

Abstract
Nociceptin (NC), alias Orphanin FQ, has been recently identified as the endogenous ligand of the opioid receptor-like 1 receptor (OP4). This new NC/OP4 receptor system belongs to the opioid family and has been characterized pharmacologically with functional and binding assays on native (mouse, rat, guinea-pig) and recombinant (human) receptors, by using specific and selective agonists (NC, NC(1–13)NH2) and a pure and competitive antagonist, [Nphe1]NC(1–13)NH2. The similar order of potency of agonists and affinity values of the antagonist indicate that the same receptor is present in the four species. OP4 is expressed in neurons, where it reduces activation of adenylyl cyclase and Ca2+ channels while activating K+ channels in a manner similar to opioids. In this way, OP4 mediates inhibitory effects in the autonomic nervous system, but its activities in the central nervous system can be either similar or opposite to those of opioids. In vivo experiments have demonstrated that NC modulates a variety of biological functions ranging from nociception to food intake, from memory processes to cardiovascular and renal functions, from spontaneous locomotor activity to gastrointestinal motility, from anxiety to the control of neurotransmitter release at peripheral and central sites. These actions have been demonstrated using NC and various pharmacological tools, as antisense oligonucleotides targeting OP4 or the peptide precursor genes, antibodies against NC, an OP4 receptor selective antagonist and with data obtained from animals in which the receptor or the peptide precursor genes were knocked out. These new advances have contributed to better understanding of the pathophysiological role of the NC/OP4 system, and ultimately will help to identify the therapeutic potential of new OP4 receptor ligands.
Keywords: Nociceptin/orphanin FQ, nociceptin receptor, recombinant and native receptors, agonists, antagonists, receptor characterization and classification
Introduction
The history of the NC/OP4 receptor system began in 1992, with the cloning of the OP1 (δ) receptor (Evans et al., 1992; Kieffer et al., 1992), followed shortly by that of the OP2 (κ) and OP3 (μ) receptors (Chen et al., 1993; Yasuda et al., 1993). These three opioid receptors show about 60% homology. Further screening of cDNA libraries with low stringency oligonucleotide probes led, in 1994, to the discovery of an ‘opioid receptor like' (ORL1) sequence by several investigators (Bunzow et al., 1994; Fukuda et al., 1994; Mollereau et al., 1994; Nishi et al., 1994; Wang et al., 1994). This new receptor shows overall 60% homology with the opioid receptors (80% in the 2nd, 3rd and 7th TM domains), much less in the N-terminal and some extracellular loops and again high homology in some intracellular loops. ORL1 shows substantial sequence identities (>90%) between species variants, namely the human (Mollereau et al., 1994), rat (Bunzow et al., 1994; Chen et al., 1994; Lachowicz et al., 1995; Wang et al., 1994), mouse (Nishi et al., 1994) and pig (Osinski et al., 1999a). Therefore, on structural grounds, the ORL1 and opioid receptors belong to the same family. Initial pharmacology (before NC was actually discovered) showed that the ORL1 receptor, stably transfected into Chinese hamster ovary (CHO) cells, could be activated by the nonselective opioid receptor agonist ethorphine and blocked by the opioid receptor antagonist diprenorphine, each at micromolar concentrations (Mollereau et al., 1994). Naloxone however, a nonselective opioid receptor antagonist showed very little affinity, if any, for ORL1 (Mollereau et al., 1994). ORL1 is considered by some experts as an opioid receptor and has been given (according to the new nomenclature for opioid receptors: Dhawan et al., (1996)) the name OP4 (Hamon, 1998), while in other classifications ORL1 has been left out from the opioid family of receptors (Dhawan et al., 1998), or included as ORL1 (Receptors and ion channel nomenclature supplement, Trends Pharmacol. Sci., 1999). In peripheral tissues, the distribution of ORL1 (intestine, vas deferens, spleen, etc.), the cell type(s) that express ORL1 (predominantly neurons) and the cellular mechanisms of action (activation of K+ and inhibition of Ca2+ channels, inhibition of cyclic AMP accumulation) are probably the same as for opioid receptors and may derive from the activation of the same type of G proteins (see Meunier, 1997 for a review). The actions of ORL1 receptors in peripheral tissues are indeed very similar to those evoked by opioid receptors. The distribution of ORL1 in the brain (rat, mouse) (Anton et al., 1996; Ikeda et al., 1998; Monteillet-Agius et al., 1998; Neal et al., 1999), as evaluated by immunohystochemistry and in situ hybridization, is widespread but expression is particularly high in cortico-limbic areas, hypothalamus, various brain stem nuclei, and spinal cord. The distribution of ORL1 appear to be similar in some and different in other neuronal circuits as the opioids receptors. These anatomical similarities and differences may help understanding why ORL1 and opioid receptors mediate similar, different and even opposite effects in the central nervous system (see section on ‘Biological activities').
ORL1 did not remain an orphan receptor for long, since only a year after its identification, towards the end of 1995, the endogenous ligand was identified, simultaneously, by two groups of investigators. Whereas the French group named it nociceptin (Meunier et al., 1995), the Swiss group called it orphanin FQ (Reinscheid et al., 1995). It is a heptadecapeptide containing several cationic residues (see primary structure in Table 1), which has rapidly become the target of numerous investigators. The current review attempts to focus on the pharmacological issues revealed by the already more than 350 papers published so far on the NC/OP4 system.
Table 1.
Chemical names and structures of opoid receptor ligands

Peptide and receptor nomenclature
In 1987 the International Union of Pharmacology (IUPHAR) established a Nomenclature Committee for mammalian receptors, recommending that a given receptor should be named after its endogenous ligand(s), attributed a progressive number (1, 2, etc) to identify the different receptor types, and that the use of Greek symbols as well as of the letter R (to designate receptor) should be avoided (see Girlestone, 1998). In 1996, the Opioid Receptor Sub Committee proposed a new nomenclature consistent with the IUPHAR guidelines (Dhawan et al., 1996). Thus the members of the opioid receptor family should be indicated as OP (for Opioid Peptides) with numeric subscripts indicating the chronological cloning of the receptor types, i.e. OP1 OP2 and OP3 corresponding to the δ-, κ-, and μ-opioid receptor types, respectively. This proposal has been ignored, however, by most of the authors publishing in this field. The situation has been further complicated after the identification of NC and its receptor. In fact, on structural and transductional grounds, the receptor for NC should be viewed as a member of the opioid receptor family which, however, is insensitive to naloxone while interancting with other opioid receptor ligands, such as naloxone benzoylhydrazone (NalBzOH) and buprenorphine. To date, the following names and abbreviations have been used for the peptide: nociceptin (NC, Noc, Noci), orphanin FQ (OFQ or oFQ), nociceptin/orphanin FQ (N/OFQ). A discussion of the advantage and disadvantages of the name nociceptin vs orphanin FQ may be found in a recent review by Civelli et al. (1998). As for its receptor, it has been differently named opioid like receptor 1 (ORL1), nociceptin receptor (NCR or NocR), orphanin FQ receptor (OFQR), nociceptin-orphanin FQ receptor (NOR, in analogy with the MOR, DOR and KOR terminology) and finally OP4 (in line with the recent IUPHAR recommendations, Hamon (1998).
These issues have recently been debated at the International Narcotics Research Conference meeting in Saratoga Springs (July 10–15, 1999) during a short symposium on opioid receptor nomenclature chaired by B. Cox and C. Chavkin, in order to collect suggestions for a unified nomenclature in line with the new edition of the IUPHAR Compendium of receptor characterisation and classification, which will be published in the near future. It is hoped that the experts in the field will accept the NC/OP4 nomenclature that is utilized throughout this article.
Basic pharmacological tools and features of OP1, OP2, OP3, and OP4 receptors
The three opioid receptors are indicated as OP1, OP2, and OP3 (Dhawan et al., 1996) by the current nomenclature recommended by IUPHAR, and by the former nomenclature in Greek letter (δ, κ, and μ); OP4 is also indicated by the initial name ORL1. The cloned human receptors are identified by their genetic code; they are 7TM receptors coupled with Gi/o proteins. Endogenous ligands that show some selectivity for one or the other receptor are listed, together with the shortest sequences of natural peptides that maintain consistent biological activities (Table 2). While enkephalins (OP1) and endomorphins (OP3) do not contain cationic residues, the shortest active sequences that activate OP2 and OP4, namely Dyn(1–8) and NC(1–13)NH2, have two (Arg6–Arg7) and four (Arg8, Lys9, Arg12, Lys13) positively charged groups respectively that are pivotal for peptide-receptor interaction. While all natural ligands for OP1, OP2 and OP3 receptors have Tyr1, which is instrumental for the opioid receptor activation, ligands of the OP4 receptor have a Phe at the N-terminal end (Table 1). For all receptors, at least one synthetic selective agonist (a few non-peptide or pseudopeptides) is available (see chemical names and structures in Table 1). For each receptor, at least one antagonist is listed: these compounds are non peptides (natrindole and nor-BNI) or pseudopeptides (CTOP and [Nphe1]NC(1–13)NH2). Non peptide ligands (both agonist and antagonists) for OP4 receptors have already been identified (see chemical names and structures in Table 1).
Table 2.
Opoid receptors and their ligands

The potencies of all compounds included in Table 2 have been established with biological assays and are indicated in brackets. Naloxone acts as an antagonist of OP1, OP2, and OP3 receptors with different potencies (pA2), but is inactive on OP4 receptor.
The endogenous ligand for the OP4 receptor
The same heptadecapeptide isolated by Meunier et al. (1995) and Reinscheid et al. (1995) is encoded in the gene sequences of several species, even though there are differences in the NC precursor identified in the mouse (Houtani et al., 1996; Mollereau et al., 1996; Pan et al., 1996), rat (Mollereau et al., 1996; Nothacker et al., 1996) and man (Mollereau et al., 1996; Nothacker et al., 1996). The NC precursor consists of a 181 amino acids in the rat, 176 amino acids in humans, and 187 amino acids in the mouse (Mollereau et al., 1996; Nothacker et al., 1996). Analysis of the nucleotide sequence of the preproNC gene revealed structural and organisational characteristics very similar to those of the opioid peptide precursors, in particular preproenkephalin and preprodynorphin, suggesting that these peptide precursors may derive from a common ancestor (Mollereau et al., 1996; Nothacker et al., 1996). In the preproNC sequence there are several pairs of basic amino acids that represent possible sites of cleavage for precursor maturation. Therefore, several biologically relevant peptides may derive from the NC precursor. The structure of the NC precursor and its components is given in Figure 1A. In particular, two peptides which are potentially derived from the NC precursor have been synthesized and evaluated for biological activity. Neither of them bind to the OP4 receptor (Mollereau et al., 1996; Nothacker et al., 1996). The first is, like NC, an heptadecapeptide terminating with the couple FG (orphanin FQ2), which has been found to be biologically active, stimulating locomotor activity in mice (Florin et al., 1997), inducing antinociception both spinally and supraspinally (Rossi et al., 1998), and inhibiting gastrointestinal transit (Rossi et al., 1998). The second peptide, named nocistatin, has been reported to act as a functional antagonist of NC (Okuda-Ashitaka et al., 1998). In most studies, nocistatin was found to be inactive per se, but was able to reverse several effects of NC, such as induction of allodynia after spinal administration in mice (Minami et al., 1998; Okuda-Ashitaka et al., 1998), inhibition of glutamate release from rat brain slices (Nicol et al., 1998a), impairment of learning and memory in mice (Hiramatsu & Inoue, 1999). Moreover, nocistatin can, per se, cause antinociception after i.c.v. administration in the rat carrageenan test (Nakagawa et al., 1999) or after i.t. administration in the rat formalin test (Yamamoto & Sakashita, 1999).
Figure 1.

(A) the NC precursor. (B) NC metabolism. APN, aminopeptidase N; EP endopeptidase.
Biologically active peptides are brought to maturation by enzymes of the furin group, which cut the amino acid sequence at pairs of cationic residues. Once released from the neurons, NC is further metabolized to inactive fragments by different types of proteolytic enzymes. Montiel et al. (1997) studied NC metabolism in mouse brain cortical slices and reported that NC inactivation is due to the action of aminopeptidase N (APN) which releases NC(2–17), a C-terminal fragment which does not bind to OP4 receptor sites (Dooley & Houghten, 1996), and of endopeptidase 24.15 (EP 24.15) which by acting at the peptide bonds Ala7–Arg8, Ala11–Arg12, and Arg12–Lys13, also releases inactive compounds. On the other hand, endopeptidase 24.11 (enkephalinase) seems to not be involved in the degradation of NC. These findings were later confirmed by showing that a mixture of inhibitors of endopeptidase 24.15 and APN potentiated the behavioural effects of NC in mice (Noble & Roques, 1997). The critical role of aminopeptidase in NC metabolism has also been suggested by Yu et al. (1996), who studied NC biotransformation in human blood. NC metabolism has also been evaluated in vivo in the rat hippocampus (Sandin et al., 1999), where the peptide is first converted to NC(1–13) and then, probably by the same enzyme, to NC(1–9). Similar results were also obtained in vitro using enzymes from different cell cultures, namely U1690 human lung carcinoma cells, SHSY5Y human neuroblastoma cells, and primary culture from rat brain cortex cells (Vlaskovska et al., 1999). Metabolic pathways of NC are indicated in Figure 1B.
Pharmacological characterization of the NC/OP4 receptor system
Basic pharmacological parameters for receptor characterisation include pKi and pEC50/pA2 (for agonists and antagonists, respectively) values determined in binding and functional assays on recombinant and native receptors. An example is presented in Table 3, where NC, NC(1–13)NH2 (the template we have adopted for structure-activity studies, see below), NC(1–9)NH2 and a recently identified OP4 antagonist, [Nphe1]NC(1–13)NH2 (Calo' et al., 2000; Guerrini et al., 1999) have been studied in CHO cells expressing the human recombinant OP4 receptor (CHOhOP4) and in native mouse OP4 receptors. Using a few agonists and one antagonist, we were able to characterize OP4 receptor with the two classical pharmacological criteria: the order of potency of agonists and the affinity of a competitive antagonist (Schild, 1973). The two receptors analysed in Table 3 appear to be of the same type since both criteria are fulfilled and, in particular, the absolute value of the antagonist affinity is the same in the two functional assays. Major differences (>1.0 log units) are observed for agonist affinities and activities: they may be attributed to differences in transduction coupling between receptors expressed in transfected cells and in the isolated organ. In the former preparation, agonists are more potent by at least 1.5 log units. Methodological details of these assays are to be found in the publications quoted in the footnotes of Table 3, which also provide definitions of the three pharmacological parameters, according to the IUPHAR recommendations (Jenkinson et al., 1995).
Table 3.
Affinities and potencies of OP4 receptor agonists and antagonists on native and recombinant receptors

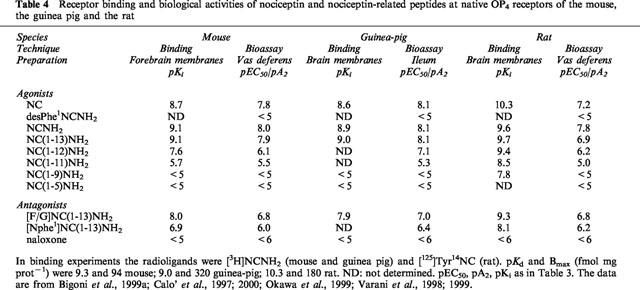
A detailed characterization of native OP4 receptors from various species (mouse, guinea-pig, and rat) is presented in Table 4. With minor discrepancies, the three couples of binding and functional assays depicted show the same order of potency of agonists, namely
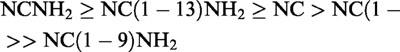 |
and very similar pA2 values for [Nphe1]NC(1–13)NH2 (6.0–6.4) and for [F/G]NC(1–13)NH2 (6.8–7.0). In contrast to [Nphe1]NC(1–13)NH2, which did not show any residual agonist activity in these preparations, [F/G]NC(1–13)NH2 displayed a small but consistent residual agonistic activity, especially in the rat vas deferens (Bigoni et al., 1999a; Okawa et al., 1999). It is therefore proposed that the OP4 receptor is the same in the mouse, the guinea-pig, and the rat.
Table 4.
Receptor binding and biological activities of nociceptin and nociceptin-related peptides at native OP4 receptors of the mouse, the guinea pig and the rat
Results obtained on the human OP4 receptor transfected into CHO cells are presented in Table 5. With minor discrepancies, the order of potency of agonists and the value of antagonistic potency of [Nphe1]NC(1–13)NH2 (pA2 6.0) evaluated in the functional assay are the same as those determined in other animal tissues. Interestingly, [F/G]NC(1–13)NH2 acts as a full agonist in CHOhOP4 cells (Butour et al., 1998; Okawa et al., 1999; Wnendt et al., 1999). A detailed discussion of the dual behaviour of this compound is presented in the section ‘The OP4 receptor ligand [F/G]NC(1–13)NH2'. Taken together these data are consistent with the proposal that the OP4 receptors present in four different species are the same pharmacological entity.
Table 5.
Receptor binding and biological activities of nociceptin and nociceptin-related peptides at recombinant human OP4 receptors expressed in CHO cells
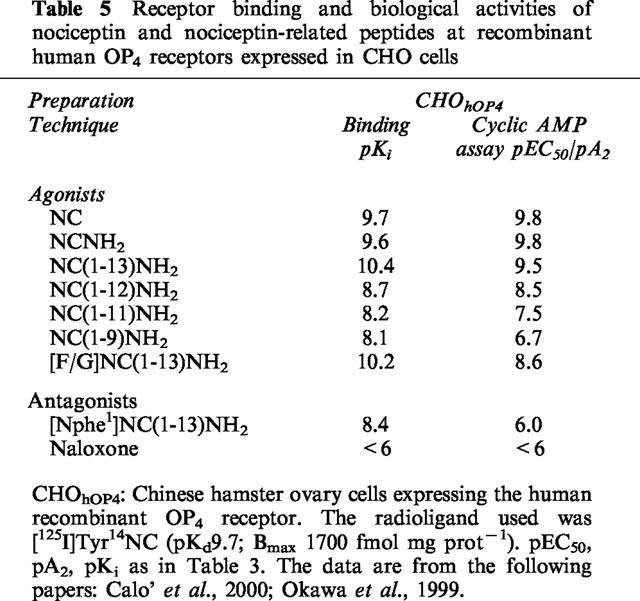
The comparison of the absolute values obtained in binding and functional assays reveals major differences between the affinity constants (pKi values) measured in binding assays and the pEC50/pA2 values determined in functional assays. The pKi values are consistently higher by 0.5–1.5 log units in the mouse and guinea-pig preparations, where a tritiated ligand has been used, whereas they were 1.8–3.5 log units higher in the rat and human preparations, where binding was measured with an iodinated ligand. The pKi values of near ten obtained in the rat and human receptor with NC are at least 1.5 log units higher than those estimated in the mouse and guinea-pig preparations. Further studies and the use of the tritiated ligands in the human and rat receptors are needed before considering the possibility of any species-related OP4 receptor subtypes.
