

Abstract
Selective inhibitors of cyclo-oxygenase-2 have been shown to be effective anti-inflammatory drugs with reduced gastrointestinal toxicity relative to conventional nonsteroidal anti-inflammatory drugs (NSAIDs). In the present study, we examined the possibility that selective COX-2 inhibition, by blocking prostacyclin synthesis, would increase blood pressure and cause leukocyte adherence and platelet aggregation.
Normal rats and rats with hypertension induced by chronic administration of Nω-nitro-L-arginine methylester were given celecoxib (10 mg kg−1) daily for 3 weeks. Celecoxib significantly elevated of blood pressure in both the normal and hypertensive rats (mean increase of >33 mm Hg after 3 weeks).
In normal rats, celecoxib had no effect on serum 6-keto prostaglandin (PG)F1α levels. Hypertensive rats exhibited a significant increase (82%) in serum 6-keto PGF1α levels, and this was reduced to the levels of normal rats by treatment with celecoxib.
Rats treated with celecoxib exhibited significant increases in weight gain (20%), plasma arginine-vasopressin levels (148%) and plasma urea (69%) relative to vehicle-treated controls. Plasma creatinine levels were unaffected by treatment with celecoxib, while plasma renin levels were significantly decreased (30%) relative to controls.
Superfusion of mesenteric venules with celecoxib (3 μM) in vivo resulted in significant increases in leukocyte adherence to the endothelium in both normal and hypertensive rats.
These studies suggest that suppression of COX-2 significantly influences vascular and/or renal function, leading to elevated blood pressure and leukocyte adherence.
Keywords: Hypertension, cyclo-oxygenase-2, neutrophil adherence, renal function, prostacyclin, endothelium
Introduction
The existence of multiple forms of cyclo-oxygenase, the key enzyme in the biosynthesis of prostaglandins, was first suggested in 1972 (Flower & Vane, 1972). The molecular characterization of two distinct isoforms of this enzyme in 1991 (Kujubu et al., 1991; Xie et al., 1991) led to a burst of activity in the pharmaceutical industry aimed at the development of selective inhibitors of COX-2, the isoform found to be highly expressed at sites of inflammation (Vane et al., 1994; Seibert et al. 1997). Inhibition of COX-2, it was proposed, would result in anti-inflammatory and analgesic properties similar to what can be achieved with conventional nonsteroidal anti-inflammatory drugs (NSAIDs), which block both COX-1 and COX-2 activity. However, by sparing COX-1 activity, the selective COX-2 inhibitors would have greatly reduced toxicity, particularly in the gastrointestinal tract (Xie et al., 1992).
Selective COX-2 inhibitors have now reached the marketplace and data from clinical trials suggest that these agents are effective in treating the symptoms of arthritis while causing less gastrointestinal injury than conventional NSAIDs (Simon et al., 1998). Concerns have been raised, however, that selective COX-2 inhibitors will exacerbate inflammatory conditions in the gastrointestinal tract as well as delay ulcer healing (Reuter et al., 1996; Mizuno et al., 1997; Wallace, 1999). Moreover, there is emerging evidence for important physiological roles of COX-2 in many circumstances, raising the possibility that selective COX-2 inhibition will result in adverse effects that were not initially predicted. For example, McAdam et al. (1999) and Catella-Lawson et al. (1999) recently demonstrated that the selective COX-2 inhibitors, celecoxib and rofecoxib, markedly suppressed prostacyclin synthesis in healthy volunteers when administered at doses in the range presently used by patients with osteoarthritis or rheumatoid arthritis. Prostacyclin is produced mainly by the vascular endothelium and the kidney (Bunting et al., 1983). Its potent vasorelaxant properties contribute to the maintenance of normal vascular tone. Moreover, prostacyclin is a powerful inhibitor of leukocyte and platelet adherence and aggregation (Bunting et al., 1983). Counteracting the effects of endothelial-derived prostacyclin synthesis is the production of thromboxane, via COX-1, by the platelet. Thromboxane is a potent vasoconstrictor and stimulus for platelet aggregation (Bunting et al., 1983). Based on the results of McAdam et al. (1999) and Catella-Lawson et al. (1999), one might speculate that inhibition of endothelial prostacyclin synthesis (COX-2), while sparing platelet thromboxane synthesis (COX-1), could lead to increased vascular tone and increased leukocyte and platelet aggregation.
