 293 cells transfected with the human Trp3 (hTrp3) gene, in which expression of the hTrp3 gene formed a nonselective cation channel that opened after activation of phospholipase C, but not after store depletion (Zhu et al., 1998). Secondly, both the ATP-induced [Ca2+]i increase and the Mn2+ influx were not affected by the SOC inhibitor, SK&F96365 (Figures 3C and 4B), ruling out involvement of SOC in the ATP-induced Ca2+ influx. Thirdly, pretreatment of cells with U-73122 or thapsigargin inhibits the phospholipase C signalling cascade; U-73122 inhibits phospholipase C, while thapsigargin depletes the intracellular Ca2+ stores and may cause SOC-induced Ca2+ influx. Thus, the ATP-induced [Ca2+]i increases seen in the presence of U-73122 or thapsigargin reflect, respectively, the activity of VOC and ROC, or VOC, ROC and SOC. The amplitudes of the [Ca2+]i increases on multiple stimulation were identical in the presence of these two inhibitors and were not affected by the SOC inhibitor, SK&F96365 (Figure 5), suggesting that, in porcine aortic smooth muscle cells, the contribution of SOC to the ATP-induced Ca2+ influx is negligible. Thus, the possible mechanisms responsible for the ATP-induced Ca2+ influx are ROC and VOC.
According to the classical classification, both P2x and P2z purinoceptors (ROC) may be involved in the ATP-induced Ca2+ influx. The fact that BzATP, a P2z (P2X7) purinoceptor-selective agonist, could not induce a [Ca2+]i increase or Mn2+ influx ruled out involvement of the P2z purinoceptor in porcine aortic smooth muscle cells. The effectiveness of 2-MeSATP, the ineffectiveness of AMPCPP and ATPγS, and the blockade of the [Ca2+]i increase by 30 μM PPADS further suggest that activation of the P2X5 purinoceptor is responsible for the ATP-induced Ca2+ influx (Figure 6) (North & Barnard, 1997; Kunapuli & Daniel, 1998; Boarder & Hourani, 1998). Opening of ROC allows a Na+ influx, which may subsequently depolarize the membrane potential and cause a VOC-mediated Ca2+ influx. Indeed, in pituitary gonadotrophs, the ATP-induced [Ca2+]i response is partially inhibited by nifedipine (Tomic et al., 1996). Similar results are seen in NG108-15 cells, in which the ATP-induced Ca2+ influx via P2x or P2z is inhibited approximately 34–76% by VOC blockers, including ω-conotoxin, nifedipine, and verapamil (Chueh & Kao, 1993). In the present study, the insensitivity of the ATP-induced [Ca2+]i increase to nifedipine, verapamil, and ω-conotoxin suggests that, in porcine aortic smooth muscle cells, VOC is not involved (Figure 7).
It is possible that the ATP-induced [Ca2+]i increase may be partially due to the release of arachidonic acid, since phospholipase A2 is reported to be stimulated by ATP (Winitz et al., 1994) and arachidonic acid mobilizes Ca2+ by either activating Ca2+ influx or triggering Ca2+ release (Vacher et al., 1989). A recent study has also shown that depletion of Ca2+ stores by arginine vasopressin or thapsigargin can induce arachidonic acid release in A-10 smooth muscle cells (Wolf et al., 1997). In the present study, the ATP-induced [Ca2+]i increase was not blocked by a phospholipase A2 inhibitor, suggesting that arachidonic acid is not involved in this process in porcine aortic smooth muscle cells.
The inability of caffeine or ryanodine to produce a [Ca2+]i increase suggests that Ca2+ release from the intracellular Ca2+ stores is mainly mediated by activation of the IP3 receptor, rather than by Ca2+-induced Ca2+ release. The reduced responses seen on repetitive ATP stimulation in the absence of extracellular Ca2+ suggest either that the receptor-G protein-phospholipase C coupling system desensitizes or that Ca2+ contained in the intracellular Ca2+ stores is not available after several stimulations due to store depletion (Figure 2). Indeed, the IP3 generation induced by ATP or SPC, even in the presence of extracellular Ca2+, was transient, with a rapidly declining peak (Figure 9) and, in the absence of extracellular Ca2+, the fullness of the Ca2+ stores was significantly reduced after repetitive ATP stimulation (Figure 3). The facts that Ca2+ levels in the intracellular Ca2+ stores were not reduced following multiple stimulations in the presence of extracellular Ca2+ (Figure 3) and the ATP responses fully recovered on reintroduction of Ca2+ suggest that to have reproducible ATP effects on the [Ca2+]i increase the filling state of the intracellular Ca2+ stores is critical. The decrease in the ATP responses following thapsigargin or U-73122 pretreatment (Figure 5) suggests that the ATP-induced Ca2+ influx desensitizes. It appears that, under physiological conditions, following repetitive ATP stimulation, even the reduced Ca2+ influx seen in a desensitized state is sufficient to refill the partially depleted intracellular Ca2+ stores and leads to a reproducible [Ca2+]i increase. The recovery of the SPC-induced Ca2+ response due to inhibition of protein kinase C-induced phosphorylation is mainly attributable to Ca2+ influx, rather than to IP3 generation (Figures 9 and 10), further indicating the pivotal role of the intracellular Ca2+ stores in the Ca2+ signalling pathway.
