

Abstract
Amitriptyline has been known to induce QT prolongation and torsades de pointes which causes sudden death. We studied the effects of amitriptyline on the human ether-a-go-go-related gene (HERG) channel expressed in Xenopus oocytes and on the rapidly activating delayed rectifier K+ current (IKr) in rat atrial myocytes.
The amplitudes of steady-state currents and tail currents of HERG were decreased by amitriptyline dose-dependently. The decrease became more pronounced at more positive potential, suggesting that the block of HERG by amitriptyline is voltage dependent. IC50 for amitriptyline block of HERG current was progressively decreased according to depolarization: IC50 values at −30, −10, +10 and +30 mV were 23.0, 8.71, 5.96 and 4.66 μM, respectively.
Block of HERG by amitriptyline was use dependent: exhibiting a much faster block at higher activation frequency. Subsequent decrease in frequency after high activation frequency resulted in a partial relief of HERG blockade.
Steady-state block by amitriptyline was obtained while depolarization to +20 mV for 0.5 s was applied at 0.5 Hz: IC50 was 3.26 μM in 2 mM [K+]o. It was increased to 4.78 μM in 4 mM [K+]o, suggesting that the affinity of amitriptyline on HERG was decreased by external K+.
In rat atrial myocytes bathed in 35°C, 5 μM amitriptyline blocked IKr by 55%. However, transient outward K+ current (Ito) was not significantly affected.
In summary, the data suggest that the block of HERG currents may contribute to arrhythmogenic side effects of amitriptyline.
Keywords: Amitriptyline, rapidly activating delayed rectifier K+ current, HERG channel, LQT, torsades de pointes, use dependence, voltage dependence
Introduction
Tricyclic antidepressants (TCAs) are commonly used for many different psychiatric and nonpsychiatric disorders. Intentional overdose for suicide with these agents is common, resulting in substantial morbidity and mortality owing to ventricular tachyarrhythmias (Biggs et al., 1977; Giardina et al., 1979; Marshall & Forker, 1982). Clinical observational data (ECG data including QRS and QT prolongation) suggest that TCA-induced ventricular tachyarrhythmias is like ‘quinidine toxicity' (Vohra et al., 1975; Thorstrand, 1976), namely, facilitation of the reentrant excitation as a result of decreased conduction velocity and temporal dispersion of action potential duration (Fowler et al., 1976; Arita & Surawicz, 1973). Amitriptyline, a TCA, which is widely used to treat manic depression, has been shown to prolong QT interval (Nishimoto et al., 1994; Alderton, 1995) and to induce torsades de pointes in human hearts (Davison, 1985). However, the electrophysiological basis for the arrhythmogenic effects of the drug has not been reported.
Warmke & Ganetzky (1994) identified the human ether-a-go-go-related gene (HERG), and it was subsequently shown that when the gene is expressed in Xenopus oocytes and HEK293 cells, it encodes a K+ channel whose property is similar to that of cardiac rapidly activating delayed rectifier K+ current (IKr) (Sanguinetti et al., 1995; Trudeau et al., 1995; Snyders & Chaudhary, 1996). HERG current exhibits nanomolar sensitivity to methanesulfonanilides, dependence on external K+ concentration, and inward rectification. Mutations in HERG have been shown to cause chromosome 7-linked inherited long QT syndrome (LQT, Curran et al., 1995), and several drugs that block IKr and HERG cause acquired LQT and torsades de pointes (Snyders & Chaudhary, 1996; Roy et al., 1996; Suessbrich et al., 1996). Thus HERG is an important target in both acquired and inherited forms of LQT.
Amitriptyline was recently reported to increase action potential duration and to induce early afterdepolarizations in canine Purkinje fibres (Wyse et al., 1996). This raises the possibility that amitriptyline may prolong action potential duration in vivo and cause LQT by inhibiting IKr or HERG channel. We used HERG channel expressed in Xenopus oocytes to test the hypothesis that amitriptyline is a blocker of HERG channel current.
Methods
Expression of HERG in oocytes
Complementary RNA of HERG was synthesized by in vitro transcription from 1 μg of linearized cDNA using T7 message machine kits (Ambion, Austin, TX, U.S.A.) and stored in 10 mM Tris-HCl (pH 7.4) at −80°C. Stage V-VI oocytes were surgically removed from female Xenopus laevis (Nasco, Modesto, CA, U.S.A.) anaesthetized with 0.17% tricane methanesulphonate (Sigma). Theca and follicle layers were manually removed from the oocytes by using fine forceps. Oocytes were then injected with 40 nl of cRNA (0.1–0.5 μg.μl−1). After injection, oocytes were maintained in modified Barth's solution containing (mM): NaCl 88, KCl 1, CaCl2 0.4, Ca(NO3)2 0.33, MgSO4 1, NaHCO3 2.4, HEPES 10 (pH 7.4), supplemented with 50 μg ml−1 gentamicin sulphonate. Currents were studied 2–7 days after injection.