In vitro functional assays in isolated organs
In experiments designed to (a) characterize the myotropic responses of isolated organs to NC and related peptides, (b) determine the type of action (direct, indirect), (c) identify the endogenous mediator if any of the recorded effect, and (d) obtain indications of the anatomical localizations and functional characteristics of OP4 receptor, NC and NC(1–13)NH2 were used as reference agonists. The three isolated tissues analysed in Table 4 are induced to contract by electrical stimulation, using stimulus intensity and characteristics that are suitable to field stimulate neurons and nerve terminals but not the muscle cells. They have been described and used before in opioid receptor pharmacology: thus, the guinea-pig ileum, whose myenteric neuronal network contains mainly OP3 receptors (Paton, 1957), has been shown to respond to NC and related peptides (Calo' et al., 1996; 1997; Zhang et al., 1997), the mouse vas deferens, whose nerve terminals contain mainly OP1 receptors (Hughes et al., 1975a), and the rat vas deferens, whose nerves contain an uncharacterized opioid receptor, have also been reported to be NC sensitive preparations (Berzetei-Gurske et al., 1996; Bigoni et al., 1999a; Calo' et al., 1996; 1997; Nicholson et al., 1996; Zhang et al., 1997). The twitch responses in the three preparations are due to nerve activation and subsequent release of neurotransmitter since they are blocked by tetrodotoxin. The contractions of the mouse and rat vas deferens derive largely from the release of noradrenaline from the sympathetic nerves, since they are blocked by the α1 adrenoceptor antagonist prazosin. OP4 receptors appear to be localized in the sympathetic terminals since NC inhibits twitches evoked by electrical field stimulation (EFS), but does not modify contractions to exogenous noradrenaline (Calo' et al., 1996). Similar results were also obtained in the guinea-pig ileum. In this preparation, NC inhibits atropine and tetrodotoxin sensitive neurogenic contractions without affecting responses to exogenous acetylcholine, thus demonstrating the prejunctional localization of the OP4 receptor.
In the three tissues, the inhibitory effect of NC is not influenced by naloxone, indicating that the other opioid receptor types present are not targeted by the peptide. A similar picture has been found regarding the inhibitory effects of NC on sensory fibers of the guinea-pig bronchus (Fischer et al., 1998; Rizzi et al., 1999a), renal pelvis (Giuliani & Maggi, 1996), and heart (Giuliani & Maggi, 1997). For instance, EFS of the guinea-pig isolated renal pelvis induces tetrodotoxin-sensitive contractile responses which are mediated by tachykinin release from sensory nerves, since they are blocked by a selective NK-2 receptor antagonist (MEN 10,376, Maggi et al., 1992). The contractile effect of EFS, but not that of exogenous tachykinins, was markedly reduced by NC whose effect was insensitive to naloxone (Bigoni et al., 1999a; Giuliani & Maggi, 1996).
From the above, it is evident that OP4 receptor is found in sympathetic, parasympathetic and sensory nerves, similar to the classical opioid receptors. This conclusion is also supported by the findings by Giuliani & Maggi (1997) who showed that NC modulates the complex-nerve mediated response of the guinea-pig left atrium: using adequate pharmacological tools, this response can be dissected into various components that include sympathetic, parasympathetic and CGRP-releasing sensory nerves (Giuliani & Maggi, 1997).
In all the preparations analysed above, the NC/OP4 receptor system displays a prejunctional inhibitory function, as do the classical opioid peptides/receptors, possibly through the same basic cellular mechanisms, namely: (a) activation of K+ conductance, as demonstrated in central neurons of the rat (Connor et al., 1996; Vaughan & Christie, 1996; Vaughan et al., 1997); (b) inhibition of Ca2+ entry through voltage-sensitive Ca2+ channels, as demonstrated in isolated central neurons (Knoflach et al., 1996); (c) inhibition of cyclic AMP accumulation (Meunier et al., 1995; Reinscheid et al., 1995). However, in other preparations, such as rat or mouse colon (Corbett et al., 1998; Osinski et al., 1999b; Rizzi et al., 1999b; Taniguchi et al., 1998; Yazdani et al., 1999), NC has been shown to induce naloxone-resistant tetrodotoxin-sensitive contractions which could derive from the inhibition of the release of endogenous relaxing agents, whose nature is at present unknown (Yazdani et al., 1999).
Structure-activity study of NC-related peptides: from agonist to antagonist
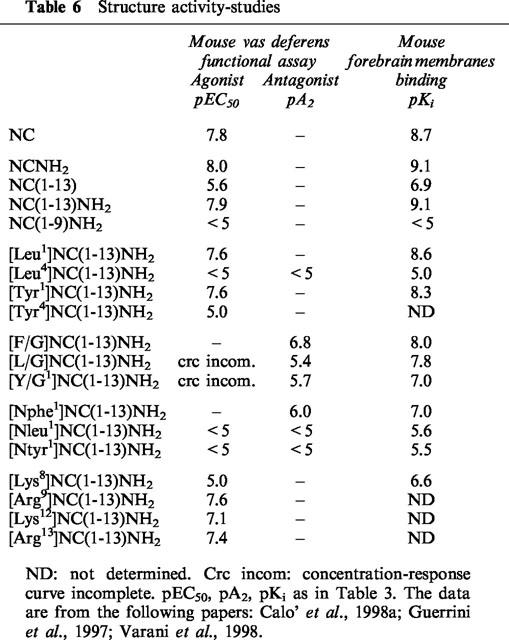
From a large series of analogues of the template NC(1–13)NH2, which were recently analysed and discussed (for a review see Salvadori et al., 1999), a number of compounds have been listed in Table 6 to enable structureactivity relationship analysis. Major purposes have been to: (a) see if the working hypothesis of dividing the peptide into ‘address' (binding) and ‘message' (activation) domains, which has been applied successfully to opioid peptides (Portoghese, 1989; Schwyzer, 1986), could also be used for NC; and (b) present and discuss the conceptual frame that led to the recent identification of a partial agonist (Calo' et al., 1998a; Guerrini et al., 1998) and an antagonist of OP4 receptors (Calo' et al., 2000; Guerrini et al., 1999). The abbreviated structures and biological activities of 19 compounds are summarized in Table 6, together with their actual affinities, as measured with binding assays on plasma membranes from mouse brain (Varani et al., 1998). The compounds were all tested for their potential agonistic activities, which are expressed in terms of pEC50, and, when found inactive or very weak, they were also tested as antagonists against NC(1–13)NH2, the reference agonist. This compound was compared to the natural peptide NC in all sets of assays. Furthermore, a binding assay using a new OP4 receptor ligand [3H]NCNH2 recently described by Varani et al. (1998), provided a fairly accurate measure of affinities for agonists, partial agonists and antagonists, allowing the evaluation of the influence of each chemical modification on OP4 receptor affinity.
Table 6.
Structure activity-studies
As shown in Figure 2, a good correlation is generally found between pEC50/pA2 and Ki values measured for the native mouse receptor using bioassay and binding data.
Figure 2.
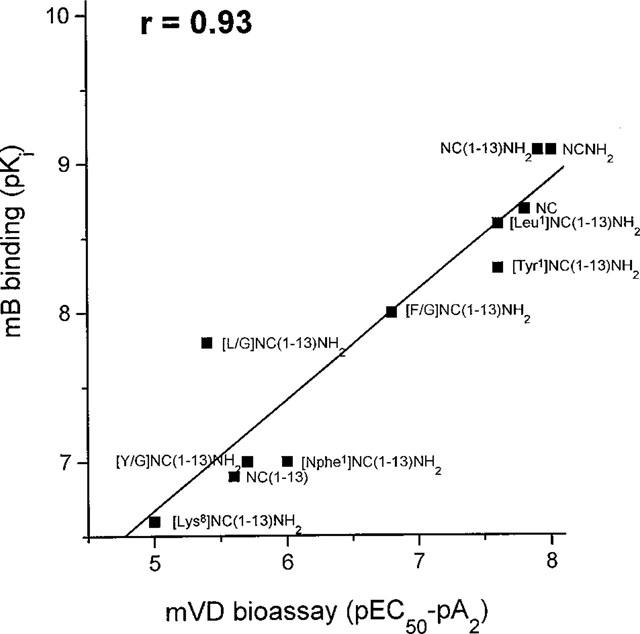
Correlation between binding and bioassay data on mouse OP4 receptors. mB, mouse brain membranes; mVD, mouse vas deferens.
These two sets of data have therefore been used to determine what alterations each chemical modifications introduced in the tridecapeptide NC(1–13)NH2, exerted on ligand affinity (address?) and activity (message?). Reducing the C-terminal end by four residues from NC(1–17) to NC(1–13) leads to a marked 2 log unit loss of potency. Further shortening to NC(1–9) leads to complete loss of activity. The lower potency of NC(1–13) appears to derive largely from metabolic degradation, perhaps by carboxypeptidases (Sandin et al., 1999), since protection of the C-terminal end with amidation is sufficient to maintain full potency and activity (see biological activities and binding affinities in Table 4 and in Bigoni et al., 1999a; Calo' et al., 1996; 1998b; Dooley & Houghten, 1996; Kapusta et al., 1999; Rizzi et al., 1999a,1999b; Varani et al., 1999; Okawa et al., 1999). Amidation of the carboxyl group in NC(1–17), to yield NC(1–17)NH2 does not significantly enhance (or very little) potency and activity in in vitro studies, but can enhance potency to values greater than for NC in some in vivo assays (Bertorelli et al., 1999a; Calo' et al., 1999). A few analogues of NC(1–13)NH2 were selected to analyse the N-terminal tetrapeptide FGGF, which has been assumed to contain the message domain of NC (Guerrini et al., 1997), in analogy with opioids. The replacement of Phe1 by Leu has no effect, while that of Phe4 eliminates activity and drastically reduces affinity by more than 3 log units. The same substitutions have the opposite effects when applied to opioid peptides; those in position 1 leading to inactivity and those in position 4 preserving binding and activity. From these data it was concluded that, since the active site of opioids is Tyr1, the corresponding site of NC should be Phe4. Indeed, the replacement of Phe1 by Tyr gives a compound that acts on OP4 as well as on OP2 and OP3 receptors (Calo' et al., 1997; Shimohigashi et al., 1996; Varani et al., 1999). The Phe4 appears to be instrumental for activation of OP4 receptor, as indeed Reinscheid et al. (1996) showed that [Tyr4]NC displays only a small fraction of the activity of NC, indicating that minor changes of Phe4 may be associated to drastic loss of potency. This has been confirmed in the sequence NC(1–13)NH2 (Table 6). In other compounds, changes were made at the N-terminal to simultaneously modify the spatial orientation of Phe1 and prevent peptide degradation by aminopeptidases. Reduction of the bond Phe1-Gly2 from CONH to CH2NH yielded a new compound, [F/G]NC(1–13)NH2, which was found to exert antagonistic activities in several pharmacological preparations, while acting as a partial or even full agonist in others (Table 9 and 10). A detailed discussion of the data obtained with [F/G]NC(1–13)NH2 by several laboratories may be found in the section ‘The OP4 receptor ligand [F/G]NC(1–13)NH2'.
Table 9.
Effects of [F/G]NC(1-13)NH2: in vitro studies
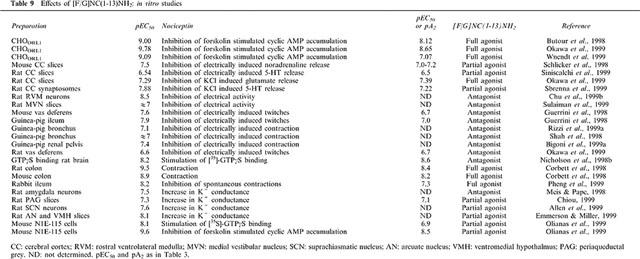
Table 10.
Effects of [F/G]NC(1-13)NH2: in vivo studies

The presence of a phenyl ring in the first position is not required for agonists, while it appears to be required for antagonism. Indeed, Phe1 cannot be replaced by Leu in [F/G]NC(1–13)NH2, because the new compound still binds to the receptor but does not block it. A strong reduction of potency is also observed when F/G is replaced by Y/G. Further modifications in position 1, particularly the displacement of the phenyl ring to the exterior by one atom, as in [Nphe1]NC(1–13)NH2, led to a compound that shows low potency, but appears to act as a pure antagonist (Rizzi et al., 1999b; Calo' et al., 2000). In fact, this new compound is devoid of residual agonistic activities in a variety of bioassays, antagonizes NC effects in both peripheral and central nervous system as well as in a variety of in vivo assays (see Table 7).
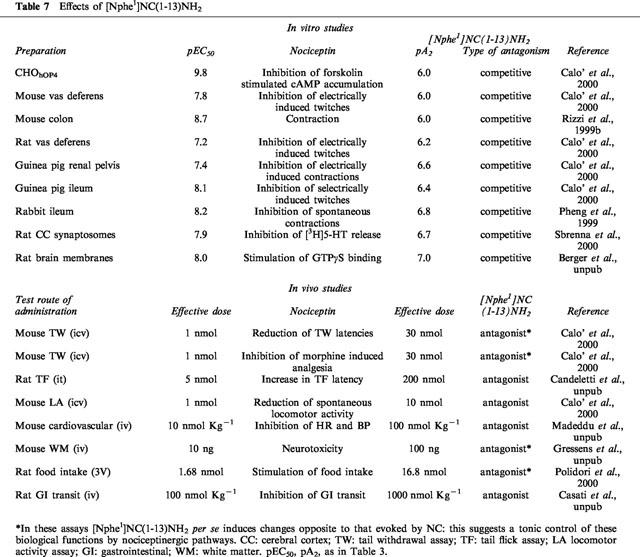
Table 7.
Effects of [Nphe1]NC(1-13)NH2
In addition, [Nphe1]NC(1–13)NH2 antagonizes the agonistic effects of [F/G]NC(1–13)NH2 in some preparations, such as CHOhOP4 (Hashimoto et al., 2000), the isolated mouse colon (Rizzi et al., 1999b) and rabbit ileum (Pheng et al., manuscript in preparation) or in vivo in the mouse tail withdrawal assay (Calo' G, Rizzi A and Regoli D., unpublished observation).
The last four compounds of Table 6 were prepared to explore the role of cationic residues in the middle and at the C-terminal end of NC(1–13)NH2. Essential for receptor occupation is Arg8, which cannot be replaced, even with Lys, while in the other positions (9, 12 and 13) Arg and Lys are equally accepted (interchangeable).
In the next two sections, we will discuss in more detail the properties of the most important new OP4 receptor ligands we have identified: [F/G]NC(1–13)NH2 and [Nphe1]NC(1–13)NH2. However, new ligands for the OP4 receptors have also been identified by other investigators. Kobayashi et al. (1997) found that some sigma receptor ligands (carbetapentane and rimcazole) act as antagonists of the OP4 receptor, albeit non-selectively and with low potency. From a combinatorial library containing more than 52 million different hexapeptides, Dooley et al. (1997) have identified 15 compounds that show high affinities for the OP4 receptor but also display partial agonistic activities which limits their usefulness as pharmacological tools. Naloxone benzoylhydrazone (NalBzOH), a non-selective opioid receptor ligand (Paul et al., 1990), was also reported to competitively block some effects of NC, with low potency (pA2 values 6.0–6.5) (Bigoni et al., 1999b; Bucher, 1998; Mamiya et al., 1999; Nicholson et al., 1998a; Noda et al., 1998; Sbrenna et al., 1999; Schlicker et al., 1998; Seki et al., 1999), but this compound also binds with high affinity to OP2 and OP3 receptors (Paul et al., 1990). Very recently, new ligands with antagonistic properties on the OP4 receptor have been identified from a conformationally constrained peptide combinatorial library (Becker et al., 1999). Although poorly selective, these novel ligands provide good templates for the development of non-peptidic ligands. Other non-selective ligands for the OP4 receptor include Mr 2266 (Bauer et al., 1999), TRK820 (Seki et al., 1999), and buprenorphine (Wnendt et al., 1999). Detailed pharmacological analyses of these compounds are outside the purpose of the present review and will be presented in a special issue of Peptides dedicated to the NC/OP4 system (Calo' et al., manuscript in preparation).
The OP4 antagonist [Nphe1]NC(1–13)NH2: characterization and uses
[Nphe1]NC(1–13)NH2 has been found to act as antagonist in a variety of in vitro and in vivo assays. The pA2 values obtained in functional in vitro assays and the effective doses estimated in in vivo assays for this ligand are presented in Table 7.
[Nphe1]NC(1–13)NH2 displays low potency (pA2 6.0–6.6). However, as it is the first pure OP4 receptor antagonist to be identified thus far, this compound has allowed us to characterize several biological effects of NC clearly mediated through OP4 receptors. The type of data that were utilised for quantification of the antagonistic effects of [Nphe1]NC(1–13)NH2 and determining the type of antagonism exerted by this compound is exemplified in Figure 3.
Figure 3.
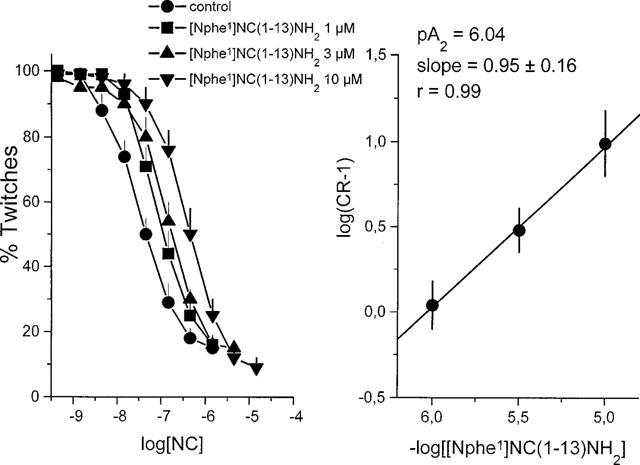
Schild plot of the antagonistic effect of [Nphe1]NC(1–13)NH2 on NC-induced depression of neurogenic contractions in the electrically stimulated mouse vas deferens.
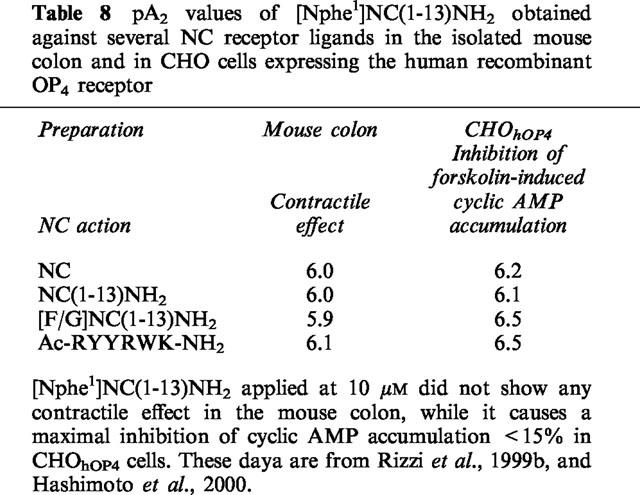
NC concentration-dependently reduces twitch responses of the mVD elicited by electrical field stimulation. Increasing concentrations of [Nphe1]NC(1–13)NH2, induce a gradual displacement of the curve to NC, yielding a linear Schild plot, with a slope close to 1.0, demonstrating that the compound acts as a competitive antagonist. Similar results have been obtained using human recombinant receptors expressed in CHO cells for NC-induced inhibition of cyclic AMP accumulation (Calo' et al., 2000) or on native receptors inhibiting the release of 5-HT from rat cerebral cortex slices (Sbrenna et al., 2000). In addition, [Nphe1]NC(1–13)NH2 acts as a pure NC receptor antagonist also in mouse brain slices, where it prevents NC-induced GTPgammaS binding without showing any residual agonistic activity (Berger et al., manuscript in preparation). In the mouse colon (Rizzi et al., 1999b) and in CHOhOP4 cells (Hashimoto et al., 2000), [Nphe1]NC(1–13)NH2 was also tested against several ligands (NC(1–13)NH2, [F/G]NC(1–13)NH2, AcRYYRWK-NH2) that are expected to act on the OP4 receptor. Results presented in Table 8 indicate that the different compounds act indeed on OP4 receptor since their effects are inhibited by [Nphe1]NC(1–13)NH2, but not by naloxone. In contrast, the effect elicited by endomorphin 1 in the mouse colon was not affected by [Nphe1]NC(1–13)NH2, while it was fully prevented by naloxone (Rizzi et al., 1999b). As expected, [Nphe1]NC(1–13)NH2 showed very similar pA2 values when tested against different OP4 receptor agonists.