In conclusion, we have demonstrated that, in porcine aortic smooth muscle cells, activation of P2 purinoceptors by ATP induces an increase in [Ca2+]i which is insensitive to pertussis toxin and cholera toxin. In addition to triggering intracellular Ca2+ release via phospholipase C activation, ATP also stimulates Ca2+ influx via P2X5 receptors. Although the activity of P2X5 receptor progressively decreases following repetitive stimulation with ATP, the overall Ca2+ signal induced by ATP remains the same if the releasing and refilling pathways of the intracellular Ca2+ stores are not inhibited, suggesting involvement of the intracellular Ca2+ stores in the Ca2+ signalling pathway. Our data further show that depletion of the intracellular Ca2+ stores alone does not alter the plasma membrane permeability to Ca2+. In contrast, the SPC-activated Ca2+ signalling pathway is solely mediated by the phospholipas C cascade, is distinct from that activated by ATP, and desensitizes. The desensitization of the SPC-induced Ca2+ signal can be reversed by inhibition of protein kinase C, which stimulates Ca2+ influx, further supporting the importance of the refilling of intracellular Ca2+ stores by influxed Ca2+ in the Ca2+ signalling pathway.
Acknowledgments
We thank Dr Thomas Barkas for helpful discussion. This work was supported by grants from the National Science Council (NSC89-2316-B016-001) and the National Defense Medical Center (DOD-89-22), Republic of China.
Abbreviations
- AACOCF3
arachidonyl trifluoromethyl ketone
- AMPCPP
α,β-methylene adenosine 5′-triphosphate
- ATPγS
adenosine 5′-O-(3-thiotriphosphate)
- BzATP
2′- and 3′-O-(4-benzoylbenzoyl)-adenosine 5′-triphosphate
- [Ca2+]i
cytosolic Ca2+ concentration
- DIDS
4,4′-diisothiocyano-2,2′-disulfonic acid stilbene
- IP3
inositol 1,4,5-trisphosphate
- KN-62
{1-[N,O-bis-(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine}
- 2-MeSATP
2-methylthioadenosine 5′-triphosphate
- PPADS
pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid
- ROC
receptor-operated Ca2+ channel
- Rp-cAMPS
Rp-adenosine 3′,5′-cyclic monophosphothioate
- SK&F96365
1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole HCl
- SPC
sphingosylphosphorylcholine
- SOC
store-operated Ca2+ channel
- U-73122
1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]-amino]hexyl]-1H-pyrrole-2,5-dione
- VOC
voltage-operated Ca2+ channel
References
- BARNARD E.A. The transmitter-gated channels: a range of receptor types and structures. Trends Pharmacol. Sci. 1996;17:305–309. [PubMed] [Google Scholar]
- BERRIDGE M.J. Elementary and global aspects of calcium signalling. J. Physiol. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIRNBAUMER L., ZHU X., JIANG M., BOULAY G., PEYTON M., VANNIER B., BROWN D., PLATANO D., SADEGHI H., STEFANI E., BIRNBAUMER M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proc. Natl. Acad. Sci. U.S.A. 1996;93:15195–15202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOARDER M.R., HOURANI S.M.O. The regulation of vascular function by P2 receptors: multiple sites and multiple receptors. Trends Pharmacol. Sci. 1998;19:99–107. doi: 10.1016/s0165-6147(98)01170-5. [DOI] [PubMed] [Google Scholar]
- CHIN T.Y., CHUEH S.H. Sphingosylphosphorylcholine stimulates mitogen-activated protein kinase via a Ca2+-dependent pathway. Am. J. Physiol. 1998;275:C1255–C1263. doi: 10.1152/ajpcell.1998.275.5.C1255. [DOI] [PubMed] [Google Scholar]
- CHUEH S.H., GILL D.L. Inositol 1,4,5-trisphosphate and guanine nucleotides activate calcium release from endoplasmic reticulum via distinct mechanisms. J. Biol. Chem. 1986;261:13883–13886. [PubMed] [Google Scholar]
- CHUEH S.H., KAO L.S. Extracellular ATP stimulates calcium influx in neuroblastoma×glioma hybrid NG108-15 cells. J. Neurochem. 1993;61:1782–1788. doi: 10.1111/j.1471-4159.1993.tb09816.x. [DOI] [PubMed] [Google Scholar]