Solutions and voltage clamp recording from oocytes
Normal Ringer solution contained (mM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 10 (pH adjusted to 7.4 with NaOH). In some experiments, [KCl] was changed to 4 mmol 1−1; osmolarity was maintained by an equimolar change in [NaCl]. The antidepressant drug, amitriptyline and all salts were purchased from Sigma (St. Louis, MO, U.S.A.). Stock solution of amitriptyline was made up in distilled water, then added to the external solutions at suitable concentrations shortly before the experiment. Solutions were applied to the oocyte by continuous perfusion of the chamber during recording. Solution exchanges were completed within 3 min, and we start to record HERG current after 5 min in which solution exchange was completed. We examined the effects of several concentrations of amitriptyline on HERG current after observing the reversibility of current by washing with normal Ringer solution. It took about 10 min for washout of ⩽5 μM drug and about 20 min for washout of ⩾10 μM drug. We rejected the oocyte if it did not recover after washing with normal Ringer for 30 min. Usually, we examined 5–6 concentrations of amitriptyline in one oocyte. Currents were measured at room temperature (21–23°C) with a two-microelectrode voltage clamp amplifier (Warner Instruments, Hamden, CT, U.S.A.). Electrodes were filled with 3 M KCl and had a resistance of 2–4 MΩ for voltage-recording electrodes and 0.6–1 MΩ for current-passing electrodes. Stimulation and data acquisition were controlled with Digidata and pCLAMP software (Axon Instruments). Data were expressed as mean values±s.e.mean.
Solutions and voltage clamp recording from rat atrial myocytes
Hearts from young adult Sprague-Dawley rats (200–250 g) were removed under cervical dislocation and perfused quickly via the aorta onto a Langendorff retrograde perfusion apparatus. Hearts were initially perfused with normal Tyrode solution (100 ml), and then perfused with a nominally Ca2+ free Tyrode solution (50 ml), followed by the same solution containing 0.12 mg ml−1 collagenase (Yakult, Japan) and 50 μM CaCl2. After perfusing enzyme solution for 14 min, the heart was removed from the Langendorff perfusion apparatus and the collagenase was washed out by rinsing with high K+, low Cl− storage medium. The atria and its appendages were then mechanically dispersed with a fire polished Pasteur pipette and stored in the same solution at 4°C until used in experiments.
Currents were recorded by the standard whole cell voltage clamp methods (Hamill et al., 1981) using Axopatch-200A (Axon instruments) amplifier. Recording pipettes (Clark Electromedical, U.K.) with resistance of 2–4 MΩ were used. The current signals were filtered via 1–10 kHz, 8-pole Bessel-type low-pass filter and digitized by an AD-DA converter (Digidata 1200, Axon Instruments) for subsequent analysis (pCLAMP software 6.0.3.).
Normal Tyrode solution contained (mM): NaCl 143, KCl 5.4, MgCl2 0.5, CaCl2 1.8, glucose 10, HEPES 5 (pH 7.4). High K solution for recording IKr contained (mM): KCl 140, MgCl2 2, HEPES 10, glucose 10 (pH 7.4). Recording pipettes contained (mM): KCl 140, EGTA 5, HEPES 10, MgATP 5, diTris-phosphocreatine 2.5 and disodium phosphocreatine 2.5 (pH 7.2). All chemicals were from Sigma, except E-4031 which was kindly provided by Eisai Co. (Japan).
Results
The effect of amitriptyline on HERG current was studied using Xenopus oocyte expression system. Throughout the experiments, holding potential was adjusted between −60 and −70 mV to obtain the minimum leak current, but the repolarization potential was made constant at −60 mV for analysing tail currents at the constant condition. Figure 1 shows voltage-clamp data obtained with 5 μM amitriptyline. Families of current traces from one cell are shown for control condition (Figure 1A, middle panel) and after exposure to amitriptyline (Figure 1A, lower panel), with the voltage-clamp protocol shown in upper panel of Figure 1A. The voltage pulses were given at 60 s intervals. In the control condition, depolarizing steps activated time-dependent outward currents. The amplitude of outward currents measured at the end of 4 s pulse (steady-state current, Iss) was increased with more positive voltage steps, and reached a maximum at −20 mV. Depolarizing steps to more positive voltages caused a decrease of the current, resulting in a negative slope in the IV curve (Figure 1B). Current-voltage relationships for Iss obtained at various concentrations of amitriptyline are plotted in Figure 1B. As concentration of amitriptyline was progressively increased, the amplitude of Iss decreased dose-dependently.