The aim of the present study was to test the hypothesis that chronic suppression of COX-2 activity through daily administration of celecoxib would lead to increased systemic blood pressure, increased leukocyte adherence and increased platelet aggregation. Studies were carried out in both normal and hypertensive rats. Hypertension was induced through the addition of a nitric oxide synthase inhibitor to the drinking water of the rats. As nitric oxide has several properties that are similar to those of prostacyclin, such as the ability to relax vascular smooth muscle, inhibit platelet aggregation and inhibit leukocyte adherence, it was possible that rats chronically treated with a nitric oxide synthase inhibitor would exhibit increased dependency on the vascular actions of prostacyclin.
Methods
Male, Wistar rats weighing 175–200 g were obtained from Charles River Breeding Farms (Montreal, PQ, Canada). All experimental procedures described below were approved by the Animal Care Committee of the University of Calgary.
Selection of dose of celecoxib
In order to determine a dose of celecoxib (Monsanto, St. Louis, MO, U.S.A) for use in the in vivo studies that exhibited selectivity for COX-2 over COX-1 and was capable of suppressing inflammatory prostaglandin synthesis, the following experiment was performed. An airpouch was induced as described in detail previously (Edwards et al., 1981; Sedgwick et al., 1985; Seibert et al., 1994). Carrageenan (2 ml of a 1% wt vol−1 solution in sterile saline) was injected into the airpouch. Six hours later, the rats were anaesthetized with 5% (vol vol−1) halothane, the pouch opened by a small incision and the exudate was collected. PGE2 concentrations in the exudate were measured using a specific enzyme-linked immunosorbent assay (Wallace et al., 1999). A sample of blood was drawn from the inferior vena cava for measurement of whole blood thromboxane synthesis, as an index of COX-1 activity (Wallace et al., 1999). Groups of five rats each were treated orally with vehicle or celecoxib (1–45 mg kg−1) 1 h prior to the injection of carrageenan into the airpouch. We have previously demonstrated that virtually all of the PGE2 produced in this model is derived from COX-2 (Wallace et al., 1999).
Effects of celecoxib in normotensive rats
Groups of five rats each were orally treated each day with celecoxib (10 mg kg−1) or vehicle. Blood pressure measurements were performed using the tail cuff method (Muscará et al., 1998) prior to initiating treatment and at 1-week intervals for a total of 3 weeks. The rats were conscious during the measurement of blood pressure. Blood pressure measurements were always performed 16–20 h after administration of celecoxib or vehicle. At the end of the study period, 2 h after the final dose of celecoxib or vehicle, the rats were anaesthetized with sodium pentobarbital (65 mg kg−1 i.p.) and a blood sample was drawn from the inferior vena cava for measurement of thromboxane B2 (as an index of COX-1 activity) and serum 6-keto PGF1α levels using specific enzyme-linked immunosorbent assays (Wallace et al., 1999). Serum nitrite and nitrate concentrations were measured by high performance liquid chromatography (Muscará & de Nucci, 1996). Additional rats underwent exactly the same treatment, but blood samples were drawn by cardiac puncture and were used for measurement of plasma renin activity, and plasma levels of endothelin-1 and arginine-vasopressin. For plasma renin activity, blood samples (1 ml) were collected in pre-cooled tubes containing 0.8 mg of EDTA and the plasma was immediately separated by centrifugation (2000×g, 10 min, 4°C). Plasma renin activity was estimated by the generation of angiotensin I from endogenous angiotensinogen (pH 6.5, 37°C, over 4 h), and angiotensin I concentrations were measured by radioimmunoassay. Plasma endothelin-1 and arginine-vasopressin were also measured by radioimmunoassay (all kits from Peninsula Labs, San Diego, CA, U.S.A). Plasma creatinine and urea levels were measured spectrophotometrically (Sigma Chemical Co., St. Louis, MO, U.S.A.).