Table 8.
pA2 values of [Nphe1]NC(1-13)NH2 obtained against several NC receptor ligands in the isolated mouse colon and in CHO cells expressing the human recombinant OP4 receptor
[Nphe1]NC(1–13)NH2 has been also demonstrated to be active in vivo. Indeed, this compound antagonizes: (i) NC (i.c.v.) induced reduction of latencies and inhibition of morphine evoked analgesia in the tail withdrawal test in the mouse (Calo' et al., 2000); (ii) NC (i.t.) evoked analgesia in the rat (Candeletti S., personal communication); NC (i.v.) induced bradycardia and hypotension in the mouse (Madeddu P., personal communication); and NC (i.c.v.) stimulated food intake in the rat (Polidori et al., 2000). In some cases, as for instance in the studies on pain threshold in the mouse (Calo' et al., 2000) or on food intake in the rat (Polidori et al., 2000), [Nphe1]NC(1–13)NH2 was found to induce per se changes opposite to those evoked by NC. This may well suggest a tonic control of these biological functions by nociceptinergic pathways. A detailed discussion of these findings, as well as their relevance for the understanding of the physiological role of the NC/OP4 system, can be found in the section dedicated to the biological actions of NC.
The OP4 receptor ligand [F/G]NC(1–13)NH2
[F/G]NC(1–13)NH2 has been identified in the frame of a SAR study (Calo' et al., 1998a) and is reported to act as a selective NC receptor antagonist in vitro in the electrically stimulated mouse vas deferens and guinea pig ileum (Guerrini et al., 1998). Since, various studies have been published on the actions of this pseudopeptide in a variety of in vitro and in vivo assays. The results of these studies have been summarized in Table 9 (in vitro studies) and 10 (in vivo studies).
From these data it is evident that [F/G]NC(1–13)NH2 may act as an antagonist, as a partial agonist or even as a full agonist, depending on the preparation. In in vitro studies performed in isolated tissues, [F/G]NC(1–13)NH2 mainly acts as an antagonist (pA2 around 7) but there are exceptions, such as the rat and mouse colon (Corbett et al., 1998; Rizzi et al., 1999b) or the rabbit ileum (Pheng et al., 1999), where the pseudopeptide acts as a full agonist and is subject to antagonism by [Nphe1]NC(1–13)NH2 (Pheng et al., 1999; Rizzi et al., 1999b). In recombinant systems showing high level of expression of the OP4 receptor, [F/G]NC(1–13)NH2 consistently behaves as a full agonist (Butour et al., 1998; Okawa et al., 1999; Wnendt et al., 1999). Again, its effects are antagonized by [Nphe1]NC(1–13)NH2 (Hashimoto et al., 2000). However, in N1E-115 cells expressing low levels of native OP4 receptors, [F/G]NC(1–13)NH2 acts as a partial agonist, either when stimulating [35S]-GTPgammaS binding or when inhibiting forskolin-stimulated [3H]-cyclic AMP formation (Olianas et al., 1999). Partial agonistic activity of [F/G]NC(1–13)NH2 has been also demonstrated in studies in which neurotransmitter release was measured (noradrenaline, Schlicker et al., 1998; or 5HT, Sbrenna et al., 2000); Siniscalchi et al., 1999) and in electrophysiological studies in which K+ currents were monitored (Allen et al., 1999; Chiou, 1999). In in vivo assays, [F/G]NC(1–13)NH2 mainly acts as a full agonist, mimicking several effects elicited by NC, when injected i.t. or i.c.v. (see Table 10). However, in few cases, the pseudopeptide behaves as a partial agonist (Bigoni et al., 1999a; Calo' et al., 1999) or as a pure antagonist (Armstead, 1999; Chu et al., 1999a,1999b; Gressens et al., 1999; Madeddu et al., 1999).
Interpretation of these data has been attempted by assuming the existence of multiple OP4 receptor types (Butour et al., 1998; Calo' et al., 1998b; Xu et al., 1998). However, this interpretation is ruled out by the demonstration that binding of NC completely disappears in mice in which the OP4 gene has been knocked out (Clarke et al., 1999). All NC binding sites appear to be the product of a single gene, that coding for the ORL1/OP4 receptor. Splice variants of the ORL1 gene have been described in the literature for the rat and mouse receptor (Pan et al., 1998; Peluso et al. 1998; Wang et al., 1994; Wick et al., 1994; 1995). These variants may account for the different pharmacological actions of [F/G]NC(1–13)NH2 in the various tests. However, no differences in the pharmacological profiles of these variants have been reported up to now. Alternatively, [F/G]NC(1–13)NH2 may actually be a ‘low efficacy agonist' whose final effect (antagonist, partial or full agonist) depends on the stimulus-response efficiency of the preparation under study. The findings by Toll et al. (1998) corroborate this hypothesis; [F/G]NC(1–13)NH2 acts as an antagonist in transfected cells expressing low levels of OP4 receptor, while it acts as a partial or full agonist in cells expressing high receptors levels. This elegant demonstration also illustrates the limits and the great possibilities of the molecular biology approach applied to pharmacological investigations.
Independently from the pharmacological behaviour (full or partial agonist, antagonist) of [F/G]NC(1–13)NH2 in the different assays, the pseudopeptide is on average 10 fold less potent than NC in vitro (Table 9). This [F/G]NC(1–13)NH2/NC ratio of potency perfectly matches with the ratio of actual affinities obtained in receptor binding experiments (Okawa et al., 1999; Varani et al., 1998; 1999). However, the ratio of potency [F/G]NC(1–13)NH2/NC obtained in in vivo studies (Table 9) is near 1 or even less. This difference can be explained assuming a higher metabolic stability of the pseudopeptide as compared to the naturally occurring peptide. Interestingly, already in the first description of [F/G]NC(1–13)NH2 (Guerrini et al., 1998) we suggested that, due to the presence of the amide at the C-terminus and of the pseudopeptide bond at the N-terminus, [F/G]NC(1–13)NH2 should be more stable than NC: this was confirmed by the following line of evidences: (i) the potency ratio [F/G]NC(1–13)NH2/NC is 10 fold higher in vitro (where peptidase activity is probably less relevant) than in vivo; (ii) in some in vivo studies [F/G]NC(1–13)NH2 shows a longer duration of action compared to NC (Bertorelli et al., 1999a; Kapusta et al., 1999; Wang et al., 1999a; Xu et al., 1998), (iii) in vitro in the rat vas deferens peptidase inhibitors increase the potency of NC but not that of [F/G]NC(1–13)NH2 (Okawa et al., 1999).
In the first reports [F/G]NC(1–13)NH2 was been proposed as a selective OP4 receptor ligand. This conclusion was based on the fact that the pseudopeptide does not modify the actions of opioid receptor agonists in several bioassays (Calo' et al., 1998a; Guerrini et al., 1998). These functional data have been recently confirmed in binding experiments (Varani et al., 1999) by showing that the affinity of [F/G]NC(1–13)NH2 is higher for NC sites (Ki 12 nM) than for opioid sites (OP3: Ki 800 nM; OP2: Ki 2630 nM; OP1: Ki >10000 nM) expressed in guinea-pig brain membranes. However, in recent experiments performed in rats (Carpenter & Dickenson, 1998; Sbrenna et al., 1999), it has been reported that part of the effects of [F/G]NC(1–13)NH2 can be reversed by naloxone. Therefore, we can not completely exclude (at least in the rat) some interactions of [F/G]NC(1–13)NH2 with classical opioid receptors.
Biological actions of nociceptin
The biological actions exerted by NC have been extensively covered in other review articles (see, for instance, Civelli et al., 1998; Darland et al., 1998; Henderson & McKnight, 1997; Meunier, 1997; Taylor & Dickenson, 1998). Here we will briefly review the major effects of this new peptide, focusing on the involvement of the endogenous NC/OP4 receptor system in these various actions. The role of the endogenous NC/OP4 receptor system in the different central nervous functions have been explored to date (a) with antisense oligonucleotides targeting OP4 receptor or preproNC genes, (b) with antibodies directed against NC, or (c) using mice in which the receptor or the peptide precursor genes have been genetically eliminated. While describing and discussing the findings obtained with these approaches, we will compare the actions of these various pharmacological and biological tools with those obtained with the first OP4 receptor selective antagonist, [Nphe1]NC(1–13)NH2.
The distribution of NC and its receptor in the central nervous system of rodents has been extensively and elegantly investigated with in situ hybridization and immunohystochemical techniques (Anton et al., 1996; Ikeda et al., 1998; Monteillet-Agius et al., 1998; Neal et al., 1999). In addition, Darland et al. (1998) have compiled maps that detail the expression of OP4 receptors and preproNC mRNAs in the rat brain. The reader is referred to the above mentioned papers for detailed coverage of this topic, which goes beyond the aims of the present article. Some of the major points are briefly summarized here. Both the receptor (Anton et al. 1996; Ikeda et al., 1998) and the peptide (Ikeda et al., 1998; Neal et al., 1999) are widely expressed in the central nervous system suggesting that the NC/OP4 receptor system may be involved in the modulation of a variety of central nervous system functions. The OP4 receptor is mainly expressed on nerve fibres, although in few brain areas, including the hippocampus, it is also expressed on neuronal cell bodies (Anton et al., 1996), suggesting that most of the actions of NC are likely to be presynaptic. The peptide is mostly expressed in middle-sized neurons, probably interneurons, although it is also found rarely in large projection-type neurons (Ikeda et al., 1998). Moreover, the distribution of NC generally matches that of the OP4 receptor (Darland et al., 1998). This anatomical organization implies that the NC/OP4 receptor system may modulate local circuitry. As underscored by Darland et al. (1998), in most studies aimed at investigating the effects of NC on various central nervous functions, the peptide has been administered i.c.v. or i.t. Only a few studies have investigated the actions of NC within discrete brain areas. This latter experimental paradigm, together with the development of selective OP4 receptor antagonists will lead, in the near future, to major advances in our understanding of the physiological roles of this system within the neuronal networks relevant to the various functions.
Nociceptin and pain threshold: supraspinal and spinal effects
Supraspinal level
Already in the first papers in which the identification of NC was reported (Meunier et al., 1995; Reinscheid et al., 1995), it was shown that, contrary to opioids (the classical analgesic agents), the i.c.v. injection of NC has a pronociceptive effect, i.e. it reduces the tail flick (Reinscheid et al., 1995) or hot plate response latency (Meunier et al., 1995). Similar results were subsequently obtained in the land snail (Kavaliers & Perrot-Sinal, 1996), mice (Calo' et al., 1998b; Nishi et al., 1997) and rats (Candeletti et al., 1998; Wang et al., 1999a; Zhu et al., 1996). A direct pronociceptive role of NC was questioned by Mogil et al. (1996a), who reported that i.c.v. injection of NC does not produce hyperalgesia, but rather reverses opioid-mediated stress-induced antinociception in different algesiometric assays and reverses morphine induced-analgesia. This suggestion for an anti-opioid role of NC has since been corroborated by results obtained in a variety of assays; indeed, NC has been shown to counteract the analgesic effect of the endogenous opioid system (Tian et al., 1997a; 1998; Zhu et al., 1996; 1997), or that of exogenously applied morphine (Bertorelli et al., 1999a; Calo' et al., 1998b; Grisel et al., 1996; Tian et al., 1997b; Zhu et al., 1997), or that of selective opioid receptor agonists (King et al., 1998; Mogil et al., 1996b), including endomorphins (Wang et al., 1999b). Worthy of mention is the fact that tolerance develops to the antiopioid effects of NC (Lutfy et al., 1999).
The involvement of the OP4 receptor in these effects is suggested by the following lines of evidence: (i) the pronociceptive action of NC is no longer present in OP4 receptor knockout mice (Nishi et al., 1997; Noda et al., 1998); (ii) antisense oligonucleotides targeting the OP4 receptor prevent the effect of NC (Tian et al., 1997b; Zhu et al., 1997); (iii) the pronociceptive effect of NC is reversed by NalBzOH, a non-selective opioid receptor ligand (Noda et al., 1998). These observations are corroborated by the recent findings that the selective OP4 receptor antagonist [Nphe1]NC(1–13)NH2 prevents the pronociceptive and anti-morphine actions of NC in the mouse tail withdrawal assay (Calo' et al., 2000). Moreover, this compound causes per se a robust, naloxone-resistant, antinociceptive effect and is able to potentiate by 3 fold at relatively low doses the analgesic effect of morphine, suggesting that nociceptinergic pathways controlling pain threshold are tonically activated at the supraspinal level. Interestingly, another low affinity NC receptor antagonist, retronociceptin methyl-ester (Ret-Noc-OMe), has been found to act as an analgesic following i.c.v. administration in mice (Yoshikawa et al., 1999), and similar results were also obtained using NalBzOH in wild type but not in OP4 receptor knockout mice (Noda et al., 1998).
The block of NC/OP4 signalling not only raises pain threshold and potentiates the effect of endogenous and exogenous opioids, but probably prevents at least in part the development of tolerance to the analgesic actions of opioids. This consideration is based on the following findings: (i) in mice knocked out for the OP4 receptor, a partial loss of tolerance to morphine analgesia has been demonstrated (Ueda et al., 1997); (ii) morphine tolerance can be partially reversed by i.c.v. injection of anti-NC antibodies (Tian et al., 1998); and (iii) there is an accelerated production and release of NC in the brain of chronic morphine tolerant rats (Yuan et al., 1999). These data would seem to suggest that the NC/OP4 receptor system may well subserve an important modulatory influence on development of morphine tolerance. Nevertheless, this proposal still depends on further studies, using OP4 receptor antagonists, to be validated.
Little is known about the mechanism(s) by which NC counteracts the analgesic action of opioids. Since the OP4 receptor and classical opioid receptors largely share the same transductional mechanisms, it is reasonable to speculate that their opposite effects on pain threshold are due to distinct localisations of NC and opioid peptides and their respective receptors on the neuronal networks involved in pain transmission. Several lines of evidence support this hypothesis. Although NC and opioid peptides show a similar distribution, they are not co-localized in nociceptive centres such as the dorsal horn, the sensory trigeminal complex or the periaqueductal grey (Schulz et al., 1996). The OP4 receptor and μ-opioid receptors are not co-localized in areas involved in pain processing (Monteillet-Agius et al., 1998). In addition, the anatomical distribution of the OP4 receptors is quite distinct to that seen for opioidstimulated [35S]-GTPγS binding (Sim & Childers, 1997).
The anti-opioid effect of NC has been evaluated in a few discrete brain areas. In the rostral ventromedial medulla of the rat, NC exerts a marked inhibitory action on all neurones, and can block the activation of some cells by opioids (Heinricher et al., 1997). Moreover, coadministration of NC attenuates the antinociceptive effect of DAMGO in the tail flick test, when applied within the rostral ventromedial medulla. A similar picture has also been found when studying the effects of NC within the periaqueductal grey where NC inhibits virtually all neurons present (Vaughan et al., 1997), and is also able to attenuate the analgesic effect of locally injected morphine (Morgan et al., 1997). It is therefore suggested that NC reverses opioid analgesia by inhibiting neuronal output from the periaqueductal grey (Morgan et al., 1997). Taken together, these findings corroborate the suggestion that the different actions of NC and opioids on nociception may be ascribed to a differential localization of their receptors in critical neuronal networks.
The analysis presented above contains an indication that OP4 receptor antagonists could provide a new class of supraspinally acting analgesics which could enable a reduction in morphine dosage, thus minimising the risk of tolerance and dependence.
Spinal level
The role of NC in modulating pain threshold in the spinal cord is controversial. Although some studies reported that i.t. injection of NC produces hyperalgesia/allodynia (Hara et al., 1997; Inoue et al., 1999; Minami et al., 1997; Okuda-Ashitaka et al., 1996; Sakurada et al., 1999), others found no effect (Grisel et al., 1996; Reinscheid et al., 1995). However most of the studies demonstrated that i.t. NC induces an antinociceptive effect similar to that evoked by classical opioid receptor agonists (Bertorelli et al., 1999b; Candeletti et al., 1998; Erb et al., 1997; Hao et al., 1998; Kamei et al., 1999; King et al., 1997; Tian et al., 1997b; Wang et al., 1999a; Xu et al., 1996; Yamamoto et al., 1997a,1997b). While tolerance develops to the antinociceptive effect of i.t. NC upon repeated administrations, there is no crosstolerance with morphine, suggesting that different receptors are involved in the actions of the two agents (Hao et al., 1997). The view that NC is a spinal analgesic is further substantiated by electrophysiological findings showing a major inhibitory action of NC on neuronal activity in the spinal dorsal horn (Faber et al., 1996; Liebel et al., 1997; Stanfa et al., 1996). Differences in animal species, or even in strains, which have been reported to be very important in determining the supraspinal effects of NC (Mogil et al., 1999), as well as in NC doses used, may account for the conflicting results reported with NC in the spinal cord. Worthy of mention is the work of Inoue et al. (1998; 1999), and Sakurada et al. (1999) showing that dose-response curve to NC is bell-shaped: very low doses of peptide (fmol range) cause hyperalgesia with a maximum at 1–10 fmol. At higher doses (nmol range), NC is antinociceptive and blocks the scratching, biting and licking induced by i.t. substance P (Inoue et al., 1999). Both actions of NC are surely mediated by the OP4 receptor, since they are absent in OP4−/− mice (Inoue et al., 1999). They also involve the activation of substance P signalling since (a) they are blocked by the NK-1 receptor antagonist CP 96345, or capsaicin pretreatment, (b) they disappear in mice lacking the tachykinin 1 gene. As pointed out by Inoue et al. (1999) OP4 receptors are present on both peripheral and central nerve endings (or terminals) of sensory neurons, where they can trigger release of neuropeptides, particularly substance P. Functional OP4 receptors are also found postsynaptically on spinal neurons, where they exert an antinociceptive effect by negatively modulating substance P signalling. The differential sensitivity of the pre- and post-synaptic sites to NC is of such magnitude (fmol vs nmol, respectively) to suspect the existence of two different functional sites. However, the fact that both pre- and post-synaptic actions of NC were eliminated in OP4−/− mice argues against such a proposition, even though this does not rule out the alternative possibility of splice variants of the receptor (Inoue et al., 1999).
Some evidence has also been obtained in favour of a role of endogenous spinal NC in modulating pain threshold. For instance, it has been demonstrated that i.t. injection of antibodies against NC decrease the antinociceptive effects of electroacupuncture (Tian et al., 1998).