Figure 1.
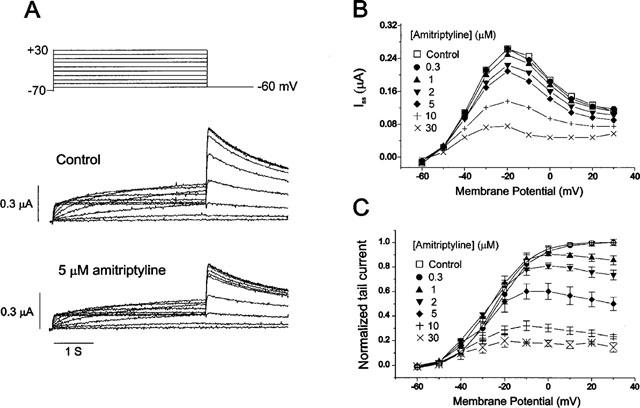
Effect of amitriptyline on HERG currents. (A) Superimposed current traces elicited by depolarizing voltage pulses (4 s) in 10 mV steps (upper panel) from a holding potential of −70 mV in the absence of amitriptyline (control, middle panel) and in the presence of 5 μM amitriptyline (lower panel). (B) Plot of the steady-state current measured at the end of depolarizing pulses against the pulse potential in different concentrations of amitriptyline (obtained from the same cell shown in A). (C) Plot of the normalized tail current measured at its peak just after repolarization. The amplitude of the tail current in the absence of the drug was taken as one. Symbols with error bars represent mean±s.e.mean: each data obtained from seven cells. Control data were fitted to the Boltzmann equation, y=1/{1+exp[(−V+V1/2)/dx]}, with V1/2 of −24.5 mV.
After the repolarizing steps to −60 mV, an outward current (tail current, Itail) was recorded. The amplitude of Itail was increased with depolarizing steps from −60 to 0 mV and was then superimposed on further depolarizing steps to +30 mV. When 5 μM amitriptyline was added to the perfusate, not only Iss but also Itail were suppressed, as shown in lower panel of Figure 1A. The amplitude of Itail was normalized to the peak amplitude obtained in the control condition at maximum depolarization, and was plotted against the potential of the step depolarization (Figure 1C). The normalized Itail can be regarded to represent the voltage dependence of activation of the HERG channels, and the data obtained in the control was well fitted by the Boltzmann equation with half-maximal activation (V1/2) at −24.5 mV, which is similar value to reports by other investigators (Wang et al., 1997). When the concentration of amitriptyline was increased, the amplitude of the peak Itail was decreased, indicating that the maximum conductance (gmax) of HERG channels is decreased by amitriptyline. It is also noticed that in the presence of amitriptyline, Itail does not reach the steady-state level, but decline at more positive potential, indicating that the blocking effect is more pronounced at positive potential.
This result may suggest that the effect of amitriptyline is voltage dependent, and we tested this possibility in Figure 2. We compared the decrease of Itail by amitriptyline at different potentials, and found a higher degree of block at more positive voltages (Figure 2A). At −30 mV, 5 μM amitriptyline did not affect the amplitude of normalized Itail significantly (from 0.31±0.06 to 0.29±0.05; n=7, P>0.05), whereas at +30 mV it reduced the amplitude of normalized Itail by 50% (from 1.00±0.01 to 0.50±0.06; n=7, P<0.05). Dose-response relationships obtained at +30 and −30 mV are demonstrated in Figure 2B. Data were fitted by Hill equations and IC50 values for amitriptyline block of HERG current were obtained at different potentials. IC50 values at −30, −10, +10, and +30 mV were 23.0, 8.71, 5.96 and 4.66 μM, respectively (n=7). These results indicate that amitriptyline block of HERG current exhibits voltage dependence.
Figure 2.