- DOUSA T.P., CHINI E.N., BEERS K.W. Adenine nucleotide diphosphates: emerging second messengers acting via intracellular Ca2+ release. Am. J. Physiol. 1996;271:C1007–C1024. doi: 10.1152/ajpcell.1996.271.4.C1007. [DOI] [PubMed] [Google Scholar]
- FURUICHI T., MIKOSHIBA D. Inositol 1,4,5-trisphosphate receptor-mediated Ca2+ signaling in the brain. J. Neurochem. 1995;64:953–960. doi: 10.1046/j.1471-4159.1995.64030953.x. [DOI] [PubMed] [Google Scholar]
- GHOSH T.K., BIAN J., GILL D.L. Intracellular calcium release mediated by sphingosine derivatives generated in cells. Science. 1990;248:1653–1656. doi: 10.1126/science.2163543. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HARDEN T.K., BOYER J.L., NICHOLAS R.A. P2-purinergic receptors: subtype-associated signaling responses and structure. Ann. Rev. Pharmacol. Toxicol. 1995;35:541–579. doi: 10.1146/annurev.pa.35.040195.002545. [DOI] [PubMed] [Google Scholar]
- HESS P. Calcium channels in vertebrate cells. Ann. Rev. Neurosci. 1990;13:337–356. doi: 10.1146/annurev.ne.13.030190.002005. [DOI] [PubMed] [Google Scholar]
- HOTH M., PENNER R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- HOURANI S.M.O., HALL D.A. Receptor for ADP on human blood platelet. Trends Pharmacol. Sci. 1994;514:103–108. doi: 10.1016/0165-6147(94)90045-0. [DOI] [PubMed] [Google Scholar]
- KUNAPULI S.P., DANIEL J.L. P2 receptor subtypes in the cardiovascular system. Biochem. J. 1998;336:513–523. doi: 10.1042/bj3360513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE M.J., VAN BROCKLYN J.R., THANGADA S., LIU D.H., HAND A.R., MENZELEEV R., SPIEGEL S., HLA T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- LEE H.C., WALSETH T.F., BRATT G.T., HAYES R.N., CLAPPER D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989;264:1608–1615. [PubMed] [Google Scholar]
- LEWIS C., NEIDHART S., HOLY C., NORTH R.A., BUELL G., SURPRENANT A. Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature. 1995;377:432–435. doi: 10.1038/377432a0. [DOI] [PubMed] [Google Scholar]
- LUCKHOFF A., CLAPHAM D.E. Inositol 1,3,4,5-tetrakisphosphate activates an endothelial Ca2+-permeable channel. Nature. 1992;355:356–358. doi: 10.1038/355356a0. [DOI] [PubMed] [Google Scholar]
- MARKS A.R. Intracellular calcium-release channels: regulators of cell life and death. Am. J. Physiol. 1997;272:H597–H605. doi: 10.1152/ajpheart.1997.272.2.H597. [DOI] [PubMed] [Google Scholar]
- MERTZ L.M., BAUN B.J., ANBUDKAR I.S. Refill status of the agonist-sensitive Ca2+ pool regulates Mn2+ influx into parotid acini. J. Biol. Chem. 1990;265:15010–15014. [PubMed] [Google Scholar]
- MEYER ZU HERINGDORF D., VAN KOPPEN C.J., JAKOBS K.H. Molecular diversity of sphingolipid signalling. FEBS Lett. 1997;410:34–38. doi: 10.1016/s0014-5793(97)00320-7. [DOI] [PubMed] [Google Scholar]
- MONTERO M., ALVARIZ J., GARCIA-SANCHO J. Uptake of Ca2+ and refilling of intracellular Ca2+ stores in Ehrlich-ascites-tumour cells and in rat thymocytes. Biochem. J. 1990;271:535–540. doi: 10.1042/bj2710535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NORTH R.A., BARNARD E.A. Nucleotide receptors. Curr. Opin. Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- OKAJIMA F., KONDO Y. Pertussis toxin inhibits phospholipase C activation and Ca2+ mobilization by sphingosylphosphorylcholine and galactosylsphingosine in HL60 leukemia cells. J. Biol. Chem. 1995;270:26332–26340. doi: 10.1074/jbc.270.44.26332. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., JR Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- ROSS R. The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J. Cell. Biol. 1971;50:172–186. doi: 10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO K., OKAJIMA F., KONDO Y. Extracellular ATP stimulates three different receptor-signal transduction systems in FRTL-5 thyroid cells. Biochem. J. 1992;283:281–287. doi: 10.1042/bj2830281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEUFFERLEIN T., ROZENGURT E. Sphingosylphosphorylcholine activation of mitogen-activated protein kinase in Swiss 3T3 cells requires protein kinase C and pertussis toxin-sensitive G protein. J. Biol. Chem. 1995;270:24334–24342. doi: 10.1074/jbc.270.41.24334. [DOI] [PubMed] [Google Scholar]