Anxiolytic-like action of nociceptin
In 1997 Jenck et al. (1997) demonstrated that NC can act as an anxiolytic, attenuating the behavioural inhibition of animals acutely exposed to stressful/anxiogenic conditions. Given i.c.v. NC, at relatively low doses (0.1–3 nmol), induced anxiolytic-like effects in several behavioural paradigms, each generating different types of anxiety (light-dark preference, elevated plus-maze, exploratory behaviour of an unfamiliar environment, pharmacological anxiogenesis, operant conflict), leading to the proposal that the peptide may act as an endogenous regulator of acute anxiety. This view has been recently corroborated by studies in knockout animals (Koster et al., 1999; Reinscheid et al. 1999) showing that genetically engineered NC precursor-deficient mice display an increased susceptibility to acute and repeated stress, as compared to their wild-type littermates. The anxiolytic-like properties of NC have also been investigated in the mouse defence test battery (Griebel et al., 1999). Unlike the classical anxiolytic drug diazepam, which affects all defensive responses, NC clearly reduced only defensive upright postures and biting reactions which are the typical reactions to highly stressful stimuli, suggesting that the peptide may not be primarily involved in all anxiety-related responses, but may actually play a role in the adaptative responses to unavoidable or extreme stress stimuli. The anxiolytic mechanisms of NC are at present largely unknown, but may be related to the inhibitory action of NC on serotoninergic mechanisms exerted at two different levels: on dorsal raphe nucleus neurons, where NC causes inhibition by increasing K+ conductance (Vaughan & Christie, 1996), and on cortical serotoninergic nerve terminals, where NC inhibits 5-HT release (Sbrenna et al., 2000; Siniscalchi et al., 1999; Werthwein et al., 1999).
Recently some nonpeptide OP4 receptor agonists have been identified by investigators at Hoffmann La Roche (Adam et al., 1998). These compounds displayed anxiolytic-like properties in the elevated plus-maze test in rats (Wichmann et al., 1999). These findings suggest that OP4 receptor agonists may provide a new class of anxiolytic drugs devoid of any motivational effects, such as abuse liability (Devine et al., 1996a). They also provide ground for an intensive research programme to be pursued in both academic and industrial laboratories (Adam et al., 1998; Wichmann et al., 1999).
Modulation of spontaneous locomotor activity
Reinscheid et al. (1995) were the first to show that i.c.v. NC administration (in the range of 1–10 nmol) inhibits spontaneous locomotor activity in mice, a finding later confirmed by several authors (Calo' et al., 1999; Devine et al., 1996b; Nishi et al., 1997; Noble & Roques, 1997; Noda et al., 1998). Repeated daily NC injections result in rapid development of tolerance to this depressor effect of the peptide on locomotion and rearing activity (Devine et al., 1996b). As is typical for various NC-induced effects, this inhibitory action of NC is insensitive to naloxone (Noble & Roques, 1997), but is reversed by NalBzOH (Noda et al., 1998), and is undetectable in OP4−/− knockout mice (Nishi et al., 1997). Nevertheless, the basal locomotor activity of OP4−/− mice is not different from that displayed by wild-type littermates which suggests that the NC/OP4 receptor system does not play a tonic role in the physiological regulation of locomotion. This view is further strengthened by the fact that: (i) a mixture of peptidase inhibitors potentiates the inhibitory effect of exogenously applied NC on locomotion by about 10 fold, but does not modify this behaviour when applied alone (Noble & Roques, 1997); and (ii) NalBzOH, at doses sufficient to prevent the inhibitory effect of NC, does not increase per se spontaneous locomotor activity.
NC has also been reported to stimulate locomotor activity and exploratory behaviour at very low doses (0.01–0.1 nmol) (Florin et al., 1996). This effect of NC has been related to the anxiolytic-like actions of the peptide (Jenck et al., 1997). Thus, NC shows a bell-shaped dose response curve for locomotor activity: stimulation at low doses (0.01–0.1 nmol), inhibition at high doses (1–10 nmol). Although bell-shaped curves are typical of conventional anxiolytic drugs such as benzodiazepines, the nature of the two opposite effects of NC has not been clarified.
Stimulation of food intake
The i.c.v. injection of NC has been shown to stimulate food intake in satiated rats (Pomonis et al., 1996). This orexigenic action is prevented by naloxone, unlike most NC-mediated effects, suggesting that the peptide stimulates food intake by activating an opioidergic neuronal pathway. Similar orexigenic effects of NC can also be detected following injection into discrete brain areas, such as the nucleus accumbens shell or the ventromedial hypothalamic nucleus (Stratford et al., 1997). It appears likely that this action involves neuronal depression within this satiety center as NC has been reported to hyperpolarize, in a concentration-dependent manner, the neurones of the ventromedial hypothalamus (Lee et al., 1997).
Polidori et al., (1999), have shown that the brain regions surrounding the third ventricle are the areas most sensitive to the orexigenic effects of NC. They also showed that [F/G]NC(1–13)NH2 is 10 fold more potent than NC for stimulation of food intake (Polidori et al., 1999). Furthemore, the orexigenic effects of NC are clearly due to OP4 receptor activation since they can be blocked either by antisense oligonucleotides targeting the receptor (Leventhal et al., 1998) or by the OP4 receptor antagonist [Nphe1]NC(1–13)NH2 (Polidori et al., 2000). While the OP4 receptor antagonist [Nphe1]NC(1–13)NH2 alone is ineffective in modifying food consumption in satiated rats it effectively reduces that induced by food deprivation. Thus, nociceptinergic pathways may well be activated by food deprivation to intervene in food intake regulation (Polidori et al., 2000). It is, therefore, conceivable that OP4 receptor antagonists may constitute new anorectic agents alongside the neuropeptide Y receptor antagonists (Rowland et al., 1996).
Nociceptin and rewarding effects of drugs
Unlike classical opioids, NC fails to produce conditioned place preference or aversion (Devine et al., 1996a). Furthermore another study, carried out in alcohol preferring rats (Ciccocioppo et al., 1999) has shown that while acute i.c.v. injection of NC just before access to ethanol increases ethanol intake, similar i.c.v. injections for 7 days result in a progressive decrease in ethanol consumption. Once the subchronic NC treatment was terminated, rats progressively recovered their usual ethanol intake. On the other hand, NC also significantly reduces the increase in time that is spent in the ethanol-paired compartment after conditioning. Thus, NC seems to negatively modulate the rewarding properties of ethanol. Likewise NC also reduces the development of place preference to morphine (Angeletti et al., 1999; Murphy et al., 1999a) as well as opioid-induced locomotion in the rat (Grandy et al., 1999). Since the mesolimbic dopaminergic system plays a pivotal role in opioid rewarding properties (Wise, 1989), it has been suggested that NC attenuates conditioned place preference to morphine by inhibiting the alkaloid stimulatory effect on mesolimbic dopamine release (Murphy et al., 1999a). In fact, i.c.v. NC effectively inhibits dopamine release (as evaluated by in vivo microdialysis) in the nucleus accumbens of the rat stimulated by systemically injected morphine (Pieretti & Di Giannuario, 1999). Notwithstanding, NC has been reported to be ineffective in altering heroin self administration rate (Walker et al., 1998) or the development of cocaine sensitization (Narayanan & Maidment, 1999). Further studies are therefore needed to elucidate the role of the endogenous NC/OP4 receptor system in regulating the mesolimbic dopaminergic system and the rewarding properties of drugs of abuse, and hence the usefulness of NC receptor agonists for controlling drug self administration.
Inhibitory effect of nociceptin on memory process
The first indications for a role of the NC/OP4 system in learning and memory came from the observations that NC injection into the hippocampus impairs spatial learning (Sandin et al., 1997) and that in vitro it inhibits synaptic transmission and longterm potentiation in rat hippocampal slices (Yu et al., 1997; Yu & Xie, 1998). In line with these findings, OP4 receptor knockout mice show greater learning ability and have better memory retention than wild-type control mice (Manabe et al., 1998). In additon, hippocampal slices of OP4 receptor-deficient mice display greater long-term potentiation in CA1 region than those of control mice. The impairment of learning induced by NC can be reversed by nocistatin (functional antagonist of NC; Hiramatsu & Inoue, 1999) or by the nonselective OP4 receptor antagonist NalBzOH (Mamiya et al., 1999). Moreover, a peptidic OP4 receptor antagonist Ret-Noc-OMe, has been reported to potentiate memory retention in a passive avoidance test in mice (Yoshikawa et al., 1999).
Collectively, these findings suggest that the NC/OP4 receptor system may play negative roles in learning and memory, and that OP4 receptor antagonists may be interesting drugs for the alleviation of memory disorders. Much remains to be done in this field.
Nociceptin/OP4 receptor system and epilepsy
NC has been shown to inhibit the release of glutamate from rat cerebral (Nicol et al., 1996) and cerebellar (Nicol et al., 1998b) cortex slices. Moreover, several studies indicate that NC counteracts glutamate-mediated excitatory transmission in various central nervous system areas (Allen et al., 1999; Faber et al., 1996; Vaughan et al., 1997; Wang et al., 1996; Yu & Xie, 1998). Since glutamate plays a key role in epilepsy (Bradford, 1995), it is reasonable to expect that NC may exert an anti-epileptic action. Indeed, NC has been reported to inhibit kindling development in rats (Gutierrez et al., 1998) and pentylenetetrazole-induced seizures in mice (Zgodzinski et al., 1999). Interestingly, pronociceptin gene expression is dramatically enhanced in the thalamic reticular nucleus during kainate-induced seizures in rats (Bregola et al., 1999). These initial studies suggest that the NC/OP4 receptor system may have a role to play in seizure/epilepsy, and also that further studies into the potential effectiveness of OP4 receptor agonists as anti-epileptic drugs should be encouraged.
Inhibition of neurotransmitter release from central neurones
NC has also been reported to inhibit neurotransmitter release in the central nervous system. This has been shown in vitro for (i) noradrenaline (mouse, rat and guinea-pig cerebral cortex slices (Schlicker et al., 1998); mouse hippocampus, hypothalamus and cerebellum (Werthwein et al., 1999)); (ii) serotonin (rat (Sbrenna et al., 2000; Siniscalchi et al., 1999) and mouse (Werthwein et al., 1999) cortex); (iii) glutamate (rat cerebral (Nicol et al., 1996) and cerebellar (Nicol et al., 1998b) cortex); (iv) dopamine (midbrain primary cultures (Murphy et al., 1999b)); and (v) acetylcholine (rabbit retina (Neal et al., 1997)). These effects of NC were not affected by naloxone, ruling out the involvement of classical opioid receptors.
NC has also been demonstrated to modulate neurotransmitter release in vivo. Thus, intracerebroventricular injection of NC in the anaesthetized rat decreased dopamine release in the nucleus accumbens (Murphy et al., 1996), an effect that could be reproduced by NC perfusion into the ventral tegmental area (Murphy & Maidment, 1999). It may therefore be related to inhibition of the mesolimbic dopaminergic pathway. In this model, NC perfusion also increased local extracellular levels of glutamate and GABA (Murphy & Maidment, 1999). Worthy of mention is the fact that NC has also been reported to inhibit dopamine release from the nucleus accumbens stimulated by morphine (Pieretti & Di Giannuario, 1999).
Dual probe microdialysis in the awake rat has revealed that NC perfusion in the substantia nigra pars reticulata increases local extracellular glutamate levels via naloxone-insensitive mechanisms (Marti et al., 1999). This stimulation was associated with a decrease in acetylcholine release in the ipsilateral striatum, an effect possibly related to inhibition of the nigrostriatal dopaminergic pathway (Marti et al., 1999).
NC perfusion in the striatum stimulated local dopamine release but this was blocked (although at very high doses) by naloxone, suggesting the involvement of classical opioid receptors (Konya et al., 1998).
The results of these studies indicate that NC is a potent modulator of neurotransmitter release in various preparations that are widely used to explore the neurochemical bases of the central actions of the drugs. For NC effects, the following combinations should be considered: (i) inhibition of glutamate release/anti-epileptic action and disruption of spatial memory; (ii) inhibition of serotonin release/anxiolytic action; (iii) inhibition of mesolimbic dopaminergic transmission/anti-rewarding properties; (iv) modulation of striatal dopamine and glutamate/effects on locomotor activity.
Inhibitory effects of nociceptin on the cardiovascular system
When given i.v. to anaesthetized rats, NC induces transient hypotension and bradycardia (Champion & Kadowitz, 1997; Giuliani et al., 1997). These effects appear to be mediated by the autonomic nervous system, because hypotension can be prevented by guanethidine pretreatment while bradycardia is reduced by bilateral cervical vagotomy and abolished by a combination of both procedures (Giuliani et al., 1997). Similar results have been obtained in conscious rats (Kapusta et al., 1997) and mice (Madeddu et al., 1999), indicating that anaesthesia does not affect the cardiovascular effects of NC and that these effects are not restricted to the rat. The cardiovascular depressor actions of NC are not modified by naloxone, but are antagonized by [F/G]NC(1–13)NH2 in both mice (Madeddu et al., 1999) and rats (Bigoni et al., 1999a). Interestingly, NC induces similar cardiovascular effects when injected i.c.v. (Kapusta et al., 1997) or into the rostral ventrolateral medulla of the rat (Chu et al., 1999a). While [F/G]NC(1–13)NH2 mimics the effects of NC when injected i.c.v. (Kapusta et al., 1999), it blocks NC-induced responses if applied on rostral ventrolateral medulla neurons both in vitro (Chu et al., 1999b) and in vivo (Chu et al., 1999a), again illustrating the dual agonistic/antagonistic nature of this OP4 receptor ligand.
These data demonstrate that NC, probably by acting at central sites, exerts a marked inhibitory action on cardiovascular parameters in rodents. On the other hand, NC has recently been shown to increase blood pressure and heart rate in sheep following i.v. administration (Arndt et al., 1999). Thus, there may be important differences in the cardiovascular effects of NC in different species.
NC also induces vasodilation in several isolated arteries of the cat (Gumusel et al., 1997) and in mesenteric resistance arteries of the rat (Champion et al., 1998). This latter study also demonstrated that vasodilator responses to NC were not prevented by naloxone, nitric oxide synthase inhibitors, atropine, phentolamine or by the CGRP receptor antagonist CGRP(8–37). Moreover, the effect of NC was unchanged in endothelium denuded vessels (Champion et al., 1998). Vasodilator effects of NC have also been described also in other vascular beds such as the hindquarters of the rat (Czapla et al., 1997), or pial arteries of the pig (Armstead, 1999). This latter effect has been demonstrated to be due to activation of the OP4 receptors coupled to increased production of cyclic AMP and subsequent activation of KATP and KCa channels (Armstead, 1999). In addition, NC has been shown to inhibit electrically-evoked noradrenaline release in the isolated rat tail artery, by acting on prejunctional OP4 receptors located on postganglionic nerve endings (Bucher, 1998). NalBzOH antagonizes this effect of NC without affecting per se noradrenaline release, suggesting that OP4 receptors are not tonically activated by endogenously-released NC.
Finally, intracavernosal injection of NC induces a pronounced and relatively long-lasting erectile response in the cat (Champion et al., 1997).
Nociceptin effects on renal functions
The first study addressing the effects of NC on renal functions was performed by Kapusta et al. (1997), who reported that the peptide induces a marked increases in water excretion and decreases in urinary sodium excretion when injected i.v. or i.c.v. These diuretic and antinatriuretic effects of NC were resistant to blockade by the selective kappa-opioid receptor antagonist, nor-binaltorphimine, at a dose which blocked dynorphinA-induced diuresis. Interestingly, i.c.v. injection of [F/G]NC(1–13)NH2 mimics the action of NC, producing similar, but longer lasting cardiovascular and renal effects (Kapusta et al., 1999). These findings indicate that [F/G]NC(1–13)NH2 acts as an agonist at OP4 receptors controlling water balance. The longer duration of action of the pseudopeptide probably reflects its higher metabolic stability (see the section on [F/G]NC(1–13)NH2). As for the mechanism(s) by which NC exerts its diuretic and antinatriuretic actions, it has been reported that NC inhibits both oxytocin and vasopressin neurons, as measured using patch-clamp recording techniques in rat supraoptic nucleus slices (Doi et al., 1998). Similarly, NC also depresses guinea-pig supraoptic nucleus neurons acting through two mechanisms, hyperpolarization subsequent to activation of inwardly rectifying K+ conductance, as well as increase in membrane resistance due to closing of low-resistance gap junctions (Slugg et al., 1999). Taken together, these results suggest that the renal actions of NC can be attributed to inhibition of vasopressin secretion from the supraoptic nucleus neurons.
These observations suggest that endogenous NC may be a novel peptide involved in the central control of water and electrolyte balance and ultimately in the regulation of arterial blood pressure. Future analogues of NC may prove to be the first clinically useful water diuretics for patients with water-retaining diseases.
Effects of nociceptin in the gut
The gastrointestinal tract is a major site of action for opioids and opium, which have been used to reduce gut motility since ancient times. Likewise, OP4 receptor are also expressed in the gut (Wang et al., 1994), but little is known about the possible functions they subserve in the tract.
Like to opioids, NC inhibits in vitro neurogenic contractions of the stomach and the small intestine in a variety of species, including guinea pigs (Calo' et al., 1996; Zhang et al., 1997), pigs (Osinski et al., 1999a); rats (Yazdani et al., 1999); rabbits (Pheng et al., 1999). The depressor effect of NC is resistant to blockade by naloxone and appears to be correlated with a reduction of acetylcholine release, at least in the rat (Yazdani et al., 1999). On the other hand, NC causes concentration-dependent contractions in colonic smooth muscle strips from rodents (Corbett et al., 1998; Osinski et al., 1999b; Rizzi et al., 1999b; Taniguchi et al., 1998; Yazdani et al., 1999). This effect is prevented by tetrodotoxin or Ca2+-free medium suggesting the involvement of nerve activation (Yazdani et al., 1999). Given the depressor role of NC in neurotransmission at various autonomic neuroeffector functions, it appears plausible to hypothesize that its contractile effect in the colon is due to decreased release of an (unknown) inhibitory neurotransmitter. Very recently, it was shown that the contractions induced by NC and a variety of OP4 receptor ligands in the mouse colon are selectively antagonized by [Nphe1]NC(1–13)NH2, and are thus mediated by such receptors (Rizzi et al., 1999b). Like opioids, central administration of NC also inhibits colonic transit in the mouse (Osinski et al., 1999b). This effect is also mimicked in the rat by i.v. administration of NC in a [Nphe1]NC(1–13)NH2 sensitive manner (Casati et al., personal communication). NC also inhibits active anion transport in the intestinal mucosa through a neural mechanism in the pig ileum (Osinski et al., 1999b). On the other hand, Taniguchi et al. (1998) reported that NC administered subcutaneously actually accelerated transit rate in the large intestine, an action opposite to that induced by morphine or selective opioid receptor agonists. This other unexpected findings raise the question of the role(s) of the NC/OP4 receptor in the gut and its relation to the opioid system. However, the body of data reported in the literature points to a new regulatory NC/OP4 receptor system in the gastrointestinal tract, which is pharmacologically distinct from opioids but functionally very similar, that could represent a new target for the development of drugs (OP4 receptor agonists) to reduce intestinal motility.
Effects in the airways
NC was found to inhibit the contractions of the guinea-pig isolated bronchus induced by electrical field stimulation (Fischer et al., 1998). This effect was found to be mediated by a prejunctional mechanism not involving the classical opioid receptors. The same study also disclosed the existence of NC immunoreactive nerve fibers in the airway wall, distinct from the tachykinin-containing fibers. These findings were later validated by the demonstration that the inhibitory effect of NC is effectively and specifically blocked by [F/G]NC(1–13)NH2 (Rizzi et al., 1999a; Shah et al., 1998). Direct evidence has been provided for inhibitory actions of NC on neurotransmitter release from cholinergic and peptidergic airways nerves (Patel et al., 1997; Shah et al., 1998; Helys et al., 1997; Nemeth et al., 1998). The effect of NC on airways in vivo has not yet been investigated.