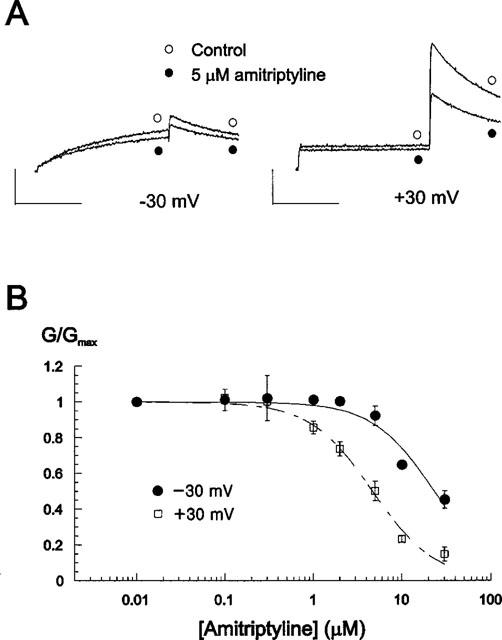
Voltage dependence of amitriptyline block on HERG current. (A) Current traces from a cell depolarized to −30 (left panel) and +30 mV (right panel) from the holding potential of −70 mV, before and after exposure to 5 μM amitriptyline. Tail currents were recorded at −60 mV. Magnitude of the decrease of the tail currents indicates that the block effect is larger at +30 mV. Calibration bars are 0.2 μA in height and 2 s in length. (B) Concentration-dependence of amitriptyline block at different membrane potentials. Conductance (G) in the presence of various concentration of amitriptyline was normalized to the maximum conductance (Gmax) obtained in control, and plotted against amitriptyline concentrations. Data were from Figure 1C. Symbols with error bars represent mean±s.e.mean: each data obtained from seven cells. Lines are the fits of data to the Hill equation, giving IC50 values of 23.0±3.24, 4.66±0.36 μM and Hill coefficients of 1.25±0.21, 1.23±0.11 at −30 and +30 mV, respectively.
The use dependence of the amitriptyline effect was examined in Figure 3. To analyse this use dependence, HERG channels were activated by 0.5 s depolarizing steps to +30 mV at intervals of 3 s, 12 s or 36 s in the presence of 3 μM amitriptyline (n=6). Figure 3A shows that the time course of the channel blockade is dramatically dependent on the activation frequency; HERG-blockade by amitriptyline occurred much faster at higher activation frequency. In Figure 3B, the data shown in Figure 3A were plotted as function of the number of test pulses. After the same number of test pulses the block of 3 μM amitriptyline was stronger at a high activation frequency than at lower activation frequency, indicating favoured binding of the drug at higher frequency. In additional experiments, steady-state HERG channel block by amitriptyline was initially obtained with depolarization at 3 s intervals. Subsequent increase of the depolarization intervals to 36 s resulted in a partial relief of HERG channel blockade (n=6, Figure 3C). These results indicate that the blockade of HERG channels by amitriptyline is strongly use-dependent.
Figure 3.
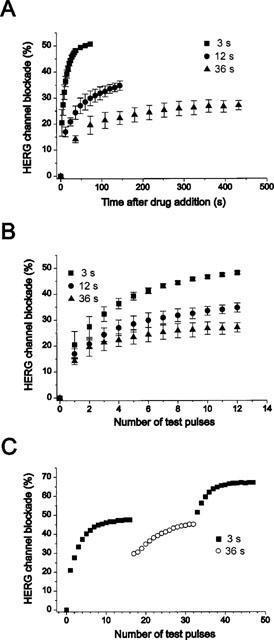
Use-dependent HERG channel blockade by 3 μM amitriptyline. (A) Tail currents were recorded at −60 mV after a 0.5 s depolarizing pre-pulse to +30 mV from a holding potential −60 mV every 3 s, 12 s, and 36 s, respectively. (B) The HERG channel blockade data from A were plotted against the number of test pulses. (C) Steady-state blockade by 3 μM amitriptyline after 16 pulses at 3 s intervals. Increasing the depolarization intervals to 36 s in the presence of amitriptyline resulted in a partial relief, and changing back to 3 s intervals increased HERG channel blockade again. Symbols with error bars in A and B represent mean±s.e.mean: each data obtained from six cells. Symbols in C is representative of six experiments.