- SHUTTLEWORTH T.J. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J. Biol. Chem. 1996;271:21720–21725. doi: 10.1074/jbc.271.36.21720. [DOI] [PubMed] [Google Scholar]
- SIPMA H., DEN HERTOG A., NELEMANS A. The phospholipase C activating P2u purinoceptor also inhibits cAMP formation in DDT1MF2 smooth muscle cells. Eur. J. Pharmacol. 1994;268:431–437. doi: 10.1016/0922-4106(94)90069-8. [DOI] [PubMed] [Google Scholar]
- SMALLRIDGE R.C., KIANG J.G., GIST I.D., FEIN H.G., GALLOWAY R.J. U-73122, an aminosteroid phospholipase C antagonist, noncompetitively inhibits thyrotropin-releasing hormone effects in GH3 rat pituitary cells. Endocrinology. 1992;131:1883–1888. doi: 10.1210/endo.131.4.1396332. [DOI] [PubMed] [Google Scholar]
- SONG S.L., CHUEH S.H. Antagonistic effect of Na+ and Mg2+ on P2z purinoceptor-associated pores in dibutyryl cyclic AMP-differentiated NG108-15 cells. J. Neurochem. 1996;67:1694–1701. doi: 10.1046/j.1471-4159.1996.67041694.x. [DOI] [PubMed] [Google Scholar]
- SURPRENANT A., BUELL G., NORTH R.A. P2x receptors bring new structure to ligand-gated ion channels. Trends Neurosci. 1995;18:224–229. doi: 10.1016/0166-2236(95)93907-f. [DOI] [PubMed] [Google Scholar]
- SURPRENANT A., RASSENDREN F., KAWASHIMA E., NORTH R.A., BUELL G. The cytolytic P2z receptor for extracellular ATP identified as a P2x receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- THASTRUP O., CULLEN P.J., DROBAK B.K., HANLEY M.R., DAWSON A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMIC M., JOBIN R.M., VERGARA L.A., STOJILKOVIC S.S. Expression of purinergic receptor channels and their role in calcium signaling and hormone release in pituitary gonadotrophs. J. Biol. Chem. 1996;271:21200–21208. doi: 10.1074/jbc.271.35.21200. [DOI] [PubMed] [Google Scholar]
- VACHER P., MCKENZIE J., DUFY B. Arachidonic acid affects membrane ionic conductances of GH3 pituitary cells. Am. J. Physiol. 1989;257:E203–E211. doi: 10.1152/ajpendo.1989.257.2.E203. [DOI] [PubMed] [Google Scholar]
- VAN KOPPEN C.J., MEYER ZU HERINGDORG D., ZHANG C., LASER K.T., JAKOBS K.H. A distinct Gi protein-coupled receptor for sphingosylphosphorylcholine in human leukemia HL-60 cells and human neutrophils. Mol. Pharmacol. 1996;19:956–961. [PubMed] [Google Scholar]
- WILKINSON G.G., PURKISS J.R., BOARDER M.R. The regulation of aortic endothelial cells by purines and pyrimidines involves co-existing P2y-purinoceptors and nucleotide receptors linked to phospholipase C. Br. J. Pharmacol. 1993;108:689–693. doi: 10.1111/j.1476-5381.1993.tb12862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WINITZ S., GUPTA S., QIAN N.X., HEASLEY L.E., NEMENOFF R.A., JOHNSON G.L. Expression of a mutant Gi2α subunit inhibits ATP and thrombin stimulation of cytoplasmic phospholipase A2-mediated arachidonic acid release independent of Ca2+ and mitogen-activated protein kinase regulation. J. Biol. Chem. 1994;269:1889–1895. [PubMed] [Google Scholar]
- WOLF M.J., WANG J., TURK J., GROSS R.W. Depletion of intracellular calcium stores activates smooth muscle cell calcium-independent phospholipase A2. J. Biol. Chem. 1997;272:522–526. doi: 10.1074/jbc.272.3.1522. [DOI] [PubMed] [Google Scholar]
- WU L., KATZ S., BROWN G.R. Inositol 1,4,5-trisphosphate-, arachidonic acid- and thapsigargin-mediated intracellular calcium movement in PANC-1 microsomes. Cell Calcium. 1994;15:228–240. doi: 10.1016/0143-4160(94)90062-0. [DOI] [PubMed] [Google Scholar]
- ZHU X., JIANG M., BIRNBAUMER L. Receptor-activated Ca2+ influx via human Trp3 stably expressed in human embryonic kidney (HEK) 293 cells. J. Biol. Chem. 1998;273:133–142. doi: 10.1074/jbc.273.1.133. [DOI] [PubMed] [Google Scholar]