Inhibition of the micturition reflex
NC has been reported to modulate several autonomic functions, among others the inhibition of the micturition reflex in rats: this effect of NC has been investigated in great detail by the Menarini group (Giuliani et al., 1998; 1999; Lecci et al., 1999). In anaesthetized rats, i.v. NC produced a dose-dependent suppression of the micturition reflex induced by distension or topical application of capsaicin (Giuliani et al., 1998). Similar results were obtained by administering the peptide i.c.v., but not i.t., indicating that NC inhibits the micturition reflex by acting at peripheral and supraspinal sites (Lecci et al., 1999). All these effect are not affected by naloxone, thus excluding the involvement of opioid receptors. Worthy of mention is the fact that NC given i.v. affords long-lasting protection to capsaicin-induced desensitization of afferent nerves, which are known to mediate the chemoceptive micturition reflex in this model. In contrast, NC did not modify the desensitization of the local response to capsaicin (efferent function) (Giuliani et al., 1999). These results suggest that NC is able to discriminate between the afferent and efferent functions of capsaicin-sensitive primary sensory neurones, by selectively protecting the afferent function of these nerves (Giuliani et al., 1999). The recent identification of OP4 receptor antagonists of either peptidic (Calo' et al., 2000; Guerrini et al., 1999) or of non-peptidic nature (Ozaki et al., 1998) should be very useful for evaluating the possible pathophysiological involvement of endogenous NC in the micturition reflex in the rat and more generally on the sensory system.
Conclusions
The recent discoveries of ORL1 and its endogenous ligand NC add another member to the already well-defined opioid receptor family. The new system has been named NC/OP4 receptor, in accord with a proposal presented at the last IUPHAR international congress (Hamon, 1998). The advantages of this nomenclature have been underlined. Structure-activity relationship studies, which have led to identification of potent agonists (NCNH2, NC(1–13)NH2), of a partial agonist ([F/G]NC(1–13)NH2), and a pure antagonist ([Nphe1]NC(1–13)NH2), have also provided the basic tools for receptor characterization and classification in terms of agonists and antagonists. These tools have been used in studying the basic pharmacology of the system in vitro, with classical biological and binding assays performed on native OP4 receptors from various species, as well as with molecular biology/biochemistry techniques on the human recombinant receptor. Several agonists and a recently discovered competitive antagonist ([Nphe1]NC(1–13)NH2) have been successfully applied to the characterisation of OP4 functional receptors and binding sites. Very similar pharmacological profiles have been observed in the various assays using the Schild criteria of agonist order of potency and antagonist affinity. These finding have led us to conclude that the same receptor mediates the actions of the system at different levels (peripheral, spinal, supraspinal) in the same species and in preparations obtained from different species (mouse, rat, guinea-pig, man).
In reviewing the numerous emerging effects of and roles played by NC in the body, we have attempted to select the most consistent effects and identify the potential application of agonists and antagonists of the OP4 receptor. A broad spectrum of possible therapeutical indications of such compounds include: (a) OP4 receptor agonists as anxiolytics, stimulants of food intake, intrathecal (spinal) analgesics, suppressants of drug abuse, anti-epileptics and for the management of hyponatremic and water-retaining syndromes; (b) OP4 receptor antagonists could be tried as anorectics, analgesics (alone or in combination with opioids) or as nootropic agents.
The recent identification of OP4 receptor agonists (Adam et al., 1998; Wichmann et al., 1999) and antagonists (Ozaki et al., 1998) of non-peptidic nature may provide the tools needed to elucidate the role of the NC/OP4 receptor system in pathophysiology and to verify the actual value of compounds that activate or inhibit this system, as new potential drugs for treatment of human diseases.
Acknowledgments
This work was supported by a 60% grant of the University of Ferrara and by a cofin99 grant of the Italian Ministry of the University (MURST). We wish to thank Dr D.G. Lambert (Dept. Anaesthesia, University of Leicester, U.K.) for helpful discussion and Dr G.A. Rae (Dept. Pharmacology, Universidade Federal de Santa Catarina, Florianopolis, Brazil) for improving the style of this paper. Thanks are also due to students and technicians (Raffaella Bigoni, Daniela Rizzi, Giuliano Marzola, Katia Varani) who have contributed substantially to the acquisition of the data quoted in tables and figures, and to collaborators who worked with us on the nociceptin project since its initial phases (D.G. Lambert, M. Massi, M. Morari, C. Bianchi, P.A. Borea, P. Madeddu, S. Candeletti, H. Berger, C.A. Maggi, E. Ongini).
Abbreviations
- CHO
Chinese hamster ovary
- EFS
electrical field stimulation
- [F/G]NC(1–13)NH2
[Phe1ψ(CH2NH)Gly2]nociceptin(1–13)NH2
- NC
nociceptin/orphanin FQ
- [Nphe1]NC(1–13)NH2
[Nphe1]nociceptin(1–13)NH2
- OP4
nociceptin receptor
References
- ADAM G., CESURA A., GALLEY G., JENCK F., MONSMA F., ROVER S., WICHMANN J. Switzerland, EP-856514-A1; 1998. New 8-substitute-1,3,8-triazaspiro[4.5]decan-4-on derivatives – useful for the treatment of diseases related to the Orphanin FQ receptor e.g. psychiatric, neurological and physiological disorders. [Google Scholar]
- ALLEN C.N., JIANG Z.G., TESHIMA K., DARLAND T., IKEDA M., NELSON C.S., QUIGLEY D.I., YOSHIOKA T., ALLEN R.G., REA M.A., GRANDY D.K. Orphanin-FQ/nociceptin (OFQ/N) modulates the activity of suprachiasmatic nucleus neurons. J. Neurosci. 1999;19:2152–2160. doi: 10.1523/JNEUROSCI.19-06-02152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANGELETTI S., PANOCKA I., CICCOCIOPPO R., GINI L., MASSI M. Effect of nociceptin on morphine-induced conditioned place preference in rats. Regulatory peptides. 1999;80:122. [Google Scholar]
- ANTON B., FEIN J., TO T., LI X., SILBERSTEIN L., EVANS C.J. Immunohistochemical localization of ORL-1 in the central nervous system of the rat. J. Comp. Neurol. 1996;368:229–251. doi: 10.1002/(SICI)1096-9861(19960429)368:2<229::AID-CNE5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- ARMSTEAD W.M. Nociceptin/orphanin FQ dilates pial arteries by K(ATP) and k(ca) channel activation. Brain Res. 1999;835:315–323. doi: 10.1016/s0006-8993(99)01623-6. [DOI] [PubMed] [Google Scholar]
- ARNDT M.L., WU D., SOONG Y., SZETO H.H. Nociceptin/orphanin FQ increases blood pressure and heart rate via sympathetic activation in sheep. Peptides. 1999;20:465–470. doi: 10.1016/s0196-9781(99)00027-3. [DOI] [PubMed] [Google Scholar]
- BAUER U., NAKAZI M., KATHMANN M., GOTHERT M., SCHLICKER E. The stereoselective kappa-opioid receptor antagonist Mr 2266 does not exhibit stereoselectivity as an antagonist at the orphan opioid (ORL1) receptor. Naunyn Schmiedebergs Arch. Pharmacol. 1999;359:17–20. doi: 10.1007/pl00005317. [DOI] [PubMed] [Google Scholar]
- BECKER J.A.J., WALLACE A., GARZON A., INGALLINELLA P., BIANCHI E., CORTESE R., SIMONIN F., KIEFFER B.L., PESSI A. Ligands for k-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial libray. J. Biolol. Chem. 1999;274:27513–27522. doi: 10.1074/jbc.274.39.27513. [DOI] [PubMed] [Google Scholar]
- BERTORELLI R., CITTERIO F., TUPPER J., ONGINI E. Further analysis of nociceptin and derivatives following intratecal administration in mice. Regul. Peptides. 1999b;80:122. [Google Scholar]
- BERTORELLI R., CORRADINI L., RAFIQ K., TUPPER J., CALO' G., ONGINI E. Nociceptin and the putative ORL-1 ligand [Phe1psi(CH2-NH)Gly2)]nociceptin(1-13)NH2 exert anti-opiod effects in the Freund's adiuvant-induced arthritis rat model of chronic pain. Br. J. Pharmacol. 1999a;128:1252–1258. doi: 10.1038/sj.bjp.0702884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERZETEI-GURSKE I.P., SCHWARTZ R.W., TOLL L. Determination of activity for nociceptin in the mouse vas deferens. Eur. J. Pharmacol. 1996;302:R1–R2. doi: 10.1016/0014-2999(96)00238-5. [DOI] [PubMed] [Google Scholar]
- BIGONI R., CALO' G., MITRI S., REGOLI D. Naloxone benzoylhydrazone acts as a non selective nociceptin receptor antagonist. Fund. Clin. Pharmacol. 1999b;13:214s. [Google Scholar]
- BIGONI R., GIULIANI S., CALO' G., RIZZI A., GUERRINI R., SALVADORI S., REGOLI D., MAGGI C.A. Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn Schmiedebergs Arch. Pharmacol. 1999a;359:160–167. doi: 10.1007/pl00005338. [DOI] [PubMed] [Google Scholar]
- BRADFORD H.F. Glutamate, GABA and epilepsy. Prog. Neurobiol. 1995;47:477–511. doi: 10.1016/0301-0082(95)00030-5. [DOI] [PubMed] [Google Scholar]
- BREGOLA G., CANDELETTI S., ROMUALDI P., SIMONATO M. Limbic seizures increase pronociceptin mRNA levels in the thalamic reticular nucleus. Neuroreport. 1999;10:541–546. doi: 10.1097/00001756-199902250-00018. [DOI] [PubMed] [Google Scholar]
- BUCHER B. ORL1 receptor-mediated inhibition by nociceptin of noradrenaline release from perivascular sympathetic nerve endings of the rat tail artery. Naunyn Schmiedebergs Arch. Pharmacol. 1998;358:682–685. doi: 10.1007/pl00005312. [DOI] [PubMed] [Google Scholar]
- BUNZOW J.R., SAEZ C., MORTRUD M., BOUVIER C., WILLIAMS J.T., LOW M., GRANDY D.K. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994;347:284–288. doi: 10.1016/0014-5793(94)00561-3. [DOI] [PubMed] [Google Scholar]
- BUTOUR J.L., MOISAND C., MOLLEREAU C., MEUNIER J.C. [Phe1psi(CH2-NH)Gly2]nociceptin(1-13)-NH2 is an agonist of the nociceptin (ORL1) receptor. Eur. J. Pharmacol. 1998;349:R5–R6. doi: 10.1016/s0014-2999(98)00273-8. [DOI] [PubMed] [Google Scholar]
- CALO' G., BIGONI R., RIZZI A., MARZOLA G., GUERRINI R., SALVADORI S., REGOLI D., BIANCHI C. Characterization of nociceptin receptors modulating locomotor activity in mice. Fund. Clin. Pharmacol. 1999;13-S1:S27.6. [Google Scholar]
- CALO' G., GUERRINI R., BIGONI R., RIZZI A., BIANCHI C., REGOLI D., SALVADORI S. Structure-Activity Study of the Nociceptin(1-13)-NH2 N-Terminal Tetrapeptide and Discovery of a Nociceptin Receptor Antagonist. J. Med. Chem. 1998a;41:3360–3366. doi: 10.1021/jm970805q. [DOI] [PubMed] [Google Scholar]
- CALO' G., GUERRINI R., BIGONI R., RIZZI A., MARZOLA G., OKAWA H., BIANCHI C., LAMBERT D.G., SALVADORI S., REGOLI D.Characterization of [Nphe1]NC(1-13)NH2, a new selective nociceptin receptor antagonist Br. J. Pharmacol. 2000129in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALO' G., RIZZI A., BODIN M., NEUGEBAUER W., SALVADORI S., GUERRINI R., BIANCHI C., REGOLI D. Pharmacological characterization of nociceptin receptor: an in vitro study. Can. J. Physiol. Pharmacol. 1997;75:713–718. [PubMed] [Google Scholar]
- CALO' G., RIZZI A., BOGONI G., NEUGEBAUER W., SALVADORI S., GUERRINI R., BIANCHI C., REGOLI D. The mouse vas deferens: a pharmacological preparation sensitive to nociceptin. Eur. J. Pharmacol. 1996;311:R3–R5. doi: 10.1016/0014-2999(96)00563-8. [DOI] [PubMed] [Google Scholar]
- CALO' G., RIZZI A., MARZOLA G., GUERRINI R., SALVADORI S., BEANI L., REGOLI D., BIANCHI C. Pharmacological characterization of the nociceptin receptor mediating hyperalgesia in the mouse tail withdrawal assay. Br. J. Pharmacol. 1998b;125:373–378. doi: 10.1038/sj.bjp.0702087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANDELETTI S., GUERRINI R., CALO' G., FERRI S. Effects of the nociceptin receptor antagonist Phe1psi(CH2NH)Gly2NC(1-13)NH2 on nociception in rats. 29th International Narcotic Research Conference 1998Garmisch-Partenkirchen, Germany; pp. A17020–25 July, 1998 [Google Scholar]
- CARPENTER K.J., DICKENSON A.H. Evidence that [Phe1 psi(CH2-NH)Gly2]nociceptin-(1-13)-NH2, a peripheral ORL-1 receptor antagonist, acts as an agonist in the rat spinal cord. Br. J. Pharmacol. 1998;125:949–951. doi: 10.1038/sj.bjp.0702188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMPION H.C., KADOWITZ P.J.Nociceptin, an endogenous ligand for the ORL1 receptor, has novel hypotensive activity in the rat Life Sci. 199760241–245.PL [DOI] [PubMed] [Google Scholar]
- CHAMPION H.C., PIERCE R.L., KADOWITZ P.J. Nociceptin, a novel endogenous ligand for the ORL1 receptor, dilates isolated resistance arteries from the rat. Regul. Pept. 1998;78:69–74. doi: 10.1016/s0167-0115(98)00117-7. [DOI] [PubMed] [Google Scholar]
- CHAMPION H.C., WANG R., HELLSTROM W.J., KADOWITZ P.J. Nociceptin, a novel endogenous ligand for the ORL1 receptor, has potent erectile activity in the cat. Am. J. Physiol. 1997;273:E214–E219. doi: 10.1152/ajpendo.1997.273.1.E214. [DOI] [PubMed] [Google Scholar]
- CHAVKIN C., JAMES I.F., GOLDSTEIN A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- CHEN Y., FAN Y., LIU J., MESTEK A., TIAN M., KOZAK C.A., YU L. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994;347:279–283. doi: 10.1016/0014-5793(94)00560-5. [DOI] [PubMed] [Google Scholar]
- CHEN Y., MESTEK A., LIU J., HURLEY J.A., YU L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- CHIOU L.C. [Phe1psi(Ch2-NH)Gly2]nociceptin(1-13)NH2 activation of an inward rectifier as a partial agonist of ORL1 receptors in rat periaqueductal gray. Br. J. Pharmacol. 1999;128:103–107. doi: 10.1038/sj.bjp.0702746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHU X., XU N., LI P., MAO L., WANG J.Q. Inhibition of cardiovascular activity following microinjection of novel opioid-like neuropeptide nociceptin (orphanin FQ) into the rat rostral ventrolateral medulla. Brain Res. 1999a;829:134–142. doi: 10.1016/s0006-8993(99)01357-8. [DOI] [PubMed] [Google Scholar]
- CHU X., XU N., LI P., WANG J.Q. The nociceptin receptor-mediated inhibition of the rat rostral ventrolateral medulla neurons in vitro. Eur. J. Pharmacol. 1999b;364:49–53. doi: 10.1016/s0014-2999(98)00816-4. [DOI] [PubMed] [Google Scholar]
- CICCOCIOPPO R., PANOCKA I., POLIDORI C., REGOLI D., MASSI M. Effect of nociceptin on alcohol intake in alcohol-preferring rats. Psychopharmacology. 1999;141:220–224. doi: 10.1007/s002130050828. [DOI] [PubMed] [Google Scholar]
- CIVELLI O., NOTHACKER H.P., REINSCHEID R. Reverse physiology: discovery of the novel neuropeptide, orphanin FQ/nociceptin. Crit. Rev. Neurobiol. 1998;12:163–176. doi: 10.1615/critrevneurobiol.v12.i3.10. [DOI] [PubMed] [Google Scholar]
- CLARKE S., CHEN Z., HSU M.S., PINTAR J.E., HILL R.G., KITCHEN I.Quantitative autoradiography of ORL1, mu, delta, and kappa opioid receptors in the brains of ORL1 or nociceptin (OFQ) knockout mice International Narcotics Research Conference 1999Saratoga Springs, NY; Sun 74July 10–15 1999 [Google Scholar]
- CONNOR M., VAUGHAN C.W., CHIENG B., CHRISTIE M.J. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurones in vitro. Br. J. Pharmacol. 1996;119:1614–1618. doi: 10.1111/j.1476-5381.1996.tb16080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORBETT A.D., MENZIES J.R.W., DAVIES M.R.P., PATERSON S.J. The pharmacological actions of nociceptin in the isolated colon of rat, mouse, and man. Naunyn Schmiedebergs Arch. Pharmacol. 1998;358 Suppl 1:P40.47. [Google Scholar]
- CORBETT A.D., PATERSON S.J., MCKNIGHT A.T., MAGNAN J., KOSTERLITZ H.W. Dynorphin and dynorphin are ligands for the kappa-subtype of opiate receptor. Nature. 1997;299:79–81. doi: 10.1038/299079a0. [DOI] [PubMed] [Google Scholar]
- CZAPLA M.A., CHAMPION H.C., KADOWITZ P.J. Decreases in systemic arterial and hindquarters perfusion pressure in response to nociceptin are not inhibited by naloxone in the rat. Peptides. 1997;18:1197–1200. doi: 10.1016/s0196-9781(97)00178-2. [DOI] [PubMed] [Google Scholar]
- DARLAND T., HEINRICHER M.M., GRANDY D.K. Orphanin FQ/nociceptin: a role in pain and analgesia, but so much more. Trends Neurosci. 1998;21:215–221. doi: 10.1016/s0166-2236(97)01204-6. [DOI] [PubMed] [Google Scholar]
- DEVINE D.P., REINSCHEID R.K., MONSMA F.J., , JR, CIVELLI O., AKIL H. The novel neuropeptide orphanin FQ fails to produce conditioned place preference or aversion. Brain Res. 1996a;727:225–229. doi: 10.1016/0006-8993(96)00476-3. [DOI] [PubMed] [Google Scholar]
- DEVINE D.P., TAYLOR L., REINSCHEID R.K., MONSMA F.J., , JR, CIVELLI O., AKIL H. Rats rapidly develop tolerance to the locomotor-inhibiting effects of the novel neuropeptide orphanin FQ. Neurochem. Res. 1996b;21:1387–1396. doi: 10.1007/BF02532380. [DOI] [PubMed] [Google Scholar]