As amitriptyline blocked HERG channels voltage- and use-dependently, it is important to examine IC50 values at frequency and voltage close to possible normal conditions which can be expected during regular action potential. Therefore, we determined the IC50 values when oocytes were depolarized from −70 to +20 mV for 0.5 s at the rate of 0.5 Hz. Also, we compared the IC50 values at different extracellular K+ concentrations ([K+]o) because [K+]o of normal Ringer solution (2 mM) was lower than that of normal extracellular fluid of mammalian (4 mM). As shown in Figure 4Ai and 4Aii, amitriptyline blocked HERG current concentration- and use-dependently, resulting in steady-state block at 16th pulse (32 s after drug addition) at each concentration. Therefore we obtained the IC50 values by fitting the data at this 16th pulse with a Hill function (Figure 4B). The fitting procedure estimated IC50 values of 3.26±0.31, 4.78±0.37 μM and Hill coefficients of 1.17±0.13, 1.43±0.16 in 2 and 4 mM [K+]o respectively (n=5) showing that the affinity of amitriptyline on HERG channel was decreased by external K+.
Figure 4.
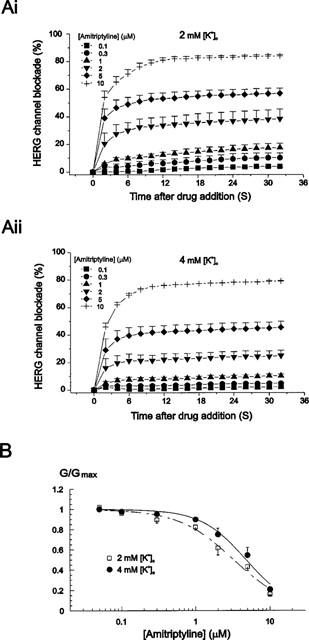
External K+ dependence of amitriptyline-induced HERG channel blockade. (Ai) Tail currents were recorded at −60 mV after a 0.5 s depolarizing pre-pulse to +20 mV from a holding potential −70 mV every 2 s at various concentrations of amitriptyline in 2 mM extracellular K+ concentration ([K+]o). (Aii) Tail currents were recorded using the same protocol in A at various concentrations of amitriptyline in 4 mM [K+]o. (B) Concentration dependence of IHERG block by amitriptyline at different [K+]o. Plot of the decrease in maximum conductance obtained from data in Ai and Aii against amitriptyline concentrations. Symbols with error bars represent mean±s.e.mean: each data obtained from five cells. Lines are the fits of data to the Hill equation, giving IC50 values of 3.26±0.31, 4.78±0.37 μM and Hill coefficients of 1.17±0.13, 1.43±0.16 in 2 and 4 mM [K+]o, respectively.
Although HERG channel is generally regarded to correspond IKr in cardiac cells in mammalian hearts, it is not certain that the result obtained from HERG channel expressed in Xenopus oocytes can be applicable to cardiac IKr. Furthermore, the difference in normal temperature between oocyte experiments and normal hearts may limit the significance of the results of above experiments. We therefore tested the effect of amitriptyline on IKr using rat atrial myocytes at 35°C. In order to isolate IKr from other K+ currents, currents were recorded in high K+ solution, as previously reported (Ho et al., 1996). Inward rectifier K+ currents were inhibited by 5 mM CsCl to minimize the instantaneous leak currents. Hyperpolarizing pulse to −100 mV induced time dependent inward current which was previously characterized as IKr (Ho et al., 1996; Song et al., 1999). We confirmed that this component is blocked by 5 μM E-4031, a selective blocker of IKr (Figure 5A, upper panel). We then tested the effect of 5 μM amitriptyline (Figure 5A, lower panel) and the result shows the inhibition of IKr by 55±3% (n=3). Also, as shown in Figure 5B, the effect of amitriptyline on transient outward K+ current (Ito) was tested by applying depolarization pulse to +40 mV, showing that the effect is insignificant.
Figure 5.
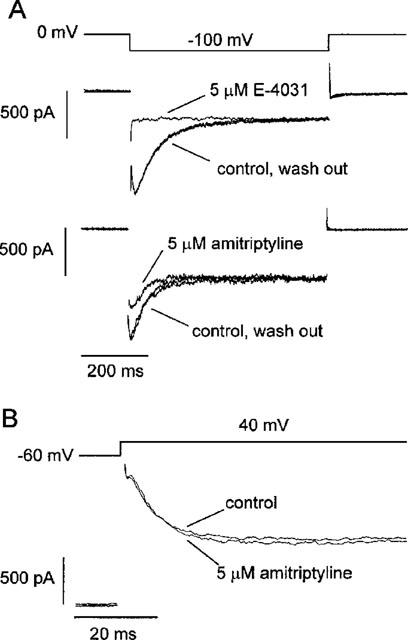
Effects of amitriptyline on rapidly activating delayed rectifier K+ current (IKr) and transient outward K+ current (Ito) in rat atrial myocytes. (A) Voltage clamp pulse protocol was shown in the upper panel. The middle panel was superimposed current traces recorded in high K+ solution (140 mM) before, in the presence, and after wash out of 5 μM E-4031. Disappearance of the time-dependent component of inward currents by E-4031 indicates that this component is IKr. In the lower panel, superimposed current traces before, in the presence, and after wash out of 5 μM amitriptyline were shown. (B) Superimposed current traces recorded in response to depolarizing pulses to +40 mV in normal Tyrode solution, before and after applying 5 μM amitriptyline. Similar observations in two more cells.