- DHAWAN B.N., CESSELIN F., RAGHUBIR R., REISINE T., BRADLEY P.B., PORTOGHESE P.S., HAMON M. International Union of Pharmacology. XII. Classification of opioid receptors. Pharmacol. Rev. 1996;48:567–592. [PubMed] [Google Scholar]
- DHAWAN B.N., RAGHUBIR R., HAMON M.Opioid receptors The IUPHAR compendium of receptor characterization and classification 1998London: IUPHAR Media Ltd; 218–226.ed. Girdlestone, D. pp [Google Scholar]
- DOI N., DUTIA M.B., RUSSELL J.A. Inhibition of rat oxytocin and vasopressin supraoptic nucleus neurons by nociceptin in vitro. Neuroscience. 1998;84:913–921. doi: 10.1016/s0306-4522(97)00547-2. [DOI] [PubMed] [Google Scholar]
- DOOLEY C.T., HOUGHTEN R.A. Orphanin FQ: receptor binding and analog structure activity relationships in rat brain. Life Sci. 1996;59:23–29. doi: 10.1016/0024-3205(96)00261-5. [DOI] [PubMed] [Google Scholar]
- DOOLEY C.T., SPAETH C.G., BERZETEI-GURSKE I.P., CRAYMER K., ADAPA I.D., BRANDT S.R., HOUGHTEN R.A., TOLL L. Binding and In vitro activities of peptides with high affinity for the Nociceptin/Orphanin FQ receptor, ORL1. J. Pharmacol. Exp. Ther. 1997;283:735–741. [PubMed] [Google Scholar]
- EMMERSON P.J., MILLER R.J. Pre- and post-synaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. J. Physiol. 1999;517:431–445. doi: 10.1111/j.1469-7793.1999.0431t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERB K., LIEBEL J.T., TEGEDER I., ZEILHOFER H.U., BRUNE K., GEISSLINGER G. Spinally delivered nociceptin/orphanin FQ reduces flinching behaviour in the rat formalin test. Neuroreport. 1997;8:1967–1970. doi: 10.1097/00001756-199705260-00034. [DOI] [PubMed] [Google Scholar]
- EVANS C.J., KEITH D.E., , JR, MORRISON H., MAGENDZO K., EDWARDS R.H. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- FABER E.S., CHAMBERS J.P., EVANS R.H., HENDERSON G. Depression of glutamatergic transmission by nociceptin in the neonatal rat hemisected spinal cord preparation in vitro. Br. J. Pharmacol. 1996;119:189–190. doi: 10.1111/j.1476-5381.1996.tb15969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISCHER A., FORSSMANN W.G., UNDEM B.J. Nociceptin-induced inhibition of tachykinergic neurotransmission in guinea pig bronchus. J. Pharmacol. Exp. Ther. 1998;285:902–907. [PubMed] [Google Scholar]
- FLORIN S., SUAUDEAU C., MEUNIER J.C., COSTENTIN J. Nociceptin stimulates locomotion and exploratory behaviour in mice. Eur. J. Pharmacol. 1996;317:9–13. doi: 10.1016/s0014-2999(96)00707-8. [DOI] [PubMed] [Google Scholar]
- FLORIN S., SUAUDEAU C., MEUNIER J.C., COSTENTIN J. Orphan neuropeptide NocII, a putative pronociceptin maturation product, stimulates locomotion in mice. Neuroreport. 1997;8:705–707. doi: 10.1097/00001756-199702100-00025. [DOI] [PubMed] [Google Scholar]
- FUKUDA K., KATO S., MORI K., NISHI M., TAKESHIMA H., IWABE N., MIYATA T., HOUTANI T., SUGIMOTO T. cDNA cloning and regional distribution of a novel memeber of the opioid receptor family. FEBS Lett. 1994;343:42–46. doi: 10.1016/0014-5793(94)80603-9. [DOI] [PubMed] [Google Scholar]
- GIRLESTONE D. London: IUPHAR Media, London, UK; 1998. The IUPHAR compendium of receptor characterization and classification. [Google Scholar]
- GIULIANI S., LECCI A., TRAMONTANA M., MAGGI C.A. The inhibitory effect of nociceptin on the micturition reflex in anaesthetized rats. Br. J. Pharmacol. 1998;124:156–672. doi: 10.1038/sj.bjp.0701983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIULIANI S., LECCI A., TRAMONTANA M., MAGGI C.A. Nociceptin protects capsaicin-sensitive afferent fibers in the rat urinary bladder from desensitization. Naunyn Schmiedeberg's Arch. Pharmacol. 1999;360:202–208. doi: 10.1007/s002109900047. [DOI] [PubMed] [Google Scholar]
- GIULIANI S., MAGGI C.A. Inhibition of tachykinin release from peripheral endings of sensory nerves by nociceptin, a novel opioid peptide. Br. J. Pharmacol. 1996;118:1567–1569. doi: 10.1111/j.1476-5381.1996.tb15576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIULIANI S., MAGGI C.A. Prejunctional modulation by nociceptin of nerve-mediated inotropic responses in guinea-pig left atrium. Eur. J. Pharmacol. 1997;332:231–236. doi: 10.1016/s0014-2999(97)01076-5. [DOI] [PubMed] [Google Scholar]
- GIULIANI S., TRAMONTANA M., LECCI A., MAGGI C.A. Effect of nociceptin on heart rate and blood pressure in anaesthetized rats. Eur. J. Pharmacol. 1997;333:177–179. doi: 10.1016/s0014-2999(97)01128-x. [DOI] [PubMed] [Google Scholar]
- GRANDY D.K., BONCI A., FIORILLO C.D., DARLAND T., RIEDL M., BUNZOW J.R., ELDE R., KALIVAS P.W., WILLIAMS J.T. International Narcotics Research Conference, 30th Meeting. Saratoga Springs, NY; 1999. Modulation of the mesolimbic dopamine reward pathway by nociceptin-orphanin FQ. [Google Scholar]
- GRESSENS P., CALO' G., EVRARD P. Nociceptin potentiates neonatal white matter excitotoxicity. Regulatory peptides. 1999;80:123. [Google Scholar]
- GRIEBEL G., PERRAULT G., SANGER D.J. Orphanin FQ, a novel neuropeptide with anti-stress-like activity. Brain Res. 1999;836:221–224. doi: 10.1016/s0006-8993(99)01684-4. [DOI] [PubMed] [Google Scholar]
- GRISEL J.E., FARRIER D.E., WILSON S.G., MOGIL J.S. [Phe1psi(CH2-NH)Gly2]nociceptin-(1-13)-NH2 acts as an agonist of the orphanin FQ/nociceptin receptor in vivo. Eur. J. Pharmacol. 1998;357:R1–R3. doi: 10.1016/s0014-2999(98)00567-6. [DOI] [PubMed] [Google Scholar]
- GRISEL J.E., MOGIL J.S., BELKNAP J.K., GRANDY D.K. Orphanin FQ acts as a supraspinal, but not a spinal, anti-opioid peptide. Neuroreport. 1996;7:2125–2129. doi: 10.1097/00001756-199609020-00012. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO' G., BIGONI R., RIZZI A., VARANI K., TOTH G., GESSI S., BOREA P.A., SALVADORI S., REGOLI D. Further studies on nociceptin related peptides: discovery of new molecules with antagonist activity on the nociceptin receptor. Regul. Peptides. 1999;80:124. doi: 10.1021/jm990075h. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO' G., RIZZI A., BIANCHI C., LAZARUS L.H., SALVADORI S., TEMUSSI P.A., REGOLI D. Address and message sequences for the nociceptin receptor: a structure- activity study of nociceptin-(1-13)-peptide amide. J. Med. Chem. 1997;40:1789–1793. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO' G., RIZZI A., BIGONI R., BIANCHI C., SALVADORI S., REGOLI D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998;123:163–165. doi: 10.1038/sj.bjp.0701640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GULLAND J.M., ROBINSON R. The morphine group. Part I. A discussion of the constitutional problem. J. Chem. Soc. 1923. pp. 980–998.
- GUMUSEL B., HAO Q., HYMAN A., CHANG J.K., KAPUSTA D.R., LIPPTON H. Nociceptin: an endogenous agonist for central opioid like1 (ORL1) receptors possesses systemic vasorelaxant properties. Life Sci. 1997;60:PL141–PL145. doi: 10.1016/s0024-3205(96)00696-0. [DOI] [PubMed] [Google Scholar]
- GUTIERREZ R., LEFF P., ANTON B. Inhibition of kindling development by nociceptin/orphanin FQ. Society for Neurosciences. 1998. p. 536.18.
- HAMON M. The new approach to opioid receptors. NS Arch. Pharmacol. 1998;358 suppl 2:SA 5.3. [Google Scholar]
- HANDA B.K., LAND A.C., LORD J.A., MORGAN B.A., RANCE M.J., SMITH C.F. Analogues of beta-LPH61-64 possessing selective agonist activity at mu- opiate receptors. Eur. J. Pharmacol. 1981;70:531–540. doi: 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]
- HAO J.X., WIESENFELD-HALLIN Z., XU X.J. Lack of cross-tolerance between the antinociceptive effect of intrathecal orphanin FQ and morphine in the rat. Neurosci. Lett. 1997;223:49–52. doi: 10.1016/s0304-3940(97)13401-2. [DOI] [PubMed] [Google Scholar]
- HAO J.X., XU I.S., WIESENFELD-HALLIN Z., XU X.J. Anti-hyperalgesic and anti-allodynic effects of intrathecal nociceptin/orphanin FQ in rats after spinal cord injury, peripheral nerve injury and inflammation. Pain. 1998;76:385–393. doi: 10.1016/S0304-3959(98)00071-2. [DOI] [PubMed] [Google Scholar]
- HARA N., MINAMI T., OKUDA-ASHITAKA E., SUGIMOTO T., SAKAI M., ONAKA M., MORI H., IMANISHI T., SHINGU K., ITO S. Characterization of nociceptin hyperalgesia and allodynia in conscious mice. Br. J. Pharmacol. 1997;121:401–408. doi: 10.1038/sj.bjp.0701146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASHIMOTO Y., CALO' G., GUERRINI R., SMITH G., LAMBERT D.G. Antagonistic effects of [Nphe1]nociceptin(1-13)NH2 on nociceptin receptor mediated inhibition of cAMP formation in Chinese ovary cells stably expressing the recombinant human nociceptin receptor. Neurosci. Lett. 2000;278:109–112. doi: 10.1016/s0304-3940(99)00915-5. [DOI] [PubMed] [Google Scholar]
- HEINRICHER M.M., MCGARAUGHTY S., GRANDY D.K. Circuitry underlying antiopioid actions of orphanin FQ in the rostral ventromedial medulla. J. Neurophysiol. 1997;78:3351–3358. doi: 10.1152/jn.1997.78.6.3351. [DOI] [PubMed] [Google Scholar]
- HELYES Z., NEMETH J., PINTER E., SZOLCSANYI J. Inhibition by nociceptin of neurogenic inflammation and the release of SP and CGRP from sensory nerve terminals. Br. J. Pharmacol. 1997;121:613–615. doi: 10.1038/sj.bjp.0701209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENDERSON G., MCKNIGHT A.T. The orphan opioid receptor and its endogenous ligand nociceptin/orphanin FQ. Trends Pharmacol. Sci. 1997;18:293–300. [PubMed] [Google Scholar]
- HIRAMATSU M., INOUE K. Effects of nocistatin on nociceptin-induced impairment of learning and memory in mice. Eur. J. Pharmacol. 1999;367:151–155. doi: 10.1016/s0014-2999(99)00003-5. [DOI] [PubMed] [Google Scholar]
- HOUTANI T., NISHI M., TAKESHIMA H., NUKADA T., SUGIMOTO T. Structure and regional distribution of nociceptin/orphanin FQ precursor. Biochem. Biophys. Res. Commun. 1996;219:714–719. doi: 10.1006/bbrc.1996.0300. [DOI] [PubMed] [Google Scholar]
- HUGHES J., KOSTERLITZ H.W., LESLIE F.M. Effect of morphine on adrenergic transmission in the mouse vas deferens. Assessment of agonist and antagonist potencies of narcotic analgesics. Br. J. Pharmacol. 1975a;53:371–381. doi: 10.1111/j.1476-5381.1975.tb07373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES J., SMITH T.W., KOSTERLITZ H.W., FOTHERGILL L.A., MORGAN B.A., MORRIS H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975b;258:577–580. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- IKEDA K., WATANABE M., ICHIKAWA T., KOBAYASHI T., YANO R., KUMANISHI T. Distribution of prepro-nociceptin/orphanin FQ mRNA and its receptor mRNA in developing and adult mouse central nervous systems. J. Comp. Neurol. 1998;399:139–151. doi: 10.1002/(sici)1096-9861(19980914)399:1<139::aid-cne11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- INOUE M., KOBAYASHI M., KOZAKI S., ZIMMER A., UEDA H. Nociceptin/orphanin FQ-induced nociceptive responses through substance P release from peripheral nerve endings in mice. Proc. Natl. Acad. Sci. U.S.A. 1998;95:10949–10953. doi: 10.1073/pnas.95.18.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INOUE M., SHIMOHIRA I., YOSHIDA A., ZIMMER A., TAKESHIMA H., SAKURADA T., UEDA H. Dose-Related Opposite Modulation by Nociceptin/Orphanin FQ of Substance P Nociception in the Nociceptors and Spinal Cord. J. Pharmacol. Exp. Ther. 1999;291:308–313. [PubMed] [Google Scholar]
- JENCK F., MOREAU J.L., MARTIN J.R., KILPATRICK G.J., REINSCHEID R.K., MONSMA F.J., JR, NOTHACKER H.P., CIVELLI O. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14854–14858. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINSON D.H., BARNARD E.A., HOYER D., HUMPHREY P.P.A., LEFF P., SHANKLEY N.P. International Union of Pharmacology Commitee on receptor nomenclature and drug classification XI Recommendations on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 1995;47:255–266. [PubMed] [Google Scholar]
- KAMEI J., OHSAWA M., KASHIWAZAKI T., NAGASE H. Antinociceptive effects of the ORL1 receptor agonist nociceptin/orphanin FQ in diabetic mice. Eur. J. Pharmacol. 1999;370:109–116. doi: 10.1016/s0014-2999(99)00112-0. [DOI] [PubMed] [Google Scholar]
- KAPUSTA D.R., CHANG J.K., KENIGS V.A. Central administration of [Phe1psi(CH2-NH)Gly2]nociceptin(1-13)-NH2 and orphanin FQ/nociceptin (OFQ/N) produce similar cardiovascular and renal responses in conscious rats. J. Pharmacol. Exp. Ther. 1999;289:173–180. [PubMed] [Google Scholar]
- KAPUSTA D.R., SEZEN S.F., CHANG J.K., LIPPTON H., KENIGS V.A. Diuretic and antinatriuretic responses produced by the endogenous opioid-like peptide, nociceptin (orphanin FQ) Life Sci. 1997;60:PL15–PL21. doi: 10.1016/s0024-3205(96)00593-0. [DOI] [PubMed] [Google Scholar]
- KAVALIERS M., PERROT-SINAL T.S. Pronociceptive effects of the neuropeptide, nociceptin, in the land snail, Cepaea nemoralis. Peptides. 1996;17:763–768. doi: 10.1016/0196-9781(96)00105-2. [DOI] [PubMed] [Google Scholar]
- KIEFFER B.L., BEFORT K., GAVERIAUX-RUFF C., HIRTH C.G. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. U.S.A. 1992;89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KING M., CHANG A., PASTERNAK G.W. Functional blockade of opioid analgesia by orphanin FQ/nociceptin. Biochem. Pharmacol. 1998;55:1537–1540. doi: 10.1016/s0006-2952(98)00023-9. [DOI] [PubMed] [Google Scholar]
- KING M.A., ROSSI G.C., CHANG A.H., WILLIAMS L., PASTERNAK G.W. Spinal analgesic activity of orphanin FQ/nociceptin and its fragments. Neurosci. Lett. 1997;223:113–116. doi: 10.1016/s0304-3940(97)13414-0. [DOI] [PubMed] [Google Scholar]
- KNOFLACH F., REINSCHEID R.K., CIVELLI O., KEMP J.A. Modulation of voltage-gated calcium channels by orphanin FQ in freshly dissociated hippocampal neurons. J. Neurosci. 1996;16:6657–6664. doi: 10.1523/JNEUROSCI.16-21-06657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOBAYASHI T., IKEDA K., TOGASHI S., ITOH N., KUMANISHI T. Effects of sigma ligands on the nociception/orphanin FQ receptor co-expressed with the G-protein-activated K+ channel in Xenopus oocytes. Br. J, Pharmacol. 1997;120:986–987. doi: 10.1038/sj.bjp.0701068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONYA H., MASUDA H., ITOH K., NAGAI K., KAKISHITA E., MATSUOKA A. Modification of dopamine release by nociceptin in conscious rat striatum. Brain Res. 1998;788:341–344. doi: 10.1016/s0006-8993(98)00075-4. [DOI] [PubMed] [Google Scholar]
- KOSTER A., MONTKOWSKI A., SCHULZ S., STUBE E.M., KNAUDT K., JENCK F., MOREAU J.L., NOTHACKER H.P., CIVELLI O., REINSCHEID R.K. Targeted disruption of the orphanin FQ/nociceptin gene increases stress susceptibility and impairs stress adaptation in mice. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10444–10449. doi: 10.1073/pnas.96.18.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACHOWICZ J.E., SHEN Y., MONSMA F.J., SIBLEY D.R. Molecular cloning of a novel G protein coupled receptor related to the opiate receptor family. Journal of Neurochemistry. 1995;64:34–40. doi: 10.1046/j.1471-4159.1995.64010034.x. [DOI] [PubMed] [Google Scholar]
- LAHTI R.A., MICKELSON M.M., MCCALL J.M., VON VOIGTLANDER P.F. [3H]U-69593 a highly selective ligand for the opioid kappa receptor. Eur. J. Pharmacol. 1985;109:281–284. doi: 10.1016/0014-2999(85)90431-5. [DOI] [PubMed] [Google Scholar]
- LECCI A., GIULIANI S., TRAMONTANA M., CRISCUOLI M., MAGGI C.A.Multiple sites of action in the inhibitory effect of nociceptin on the mictrurition reflex J. Urol. 1997. in press [PubMed]
- LEE K., NICHOLSON J.R., MCKNIGHT A.T. Nociceptin hyperpolarises neurones in the rat ventromedial hypothalamus. Neurosci. Lett. 1997;239:37–40. doi: 10.1016/s0304-3940(97)00875-6. [DOI] [PubMed] [Google Scholar]
- LEVENTHAL L., MATHIS J.P., ROSSI G.C., PASTERNAK G.W., BODNAR R.J. Orphan opioid receptor antisense probes block orphanin FQ-induced hyperphagia. Eur. J. Pharmacol. 1998;349:R1–R3. doi: 10.1016/s0014-2999(98)00272-6. [DOI] [PubMed] [Google Scholar]
- LI C.H., CHUNG D. Isolation and structure of an untriakontapeptide with opiate activity from camel pituitary glands. Proc. Natl. Acad. Sci. U.S.A. 1976;73:1145–1148. doi: 10.1073/pnas.73.4.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEBEL J.T., SWANDULLA D., ZEILHOFER H.U. Modulation of excitatory synaptic transmission by nociceptin in superficial dorsal horn neurones of the neonatal rat spinal cord. Br. J. Pharmacol. 1997;121:425–432. doi: 10.1038/sj.bjp.0701149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUKE M.C., HAHN E.F., PRICE M., PASTERNAK G.W. Irreversible opiate agonists and antagonists: V. Hydrazone and acylhydrazone derivatives of naltrexone. Life Sci. 1988;43:1249–1256. doi: 10.1016/0024-3205(88)90215-9. [DOI] [PubMed] [Google Scholar]
- LUTFY K., SHARZA S.A., MAIDMENT N.T. Tolerance develops to the inhibitory effect of orphanin FQ on morphine- induced antinociception in the rat. Neuroreport. 1999;10:103–106. doi: 10.1097/00001756-199901180-00020. [DOI] [PubMed] [Google Scholar]
- MADEDDU P., SALIS M.B., MILIA A.F., EMANUELI C., GUERRINI R., REGOLI D., CALO' G. Cardiovascular effects of nociceptin in unanesthetized mice. Hypertension. 1999;33:914–919. doi: 10.1161/01.hyp.33.3.914. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., THEODORSSON E., SANTICIOLI P., GIULIANI S. Tachykinins and calcitonin gene-related peptide as co-transmitters in local motor responses produced by sensory nerve activation in the guinea-pig isolated renal pelvis. Neuroscience. 1992;46:549–559. doi: 10.1016/0306-4522(92)90143-p. [DOI] [PubMed] [Google Scholar]
- MAMIYA T., NODA Y., NISHI M., TAKESHIMA H., NABESHIMA T. Nociceptin system plays a role in the memory retention: involvement of naloxone benzoylhydrazone binding sites. Neuroreport. 1999;10:1171–1175. doi: 10.1097/00001756-199904260-00003. [DOI] [PubMed] [Google Scholar]
- MANABE T., NODA Y., MAMIYA T., KATAGIRI H., HOUTANI T., NISHI M., NODA T., TAKAHASHI T., SUGIMOTO T., NABESHIMA T., TAKESHIMA H. Facilitation of long-term potentiation and memory in mice lacking nociceptin receptors. Nature. 1998;394:577–581. doi: 10.1038/29073. [DOI] [PubMed] [Google Scholar]
- MARTI M., SBRENNA S., CALO' G., BIANCHI C., BEANI L., MORARI M. Facilitation of glutamate release via ORL1 receptors in the sbstantia nigra. A microdialysis study in the awake rat. Regulatory Peptides. 1999;80:124. [Google Scholar]
- MEIS S., PAPE H.C. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to Nociceptin/Orphanin FQ. J. Neurosci. 1998;18:8133–8144. doi: 10.1523/JNEUROSCI.18-20-08133.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEUNIER J.C. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur. J. Pharmacol. 1997;340:1–15. doi: 10.1016/s0014-2999(97)01411-8. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSERRAT B., MAZARGUIL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MINAMI T., OKUDA-ASHITAKA E., NISHIUCHI Y., KIMURA T., TACHIBANA S., MORI H., ITO S. Anti-nociceptive responses produced by human putative counterpart of nocistatin. Br. J. Pharmacol. 1998;124:1016–1018. doi: 10.1038/sj.bjp.0701995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINAMI T., OKUDA-ASHITAKA E., NISHIZAWA M., MORI H., ITO S. Inhibition of nociceptin-induced allodynia in conscious mice by prostaglandin D2. Br. J. Pharmacol. 1997;122:605–610. doi: 10.1038/sj.bjp.0701421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOGIL J.S., GRISEL J.E., REINSCHEID R.K., CIVELLI O., BELKNAP J.K., GRANDY D.K. Orphanin FQ is a functional anti-opioid peptide. Neuroscience. 1996a;75:333–337. doi: 10.1016/0306-4522(96)00338-7. [DOI] [PubMed] [Google Scholar]
- MOGIL J.S., GRISEL J.E., ZHANGS G., BELKNAP J.K., GRANDY D.K. Functional antagonism of mu-, delta- and kappa-opioid antinociception by orphanin FQ. Neurosci. Lett. 1996b;214:131–134. doi: 10.1016/0304-3940(96)12917-7. [DOI] [PubMed] [Google Scholar]
- MOGIL J.S., NESSIM L.A., WILSON S.G. Strain-dependent effects of supraspinal orphanin FQ/nociceptin on thermal nociceptive sensitivity in mice. Neurosci. Lett. 1999;261:147–150. doi: 10.1016/s0304-3940(99)00012-9. [DOI] [PubMed] [Google Scholar]
- MOLLEREAU C., PARMENTIER M., MAILLEUX P., BUTOUR J.L., MOISAND C., CHALON P., CAPUT D., VASSART G., MEUNIER J.C. ORL1, a novel member of the opioid receptor family–Cloning, functional expression and localization. FEBS Lett. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- MOLLEREAU C., SIMONS M.J., SOULARUE P., LINERS F., VASSART G., MEUNIER J.C., PARMENTIER M. Structure, tissue distribution, and chromosomal localization of the prepronociceptin gene. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8666–8670. doi: 10.1073/pnas.93.16.8666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTEILLET-AGIUS G., FEIN J., ANTON B., EVANS C.J. ORL-1 and mu opioid receptor antisera label different fibers in areas involved in pain processing. J. Comp. Neurol. 1998;399:373–383. doi: 10.1002/(sici)1096-9861(19980928)399:3<373::aid-cne6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- MONTIEL J.L., CORNILLE F., ROQUES B.P., NOBLE F. Nociceptin/orphanin FQ metabolism: role of aminopeptidase and endopeptidase 24.15. J. Neurochem. 1997;68:354–361. doi: 10.1046/j.1471-4159.1997.68010354.x. [DOI] [PubMed] [Google Scholar]
- MORGAN M.M., GRISEL J.E., ROBBINS C.S., GRANDY D.K. Antinociception mediated by the periaqueductal gray is attenuated by orphanin FQ. Neuroreport. 1997;8:3431–3434. doi: 10.1097/00001756-199711100-00003. [DOI] [PubMed] [Google Scholar]
- MOSBERG H.I., HURST R., HRUBY V.J., GEE K., YAMAMURA H.I., GALLIGAN J.J., BURKS T.F. Bis-penicillamine enkephalins possess highly improved specificity toward delta opioid receptors. Proc. Natl. Acad. Sci. U.S.A. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY N.P., LEE Y., MAIDMENT N.T. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999a;832:168–170. doi: 10.1016/s0006-8993(99)01425-0. [DOI] [PubMed] [Google Scholar]
- MURPHY N.P., LY H.T., MAIDMENT N.T. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75:1–4. doi: 10.1016/0306-4522(96)00322-3. [DOI] [PubMed] [Google Scholar]
- MURPHY N.P., MAIDMENT N.T. Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. J. Neurochem. 1999;73:179–186. doi: 10.1046/j.1471-4159.1999.0730179.x. [DOI] [PubMed] [Google Scholar]
- MURPHY N.P., TAM A.M., LAM H.A., MAIDMENT N.T.Orphanin FQ/nociceptin modulation of dopamine release from midbrain primary cultures International Narcotics Research Conference, 30th meeting 1999bSaratoga Springs, NY; July 10–15, 1999 [Google Scholar]
- NAKAGAWA T., KANEKO M., INAMURA S., SATOH M. Intracerebroventricular administration of nocistatin reduces inflammatory hyperalgesia in rats. Neurosci. Lett. 1999;265:64–66. doi: 10.1016/s0304-3940(99)00202-5. [DOI] [PubMed] [Google Scholar]
- NARAYANAN S., MAIDMENT N.T. Orphanin FQ and behavioral sensitization to cocaine. Pharmacol. Biochem. Behav. 1999;63:271–277. doi: 10.1016/s0091-3057(98)00261-5. [DOI] [PubMed] [Google Scholar]
- NEAL C.R., , JR, MANSOUR A., REINSCHEID R., NOTHACKER H.P., CIVELLI O., WATSON S.J., , JR Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J. Comp. Neurol. 1999;406:503–547. [PubMed] [Google Scholar]
- NEAL M.J., CUNNINGHAM J.R., PATERSON S.J., MCKNIGHT A.T. Inhibition by nociceptin of the light-evoked release of ACh from retinal cholinergic neurones. Br. J. Pharmacol. 1997;120:1399–1400. doi: 10.1038/sj.bjp.0701135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEMETH J., HELYES Z., OROSZI G., THAN M., PINTER E., SZOLCSANYI J. Inhibition of nociceptin on sensory neuropeptide release and mast cell- mediated plasma extravasation in rats. Eur. J. Pharmacol. 1998;347:101–104. doi: 10.1016/s0014-2999(98)00216-7. [DOI] [PubMed] [Google Scholar]
- NICHOLSON J.R., MASON S.L., LEE K., MCKNIGHT A.T. The effect of the agonist Ac-RYYRWKNH2 and the antagonist [Phe1psi(CH2-NH)Gly2]nociceptin(1-13)NH2 at the ORL1 receptor of central and peripheral sites. 29th International Narcotics Research Conference 1998bGarmisch-Partenkirchen (Germany)A12920–25 July 1998 [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MCKNIGHT A.T. Characterization of the response in the rat vas deferens to the ORL-1 agonist nociceptin. Br. J. Pharmacol. 1996;119 Proceeding Suppl:36P. [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MENZIES J.R., CORBETT A.D., MCKNIGHT A.T. Pharmacological studies on the ‘orphan' opioid receptor in central and peripheral sites. Can. J. Physiol. Pharmacol. 1998a;76:304–313. [PubMed] [Google Scholar]
- NICOL B., LAMBERT D.G., ROWBOTHAM D.J., OKUDA-ASHITAKA E., ITO S., SMART D., MCKNIGHT A.T. Nocistatin reverses nociceptin inhibition of glutamate release from rat brain slices. Eur. J. Pharmacol. 1998a;356:R1–R3. doi: 10.1016/s0014-2999(98)00545-7. [DOI] [PubMed] [Google Scholar]
- NICOL B., LAMBERT D.G., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T. Nociceptin induced inhibition of K+ evoked glutamate release from rat cerebrocortical slices. Br. J. Pharmacol. 1996;119:1081–1083. doi: 10.1111/j.1476-5381.1996.tb16007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICOL B., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T., LAMBERT D.G. Nociceptin inhibits glutamate release from rat cerebellar slices. Br. J. Pharmacol. 1998b;123:217P. doi: 10.1111/j.1476-5381.1996.tb16007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHI M., HOUTANI T., NODA Y., MAMIYA T., SATO K., DOI T., KUNO J., TAKESHIMA H., NUKADA T., NABESHIMA T., YAMASHITA T., NODA T., SUGIMOTO T. Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J. 1997;16:1858–1864. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISHI M., TAKESHIMA H., MORI M., NAKAGAWARA K., TAKEUCHI T. Structure and chromosomal mapping of genes for the mouse κ-opioid receptor and an opioid receptor homologue (MORC) Biochem. Biophys. Res. Commun. 1994;205:1353–1357. doi: 10.1006/bbrc.1994.2814. [DOI] [PubMed] [Google Scholar]
- NOBLE F., ROQUES B.P. Association of aminopeptidase N and endopeptidase 24.15 inhibitors potentiate behavioral effects mediated by nociceptin/orphanin FQ in mice. FEBS Lett. 1997;401:227–229. doi: 10.1016/s0014-5793(96)01476-7. [DOI] [PubMed] [Google Scholar]
- NODA Y., MAMIYA T., NABESHIMA T., NISHI M., HIGASHIOKA M., TAKESHIMA H. Loss of antinociception induced by naloxone benzoylhydrazone in nociceptin receptor-knockout mice. J. Biol. Chem. 1998;273:18047–18051. doi: 10.1074/jbc.273.29.18047. [DOI] [PubMed] [Google Scholar]
- NOTHACKER H.P., REINSCHEID R.K., MANSOUR A., HENNINGSEN R.A., ARDATI A., MONSMA F.J., WATSON S.J., CIVELLI O. Primary structure and tissue distribution of the orphanin FQ precursor. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8677–8682. doi: 10.1073/pnas.93.16.8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKAWA H., NICOL B., BIGONI R., HIRST R.A., CALO' G., GUERRINI R., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T., LAMBERT D.G. Comparison of the effects of Phe1psi(CH2-NH)Gly2]nociceptin(1-13)NH2 in rat brain, rat vas deferens and CHO cells expressing recombinant human nociceptin recetors. Br. J. Pharmacol. 1999;127:123–130. doi: 10.1038/sj.bjp.0702539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKUDA-ASHITAKA E., MINAMI T., TACHIBANA S., YOSHIHARA Y., NISHIUCHI Y., KIMURA T., ITO S. Nocistatin, a peptide that blocks nociceptin action in pain transmission. Nature. 1998;392:286–289. doi: 10.1038/32660. [DOI] [PubMed] [Google Scholar]
- OKUDA-ASHITAKA E., TACHIBANA S., HOUTANI T., MINAMI T., MASU Y., NISHI M., TAKESHIMA H., SUGIMOTO T., ITO S. Identification and characterization of an endogenous ligand for opioid receptor homologue ROR-C: its involvement in allodynic response to innocuous stimulus. Brain Res. Mol. Brain Res. 1996;43:96–104. doi: 10.1016/s0169-328x(96)00165-9. [DOI] [PubMed] [Google Scholar]
- OLIANAS M.C., MAULLU C., INGIANNI A., ONALI P. [Phe1psi(CH2-NH)Gly2]nociceptin-(1-13)-NH2 acts as a partial agonist at ORL1 receptor endogenously expressed in mouse N1E-115 neuroblastoma cells. Neuroreport. 1999;10:1127–1131. doi: 10.1097/00001756-199904060-00041. [DOI] [PubMed] [Google Scholar]
- OLOFSON R.A., SCHNUR R.C., BUNES L., PEPE J.P. Selective N-dealkylation of tertiary amines with vinyl chloroformate: an improved synthesis of naloxone. Tetrahedron Letters. 1977;18:1567–1570. [Google Scholar]
- OSINSKI M.A., BASS P., GAUMNITZ E.A. Peripheral and central actions of orphanin FQ (nociceptin) on murine colon. Am. J. Physiol. 1999b;276:G125–G131. doi: 10.1152/ajpgi.1999.276.1.G125. [DOI] [PubMed] [Google Scholar]
- OSINSKI M.A., PAMPUSCH M.S., MURTAUGH M.P., BROWN D.R. Cloning, expression and functional role of a nociceptin/orphanin FQ receptor in the porcine gastrointestinal tract. Eur. J. Pharmacol. 1999a;365:281–289. doi: 10.1016/s0014-2999(98)00869-3. [DOI] [PubMed] [Google Scholar]
- OZAKI S., KAWAMOTO H., ITO Y., HAYASHI K., IWASAWA Y., HIRANO K.New 2-oxoimidazole derivatives–are nociceptin receptor binding inhibitors useful e.g. as analgesics and for amelioreting brain function 1998. Japan, WO9854168-A1
- PAN Y.X., XU J., PASTERNAK G.W. Cloning and expression of a cDNA encoding a mouse brain orphanin FQ nociceptin precursor. Biochem. J. 1996;315:11–13. doi: 10.1042/bj3150011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAN Y.X., XU J., WAN B.L., ZUCKERMAN A., PASTERNAK G.W. Identification and differential regional expression of KOR-3/ORL-1 gene splice variants in mouse brain. FEBS Lett. 1998;435:65–68. doi: 10.1016/s0014-5793(98)01039-4. [DOI] [PubMed] [Google Scholar]
- PATEL H.J., GIEMBYCZ M.A., SPICUZZA L., BARNES P.J., BELVISI M.G. Naloxone-insensitive inhibition of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by the novel opioid, nociceptin. Br. J. Pharmacol. 1997;120:735–736. doi: 10.1038/sj.bjp.0701013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATON W.D.M. The action of morphine and related substances on contraction and on acetylcholine output of coaxially stimulated guinea pig ileum. Br. J. Pharmacol. 1957;12:119–127. doi: 10.1111/j.1476-5381.1957.tb01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAUL D., LEVISON J.A., HOWARD D.H., PICK C.G., HAHN E.F., PASTERNAK G.W. Naloxone benzoylhydrazone (NalBzoH) analgesia. J. Pharmacol. Exp. Ther. 1990;255:769–774. [PubMed] [Google Scholar]
- PELTON J.T., GULYA K., HRUBY V.J., DUCKLES S.P., YAMAMURA H.I. Conformationally restricted analogs of somatostatin with high mu-opiate receptor specificity. Proc. Natl. Acad. Sci. U.S.A. 1985;82:236–239. doi: 10.1073/pnas.82.1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PELUSO J., LAFORGE K.S., MATTHES H.W., KREEK M.J., KIEFFER B.L., GAVERIAUX-RUFF C. Distribution of nociceptin/orphanin FQ receptor transcript in human central nervous system and immune cells. J. Neuroimmunol. 1998;81:184–192. doi: 10.1016/s0165-5728(97)00178-1. [DOI] [PubMed] [Google Scholar]
- PHENG L.H., CALO' G., REGOLI D.Ileon de lapin: une preparation sensible a la nociceptine 41 annual meeting of the Club de recherche clinique du Quebecz 1999Montreal, Quebec, CA; Oct 1999 [Google Scholar]
- PIERETTI S., DI GIANNUARIO A. Orphanin FQ effects on morphine-induced dopamine release in the accumbens of rats. Regulatory Peptides. 1999;80:126. [Google Scholar]
- POLIDORI C., CALO' G., CICCOCIOPPO R., GUERRINI R., REGOLI D., MASSI M.Pharmacological characterization of the nociceptin receptor mediating hyperphagia: identification of a selective antagonist Psychopharmacology 2000. in press [DOI] [PubMed]
- POLIDORI C., CALO' G., DECARO G., REGOLI D., POMPEI P., MASSI M. Sensitivity of brain sites to the orexigenic effect of nociceptin or af its analog [Phe1psi(CH2-NH)Gly2]NC(1-13)NH2. Regul. Peptides. 1999;80:126. [Google Scholar]
- POMONIS J.D., BILLINGTON C.J., LEVINE A.S. Orphanin FQ, agonist of orphan opioid receptor ORL1, stimulates feeding in rats. Neuroreport. 1996;8:369–371. doi: 10.1097/00001756-199612200-00072. [DOI] [PubMed] [Google Scholar]
- PORTOGHESE A.S., LIPKOWSKI A.W., TAKEMORI A.E. Bimorphinans as highly selective, potent kappa opioid receptor antagonists. J. Med. Chem. 1987;30:238–239. doi: 10.1021/jm00385a002. [DOI] [PubMed] [Google Scholar]
- PORTOGHESE P.S. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 1989;10:230–235. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- PORTOGHESE P.S., SULTANA M., TAKEMORI A.E. Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur. J. Pharmacol. 1988;146:185–186. doi: 10.1016/0014-2999(88)90502-x. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., ARDATI A., MONSMA F.J., JR, CIVELLI O. Structure-activity relationship studies on the novel neuropeptide orphanin FQ. J. Biol. Chem. 1996;271:14163–14168. doi: 10.1074/jbc.271.24.14163. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., KNAUDT K., KOESTER A., MONTKOWSKI A., CIVELLI O.OFQ/N and the endogenous opioids act synergistically in protecting from excessive sensation of fear and pain International Narcotics Research Conference, 30th meeting 1999Saratoga Springs, NY; July 10–15, 1999 [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J., , JR, CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- RIZZI A., BIGONI R., CALO' G., GUERRINI R., SALVADORI S., REGOLI D. [Nphe1]nociceptin(1-13)NH2 antagonizes nociceptin effects in the mouse colon. Eur. J. Pharmacol. 1999b;285:R3–R5. doi: 10.1016/s0014-2999(99)00730-x. [DOI] [PubMed] [Google Scholar]