Discussion
Several reports of ventricular arrhythmia including torsades de pointes associated with the use of amitriptyline have been documented (Davison, 1985; Ansel et al., 1993; Nishimoto et al., 1994). The fact that amitriptyline causes QT prolongation in the ECG suggests the involvement of K+ channel blockade (Alderton, 1995; Nishimoto et al., 1994), but a direct evidence is lacking. This study was therefore designed to test the hypothesis that amitriptyline might block HERG channel, a molecular equivalent of delayed rectifier K+ channel, which is important for keeping action potential duration short in heart (Sanguinetti & Jurkiewicz, 1990).
After administration of amitriptyline, total peak serum values of 0.2–0.7 μM under therapeutic conditions, rising to >3.2 μM in overdose are achieved (Amsterdam et al., 1980; Schulz et al., 1985). The drug is oxidized by microsomal enzymes in liver with an elimination half-life of ∼21 h. The half-life of amitriptyline may be prolonged in the patients with hepatic disease and in the elderly (Nies et al., 1977). Also, it is known that serum concentrations of the drug usually exceed 3.2 μM before arrhythmias develops, and the concentration is a mean of 5.4±0.6 μM for sustained ventricular tachyarrhythmias to ensue (Marshall & Forker, 1982). The result of the present study demonstrates that amitriptyline blocked HERG channels potently with IC50 value of 4.78±0.37 μM in 4 mM [K+]o pulsed at 0.5 Hz, which is the similar level to both the serum concentration achieved in overdose (Amsterdam et al., 1980; Schulz et al., 1985) and that in which the drug causes ventricular arrhythmia (Marshall & Forker, 1982). Therefore, the present study strongly suggest that blockade of HERG current may underlie the proarrhythmic effect of amitriptyline in overdose, for example, QT prolongation and torsades de pointes.
Block of HERG channel by amitriptyline was voltage- and use-dependent: exhibiting a higher degree of block at more positive voltages, as shown in Figure 2 and much faster block at higher activation frequency, as shown in Figure 3. The voltage and use dependence of amitriptyline-mediated HERG channel blockade suggests that the drug binds to the open or inactivated state of HERG channels, although our results did not distinguish between open- and inactivated-state block. Several drugs that cause acquired LQT and torsades de pointes also have been shown to block HERG channel voltage- and use-dependently (Suessbrich et al., 1996; 1997; Rampe et al., 1997). An antipsychotic drug, haloperidol (Suessbrich et al., 1997) and two histamine receptor antagonists, terfenadine and astemizole (Suessbrich et al., 1996) have been known to bind the inactivated-state of HERG preferentially. In contrast, a gastrointestinal prokinetic agent, cisapride has been shown to block the open-state of the channel (Rampe et al., 1997). This variability may suggest that HERG channels have several different binding sites for the blockade.
The present study showed that a tricyclic antidepressant (amitriptyline) blocked HERG, the molecular equivalent of IKr, which is one of the most important K+ currents in repolarization of cardiac action potential (Sanguinetti & Jurkiewicz, 1990). This effect was expected to explain the torsadogenic side effects seen during treatment of this drug in patients. However, it is possible that the increase in action potential duration by amitriptyline could be caused by the block of other K+ currents. It has been reported that amitriptyline blocked Ito in the open-state in rat ventricular myocytes (Casis & Sanchez-Chapula, 1998). Also, another tricyclic antidepressant, imipramine inhibited inward rectifier K+ currents (IK1) in bovine ventricular myocytes (Isenberg & Tamargo, 1985). However, as shown in Figure 5, amitriptyline at 5 μM preferentially blocked IKr rather than Ito in our experimental condition (e.g. 35°C), which suggest that amitriptyline may prolong action potential duration primarily by blocking of IKr rather than blocking of Ito.