- RIZZI A., CALO' G., TREVISANI M., TOGNETTO M., FABBRI L., MAPP C., GUERRINI R., SALVADORI S., REGOLI D., GEPPETTI P. Nociceptin receptor activation inhibits tachykinergic non adrenergic non cholinergic contraction of guinea pig isolated bronchus. Life Sci. 1999a;64:L157–L163. doi: 10.1016/s0024-3205(99)00045-4. [DOI] [PubMed] [Google Scholar]
- ROSSI G.C., MATHIS J.P., PASTERNAK G.W. Analgesic activity of orphanin FQ2, murine prepro-orphanin FQ141-157 in mice. Neuroreport. 1998;9:1165–1168. [PubMed] [Google Scholar]
- ROWLAND N.E., MORIEN A., LI B.H. The physiology and brain mechanisms of feeding. Nutrition. 1996;12:626–639. doi: 10.1016/s0899-9007(96)00227-4. [DOI] [PubMed] [Google Scholar]
- SAKURADA T., KATSUYAMA S., SAKURADA S., INOUE M., TAN-NO K., KISARA K., SAKURADA C., UEDA H., SASAKI J. Nociceptin-induced scratching, biting and licking in mice: involvement of spinal NK1 receptors. Br. J. Pharmacol. 1999;127:1712–1718. doi: 10.1038/sj.bjp.0702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVADORI S., GUERRINI R., CALO' G., REGOLI D. Structure activity studies on nociceptin/orphanin FQ: from full agonist, to partial agonist, to pure antagonist. Il Famaco. 1999;54:810–825. doi: 10.1016/s0014-827x(99)00108-1. [DOI] [PubMed] [Google Scholar]
- SANDIN J., GEORGIEVA J., SCHOTT P.A., OGREN S.O., TERENIUS L. Nociceptin/orphanin FQ microinjected into hippocampus impairs spatial learning in rats. Eur. J. Neurosci. 1997;9:194–197. doi: 10.1111/j.1460-9568.1997.tb01367.x. [DOI] [PubMed] [Google Scholar]
- SANDIN J., GEORGIEVA J., SILBERRING J., TERENIUS L. In vivo metabolism of nociceptin/orphanin FQ in rat hippocampus. Neuroreport. 1999;10:71–76. doi: 10.1097/00001756-199901180-00014. [DOI] [PubMed] [Google Scholar]
- SBRENNA S., MARTI M., MORARI M., CALO' G., GUERRINI R., BEANI L., BIANCHI C. 5-HT, Glu and GABA efflux from rat cerebrocortical synaptosomes: modulation by opioid receptor like 1 receptor. Regul. Peptides. 1999;80:127. [PubMed] [Google Scholar]
- SBRENNA S., MARTI M., MORARI M., CALO' G., GUERRINI R., BEANI L., BIANCHI C. Modulation of 5-HT efflux from rat cortical synaptosomes by opioids and nociceptin Br. J. Pharmacol.in press [DOI] [PMC free article] [PubMed]
- SCHILD H.O. Drug receptors. Baltimore: University Park Press; 1973. Receptor classification with special reference to beta adrenergic receptors; pp. 29–36. [Google Scholar]
- SCHILLER P.W., NGUYEN T.M., WELTROWSKA G., WILKES B.C., MARSDEN B.J., LEMIEUX C., CHUNG N.N. Differential stereochemical requirements of mu vs. delta opioid receptors for ligand binding and signal transduction: development of a class of potent and highly delta-selective peptide antagonists. Proc. Natl. Acad. Sci. U.S.A. 1992;89:11871–11875. doi: 10.1073/pnas.89.24.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLICKER E., WERTHWEIN S., KATHMANN M., BAUER U. Nociceptin inhibits noradrenaline release in the mouse brain cortex via presynaptic ORL1 receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:418–422. doi: 10.1007/pl00005273. [DOI] [PubMed] [Google Scholar]
- SCHULZ S., SCHREFF M., NUSS D., GRAMSCH C., HOLLT V. Nociceptin/orphanin FQ and opioid peptides show overlapping distribution but not colocalization in pain-modulatory brain regions. Neuroreport. 1996;7:3021–3025. doi: 10.1097/00001756-199611250-00045. [DOI] [PubMed] [Google Scholar]
- SCHWYZER R. Molecular mechanism of opioid receptor selection. Biochemistry. 1986;25:6335–6342. doi: 10.1021/bi00368a075. [DOI] [PubMed] [Google Scholar]
- SEKI T., AWAMURA S., KIMURA C., IDE S., SAKANO K., MINAMI M., NAGASE H., SATOH M. Pharmacological properties of TRK-820 on cloned mu-, delta- and kappa- opioid receptors and nociceptin receptor. Eur. J. Pharmacol. 1999;376:159–167. doi: 10.1016/s0014-2999(99)00369-6. [DOI] [PubMed] [Google Scholar]
- SHAH S., PAGE C.P., SPINA D. Nociceptin inhibits non-adrenergic non-cholinergic contraction in guinea-pig airway. Br. J. Pharmacol. 1998;125:510–516. doi: 10.1038/sj.bjp.0702068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOHIGASHI Y., HATANO R., FUJITA T., NAKASHIMA R., NOSE T., SUJAKU T., SAIGO A., SHINJO K., NAGAHISA A. Sensitivity of opioid receptor-like receptor ORL1 for chemical modification on nociceptin, a naturally occurring nociceptive peptide. J. Biol. Chem. 1996;271:23642–23645. doi: 10.1074/jbc.271.39.23642. [DOI] [PubMed] [Google Scholar]
- SIM L.J., CHILDERS S.R. Anatomical distribution of mu, delta, and kappa opioid- and nociceptin/orphanin FQstimulated [35S]guanylyl-5′-O-(gamma-thio)-triphosphate binding in guinea pig brain. J. Comp. Neurol. 1997;386:562–572. doi: 10.1002/(sici)1096-9861(19971006)386:4<562::aid-cne4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- SINISCALCHI A., RODI D., BEANI L., BIANCHI C. Inhibitory effect of nociceptin on [3H]5-HT release from the rat cerebral cortex slices. Br. J. Pharmacol. 1999;128:119–123. doi: 10.1038/sj.bjp.0702793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLUGG R.M., OK R.N., GRANDY D.K., KELLY M.J. Activation of an Inwardly Rectifying K+ Conductance by Orphanin- FQ/Nociceptin in Vasopressin-Containing Neurons. Neuroendocrinology. 1999;69:385–396. doi: 10.1159/000054441. [DOI] [PubMed] [Google Scholar]
- STANFA L.C., CHAPMAN V., KERR N., DICKENSON A.H. Inhibitory action of nociceptin on spinal dorsal horn neurones of the rat, in vivo. Br. J. Pharmacol. 1996;118:1875–1877. doi: 10.1111/j.1476-5381.1996.tb15618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRATFORD T.R., HOLAHAN M.R., KELLEY A.E. Injections of nociceptin into nucleus accumbens shell or ventromedial hypothalamic nucleus increase food intake. Neuroreport. 1997;8:423–426. doi: 10.1097/00001756-199701200-00009. [DOI] [PubMed] [Google Scholar]
- SULAIMAN M.R., NIKLASSON M., THAM R., DUTIA M.B. Modulation of vestibular function by nociceptin/orphanin FQ: an in vivo and in vitro study. Brain Res. 1999;828:74–82. doi: 10.1016/s0006-8993(99)01331-1. [DOI] [PubMed] [Google Scholar]
- TANIGUCHI H., YOMOTA E., NOGI K., ONODA Y., IKEZAWA K. The effect of nociceptin, an endogenous ligand for the ORL1 receptor, on rat colonic contraction and transit. Eur. J. Pharmacol. 1998;353:265–271. doi: 10.1016/s0014-2999(98)00411-7. [DOI] [PubMed] [Google Scholar]
- TAYLOR F., DICKENSON A. Nociceptin/orphanin FQ. A new opioid, a new analgesic. Neuroreport. 1998;9:R65–R70. [PubMed] [Google Scholar]
- TIAN J.H., XU W., FANG Y., MOGIL J.S., GRISEL J.E., GRANDY D.K., HAN J.S. Bidirectional modulatory effect of orphanin FQ on morphine-induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br. J. Pharmacol. 1997b;120:676–680. doi: 10.1038/sj.bjp.0700942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIAN J.H., XU W., ZHANG W., FANG Y., GRISEL J.E., MOGIL J.S., GRANDY D.K., HAN J.S. Involvement of endogenous orphanin FQ in electroacupuncture-induced analgesia. Neuroreport. 1997a;8:497–500. doi: 10.1097/00001756-199701200-00024. [DOI] [PubMed] [Google Scholar]
- TIAN J.H., ZHANG W., FANG Y., XU W., GRANDY D.K., HAN J.S. Endogenous orphanin FQ: evidence for a role in the modulation of electroacupuncture analgesia and the development of tolerance to analgesia produced by morphine and electroacupuncture. Br. J. Pharmacol. 1998;124:21–26. doi: 10.1038/sj.bjp.0701788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOLL L., BURNSIDE J., BERZETEI-GURSKE I.Agonist activity of ORL1 antagonists is dependent upon receptor number INRC 1998 1998Garmisch-Partenkirchen, Germany; pp. A892025 July, 1998 [Google Scholar]
- UEDA H., YAMAGUCHI T., TOKUYAMA S., INOUE M., NISHI M., TAKESHIMA H. Partial loss of tolerance liability to morphine analgesia in mice lacking the nociceptin receptor gene. Neurosci. Lett. 1997;237:136–138. doi: 10.1016/s0304-3940(97)00832-x. [DOI] [PubMed] [Google Scholar]
- VARANI K., CALO' G., RIZZI A., MERIGHI S., TOTH G., GUERRINI R., SALVADORI S., BOREA P.A., REGOLI D. Nociceptin receptor binding in mouse forebrain membranes: thermodynamic characteristics and structure activity relationships. Br. J. Pharmacol. 1998;125:1485–1490. doi: 10.1038/sj.bjp.0702226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARANI K., RIZZI A., CALO' G., BIGONI R., TOTH G., GUERRINI R., GESSI S., SALVADORI S., BOREA P.A., REGOLI D. Pharmacology of [Tyr1]nociceptin analogs: receptor binding and bioassay studies. Naunyn Schmiedebergs Arch. Pharmacol. 1999;360:270–277. doi: 10.1007/s002109900074. [DOI] [PubMed] [Google Scholar]
- VAUGHAN C.W., CHRISTIE M.J. Increase by the ORL1 receptor (opioid receptor-like1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br. J. Pharmacol. 1996;117:1609–1611. doi: 10.1111/j.1476-5381.1996.tb15329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHAN C.W., INGRAM S.L., CHRISTIE M.J. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal gray neurons in vitro. J. Neurosci. 1997;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VLASKOVSKA M., KASAKOV L., SUDER P., SILBERRING J., TERENIUS L. Biotransformation of nociceptin/orphanin FQ by enzyme activity from morphine-naive and morphine-treated cell cultures. Brain Res. 1999;818:212–220. doi: 10.1016/s0006-8993(98)01266-9. [DOI] [PubMed] [Google Scholar]
- WALKER J.R., SPINA M., TERENIUS L., KOOB G.F. Nociceptin fails to affect heroin self-administration in the rat. Neuroreport. 1998;9:2243–2247. doi: 10.1097/00001756-199807130-00017. [DOI] [PubMed] [Google Scholar]
- WANG J.B., JOHNSON P.S., IMAI Y., PERSICO A.M., OZENBERGER B.A., EPPLER C.M., UHL G.R. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994;348:75–79. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- WANG X.M., ZHANG K.M., MOKHA S.S. Nociceptin (orphanin FQ), an endogenous ligand for the QRL1 (opioid receptor-like1) receptor; modulates responses of trigeminal neurons evoked by excitatory amino acids and somatosensory stimuli. J. Neurophysiol. 1996;76:3568–3572. doi: 10.1152/jn.1996.76.5.3568. [DOI] [PubMed] [Google Scholar]
- WANG Y.Q., ZHU C.B., CAO X.D., WU G.C. Supraspinal hyperalgesia and spinal analgesia by [Phe1psi(CH2-NH)Gly2]nociceptin-(1-13)-NH2 in rat. Eur. J. Pharmacol. 1999a;376:R1–R3. doi: 10.1016/s0014-2999(99)00399-4. [DOI] [PubMed] [Google Scholar]
- WANG Y.Q., ZHU C.B., WU G.C., CAO X.D., WANG Y., CUI D.F. Effects of orphanin FQ on endomorphin-1 induced analgesia. Brain Res. 1999b;835:241–246. doi: 10.1016/s0006-8993(99)01589-9. [DOI] [PubMed] [Google Scholar]
- WERTHWEIN S., BAUER U., NAKAZI M., KATHMANN M., SCHLICKER E. Further characterization of the ORL1 receptor-mediated inhibition of noradrenaline release in the mouse brain in vitro. Br. J. Pharmacol. 1999;127:300–308. doi: 10.1038/sj.bjp.0702534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WICHMANN J., ADAM G., ROVER S., CESURA A.M., DAUTZENBERG F.M., JENCK F. 8-Acenaphthen-1-YL-1-phenyl-1,3,8-triazaspiro[4,5]decan-4-one derivatives as orphanin fq receptor agonists. Bioorg. Med. Chem. Lett. 1999;9:2343–2348. doi: 10.1016/s0960-894x(99)00385-6. [DOI] [PubMed] [Google Scholar]
- WICK M.J., MINNERATH S.R., LIN X., ELDE R., LAW P.Y., LOH H.H. Isolation of a novel cDNA encoding a putative membrane receptor with high homology to the cloned mu, delta, and kappa opioid receptors. Brain Res. Mol. Brain Res. 1994;27:37–44. doi: 10.1016/0169-328x(94)90181-3. [DOI] [PubMed] [Google Scholar]
- WICK M.J., MINNERATH S.R., ROY S., RAMAKRISHNAN S., LOH H.H. Expression of alternate forms of brain opioid ‘orphan' receptor mRNA in activated human peripheral blood lymphocytes and lymphocytic cell lines. Brain Res. Mol. Brain Res. 1995;32:342–347. doi: 10.1016/0169-328x(95)00096-b. [DOI] [PubMed] [Google Scholar]
- WISE R.A. Opiate reward: sites and substrates. Neurosci. Biobehav. Rev. 1989;13:129–133. doi: 10.1016/s0149-7634(89)80021-1. [DOI] [PubMed] [Google Scholar]
- WNENDT S., KRUGER T., JANOCHA E., HILDEBRANDT D., ENGLBERGER W. Agonistic effect of buprenorphine in a nociceptin/OFQ receptor- triggered reporter gene assay. Mol. Pharmacol. 1999;56:334–338. doi: 10.1124/mol.56.2.334. [DOI] [PubMed] [Google Scholar]
- XU I.S., WIESENFELD-HALLIN Z., XU X.J. [Phe1psi(CH2-NH)Gly2]-nociceptin-(1-13)NH2, a proposed antagonist of the nociceptin receptor, is a potent and stable agonist in the rat spinal cord. Neurosci. Lett. 1998;249:127–130. doi: 10.1016/s0304-3940(98)00411-x. [DOI] [PubMed] [Google Scholar]
- XU X.J., HAO J.X., WIESENFELD-HALLIN Z. Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport. 1996;7:2092–2094. [PubMed] [Google Scholar]
- YAMAMOTO T., NOZAKI-TAGUCHI N., KIMURA S. Analgesic effect of intrathecally administered nociceptin, an opioid receptor-like1 receptor agonist, in the rat formalin test. Neuroscience. 1997a;81:249–254. doi: 10.1016/s0306-4522(97)00166-8. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO T., NOZAKI-TAGUCHI N., KIMURA S. Effects of intrathecally administered nociceptin, an opioid receptor- like1 (ORL1) receptor agonist, on the thermal hyperalgesia induced by carageenan injection into the rat paw. Brain Res. 1997b;754:329–332. doi: 10.1016/s0006-8993(97)00186-8. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO T., SAKASHITA Y. Effect of nocistatin and its interaction with nociceptin/orphanin FQ on the rat formalin test. Neurosci. Lett. 1999;262:179–182. doi: 10.1016/s0304-3940(99)00073-7. [DOI] [PubMed] [Google Scholar]
- YASUDA K., RAYNOR K., KONG H., BREDER C.D., TAKEDA J., REISINE T., BELL G.I. Cloning and functional comparison of kappa and delta opioid receptors from mouse brain. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6736–6740. doi: 10.1073/pnas.90.14.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAZDANI A., TAKAHASHI T., BAGNOL D., WATSON S.J., OWYANG C. Functional significance of a newly discovered neuropeptide, orphanin FQ, in rat gastrointestinal motility. Gastroenterology. 1999;116:108–117. doi: 10.1016/s0016-5085(99)70234-9. [DOI] [PubMed] [Google Scholar]
- YOSHIKAWA M., TAKAHASHI M., YUNDEN J.Analgesic and cognition-enhancing activity of retronociceptin methyl-ester, a nociceptin antagonist International Narcotic Research Conference 1999Saratoga Springs, NY; July 10–15 1999. pp. Sun63 [Google Scholar]
- YU J., CHAIT B.T., TOLL L., KREEK M.J. Nociceptin in vitro biotransformation in human blood. Peptides. 1996;17:873–876. doi: 10.1016/0196-9781(96)00079-4. [DOI] [PubMed] [Google Scholar]
- YU T.P., FEIN J., PHAN T., EVANS C.J., XIE C.W. Orphanin FQ inhibits synaptic transmission and long-term potentiation in rat hippocampus. Hippocampus. 1997;7:88–94. doi: 10.1002/(SICI)1098-1063(1997)7:1<88::AID-HIPO9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- YU T.P., XIE C.W. Orphanin FQ/nociceptin inhibits synaptic transmission and long-term potentiation in rat dentate gyrus through postsynaptic mechanisms. J. Neurophysiol. 1998;80:1277–1284. doi: 10.1152/jn.1998.80.3.1277. [DOI] [PubMed] [Google Scholar]
- YUAN L., HAN Z., CHANG J.K., HAN J.S. Accelerated release and production of orphanin FQ in brain of chronic morphine tolerant rats. Brain Res. 1999;826:330–334. doi: 10.1016/s0006-8993(99)01337-2. [DOI] [PubMed] [Google Scholar]
- ZADINA J.E., HACKLER L., GE L.J., KASTIN A.J. A potent and selective endogenous agonist for the μ opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- ZGODZINSKI W., RUBAJ A., GUSTAW K., SIEKLUCKA-DZIUBA M., KLEINROK Z.Anticonvulsant effects of orphanin FQ receptor ligands International Narcotics Research Conference 1997Saratoga Springs, NY; July 10–15 1999 [Google Scholar]
- ZHANG G., MURRAY T.F., GRANDY D.K. Orphanin FQ has an inhibitory effect on the guinea pig ileum and the mouse vas deferens. Brain Res. 1999;772:102–106. doi: 10.1016/s0006-8993(97)00858-5. [DOI] [PubMed] [Google Scholar]
- ZHU C.B., CAO X.D., XU S.F., WU G.C. Orphanin FQ potentiates formalin-induced pain behavior and antagonizes morphine analgesia in rats. Neurosci. Lett. 1997;235:37–40. doi: 10.1016/s0304-3940(97)00704-0. [DOI] [PubMed] [Google Scholar]
- ZHU C.B., XU S.F., CAO X.D., WU G.C., ZHANG X.L., LI M.Y., CUI D.F., CHI C.W. Antagonistic action of orphanin FQ on acupuncture analgesia in rat brain. Acupunct. Electrother. Res. 1996;21:199–205. doi: 10.3727/036012996816356852. [DOI] [PubMed] [Google Scholar]