The present study showed that amitriptyline blocks HERG and possibly IKr more at positive voltage and at high frequency, which may not be consistent with reverse use dependent repolarization lengthening by other IKr blockers in cardiac cells. Reverse use dependence has been implicated in the bradycardia-dependent proarrhythmic effects of various class III agents and this effect has been demonstrated with various IKr blocking agents, including E-4031, dofetilide, sotalol, and WAY-123,398 (Hondeghem & Snyders, 1990; Gwilt et al., 1991). It is not known whether amitriptyline prolongs action potential duration in a frequency-dependent manner. However, the findings in the present study, the use- and voltage-dependent block of HERG by amitriptyline indicate that amitriptyline may prolong the repolarization of human cardiac muscle without reverse use dependence, which may be a model for an ideal class III drug without the risk of proarrhythmia.
Acknowledgments
We are grateful to Dr Se-Young Choi for his helpful discussion. This work was supported by grants from Korea Science and Engineering Foundation (98-0401-02) and Ministry of Health and Welfare (#HMP-97-M-2-0025).
Abbreviations
- Gmax
maximum conductance
- HERG
human ether-a-go-go-related gene
- IK1
inward rectifier K+ current
- IKr
rapidly activating delayed rectifier K+ current
- Iss
steady-state current
- Itail
tail current
- Ito
transient outward K+ current
- [K+]o
extracellular K+ concentration
- LQT
long QT syndrome
- n
number of samples
- TCAs
tricyclic antidepressants
- V1/2
potential for half-maximal activation
References
- ALDERTON H.R. Tricyclic medication in children and the QT interval: case report and discussion. Can. J. Psychiatry. 1995;40:325–329. [PubMed] [Google Scholar]
- AMSTERDAM J., BRUNSWICK D., MENDELS J. The clinical application of tricyclic antidepressant pharmacokinetics and plasma levels. Am. J. Psychiatry. 1980;137:653–662. doi: 10.1176/ajp.137.6.653. [DOI] [PubMed] [Google Scholar]
- ANSEL G.M., COYNE K., ARNOLD S., NELSON S.D. Mechanisms of ventricular arrhythmia during amitriptyline toxicity. J. Cardiovasc. Pharmacol. 1993;22:798–803. doi: 10.1097/00005344-199312000-00004. [DOI] [PubMed] [Google Scholar]
- ARITA M., SURAWICZ B. Electrophysiologic effects of phenothiazines on canine cardiac fibers. J. Pharmacol. Exp. Ther. 1973;184:619–630. [PubMed] [Google Scholar]
- BIGGS J.T., SPIKER D.G., PETIT J.M., ZIEGLER V.E. Tricyclic antidepressant overdose: incidence of symptoms. JAMA. 1977;238:135–138. [PubMed] [Google Scholar]
- CASIS O., SANCHEZ-CHAPULA J.A. Mechanism of block of cardiac transient outward K+ current (Ito) by antidepressant drugs. J. Cardiovasc. Pharmacol. 1998;32:527–534. doi: 10.1097/00005344-199810000-00004. [DOI] [PubMed] [Google Scholar]
- CURRAN M.E., SPLAWSKI I., TIMOTHY K.W., VINCENT G.M., GREEN E.D., KEATING M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- DAVISON E.T. Amitriptyline-induced torsade de pointes. Successful therapy with atrial pacing. J. Electrocardiol. 1985;18:299–301. doi: 10.1016/s0022-0736(85)80055-8. [DOI] [PubMed] [Google Scholar]
- FOWLER N.O., MCCALL D., CHOU T.C., HOLMES J.C., HANENSON I.B. Electrocardiographic changes and cardiac arrhythmias in patients receiving pyschotropic drugs. Am. J. Cardiol. 1976;37:223–230. doi: 10.1016/0002-9149(76)90316-7. [DOI] [PubMed] [Google Scholar]
- GIARDINA E.G., BIGGER J.T., JR, GLASSMAN A.H., PEREL J.M., KANTOR S.J. The electrocardiographic and antiarrhythmic effects of imipramine hydrochloride at therapeutic plasma concentrations. Circulation. 1979;60:1045–1052. doi: 10.1161/01.cir.60.5.1045. [DOI] [PubMed] [Google Scholar]
- GWILT M., ARROWSMITH J.E., BLACKBURN K.J., BURGES R.A., CROSS P.E., DALRYMPLE H.W., HIGGINS A.J. UK-68,798: a novel, potent and highly selective class III antiarrhythmic agent which blocks potassium channels in cardiac cells. J. Pharmacol. Exp. Ther. 1991;256:318–324. [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HO W.K., EARM Y.E., LEE S.H., BROWN H.F., NOBLE D. Voltage- and time-dependent block of delayed rectifier K+ current in rabbit sino-atrial node cells by external Ca2+ and Mg2+ J. Physiol. 1996;494:727–742. doi: 10.1113/jphysiol.1996.sp021528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDEGHEM L.M., SNYDERS D.J. Class III antiarrhythmic agents have a lot of potential but a long way to go: reduced effectiveness and dangers of reverse use-dependence. Circulation. 1990;81:686–690. doi: 10.1161/01.cir.81.2.686. [DOI] [PubMed] [Google Scholar]
- ISENBERG G., TAMARGO J. Effect of imipramine on calcium and potassium currents in isolated bovine ventricular myocytes. Eur. J. Pharmacol. 1985;108:121–131. doi: 10.1016/0014-2999(85)90716-2. [DOI] [PubMed] [Google Scholar]
- MARSHALL J.B., FORKER A.D. Cardiovascular effects of tricyclic antidepressant drugs: therapeutic usage, overdose, and management of complications. Am. Heart J. 1982;103:401–414. doi: 10.1016/0002-8703(82)90281-2. [DOI] [PubMed] [Google Scholar]
- NIES A., ROBINSON D.S., FRIEDMAN M.J., GREEN R., COOPER T.B., RAVARIS C.L., IVES J.O. Relationship between age and tricyclic antidepressant plasma levels. Am. J. Psychiatry. 1977;134:790–793. doi: 10.1176/ajp.134.7.790. [DOI] [PubMed] [Google Scholar]
- NISHIMOTO M., HASHIMOTO H., OZAKI T., TAGUCHI T., OHARA K., NAKASHIMA M. Effects of imipramine and amitriptyline on intraventricular conduction, effective refractory period, incidence of ventricular arrhythmias induced by programmed stimulation, and on electrocardiogram after myocardial infarction in dog. Arch. Int. Pharmacodyn. Ther. 1994;328:39–53. [PubMed] [Google Scholar]
- RAMPE D., ROY M.L., DENNIS A., BROWN A.M. A mechanism for the proarrhythmic effects of cisapride (Propulsid): high affinity blockade of the human cardiac potassium channel HERG. FEBS Lett. 1997;417:28–32. doi: 10.1016/s0014-5793(97)01249-0. [DOI] [PubMed] [Google Scholar]
- ROY M., DUMAINE R., BROWN A.M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990;96:196–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHULZ P., DICK P., BLASCHKE T.F., HOLLISTER L. Discrepancies between pharmacokinetic studies of amitriptyline. Clin. Pharmacokinet. 1985;10:257–268. doi: 10.2165/00003088-198510030-00005. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., CHAUDHARY A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol. Pharmacol. 1996;49:949–955. [PubMed] [Google Scholar]
- SONG D.K., EARM Y.E., HO W.K. Blockade of the delayed rectifier K+ currents, IKr, in rabbit sinoatrial node cells by external divalent cations. Pflugers Arch. 1999;438:147–153. doi: 10.1007/s004240050892. [DOI] [PubMed] [Google Scholar]
- SUESSBRICH H., SCHONHERR R., HEINEMANN S.H., ATTALI B., LANG F., BUSCH A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol. 1997;120:968–974. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUESSBRICH H., WALDEGGER S., LANG F., BUSCH A.E. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- THORSTRAND C. Clinical features in poisonings by tricyclic antidepressants with special reference to the ECG. Acta. Med. Scan. 1976;199:337–344. doi: 10.1111/j.0954-6820.1976.tb06745.x. [DOI] [PubMed] [Google Scholar]
- TRUDEAU M.C., WARMKE J.W., GANETZKY B., ROBERTSON G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VOHRA J., HUNT D., BURROWS G., SLOMAN G. Intercardiac conduction defects following overdose of tricyclic antidepressant drugs. Eur. J. Cardiol. 1975;2:443–452. [PubMed] [Google Scholar]
- WANG S., LIU S., MORALES M.J., STRAUSS H.C., RASMUSSON R.L. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J. Physiol. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARMKE J.W., GANETZKY B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYSE K.R., BURSILL J.A., CAMPBELL T.J. Differential effects of antiarrhythmic agents on post-pause repolarization in cardiac Purkinje fibres. Clin. Exp. Pharmacol. Physiol. 1996;23:825–829. doi: 10.1111/j.1440-1681.1996.tb01187.x. [DOI] [PubMed] [Google Scholar]