In vivo responses to endothelin-1 and norepinephrine
Six rats were anaesthetized with sodium pentobarbital (65 mg kg−1 i.p.) and a femoral vein and carotid artery were cannulated for administration of drugs and measurement of blood pressure, respectively. Each rat was given single bolus injections (i.v.) of endothelin-1 (50–20 pmol kg−1) or norepinephrine (0.25–2.5 μg kg−1). After the blood pressure had recovered to basal levels, another injection was performed. The rats were then given celecoxib (10 mg kg−1 i.p.) and 90 min later, the responses to endothelin-1 and norepinephrine were assessed. Thus, in each rat, the responses to a range of doses of each agent were assessed before and after administration of celecoxib. In control rats, which received vehicle in place of celecoxib, the responses to the second round of injections with endothelin-1 and norepinephrine were not significantly different from the responses to the initial injections.
Effects of celecoxib in hypertensive rats
Three groups of 6–7 rats each were studied. In two of the groups, hypertension was induced by addition of a nitric oxide synthase inhibitor, Nω-nitro-L-arginine methylester (L-NAME), to the drinking water at a concentration of 400 mg l−1. The third group received normal drinking water. Two weeks after starting the study, blood pressure was measured using the tail cuff method, as described above. The rats with L-NAME-induced hypertension continued to receive the supplemented drinking water for the following 3 weeks. During that time, one group was treated orally each day with celecoxib (10 mg kg−1), while the other group was treated with vehicle. The group of rats that received normal drinking water also received daily oral treatments with vehicle. Blood pressure measurements were made, as described above, at the end of the second, third, fourth and fifth weeks of the study. These measurements were always made 16–20 h after the administration of celecoxib or vehicle. After the final (fifth week) measurement of blood pressure, the rats were given the final dose of celecoxib or vehicle, and 2 h later were anaesthetized with sodium pentobarbital (65 mg kg−1 i.p.). Blood samples were drawn from the inferior vena cava for measurement of serum thromboxane B2, 6-keto PGF1α and nitrate/nitrite, as described above. The descending aorta, right kidney were excised and processed as described previously (8) for determination of COX-2 mRNA expression by RT–PCR and COX-2 protein expression by Western blotting.
Additional groups of rats (n=5) received L-NAME-supplemented or normal drinking water for 2 weeks and were then sacrificed. Aortic rings were prepared from the thoracic descending aorta and were mounted under 2 g of passive tension in 25 ml organ baths containing a physiological salt solution (37°C, bubbled with 95% O2/5% CO2). The salt solution consisted of (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 12.5 and glucose 11.1. Tissues (endothelium-intact preparations) were allowed to equilibrate for 1 h, then isometric tension generated in response to agonists was measured. Naproxen or celecoxib (10−9–10−5 M) was added cumulatively to the organ bath under different levels of phenylephrine-induced pre-contraction:(1) no phenylephrine; (2) 10–30 nM phenylephrine (10 and 20% of the maximal contractile response), 3) 1–10 μM phenylephrine (maximal contractile response).
Effects of celecoxib on leukocyte adherence
The effects of celecoxib on leukocyte adherence to the vascular endothelium in normal rats and in rats with L-NAME-induced hypertension (2 weeks of L-NAME administration) were examined using an intravital microscopy preparation, as described previously (Muscará et al., 1998). Rats (n=5 per group) were anaesthetized with sodium pentobarbital (65 mg kg−1 i.p.). Images of the mesenteric microcirculation were recorded for 5 min after a 15-min equilibration period. The mesentery was then superfused with bicarbonate-buffered saline containing 3 μM celecoxib or buffer alone and the images were recorded for 5 min beginning 15, 30, 45 and 60 min after the start of the superfusion. This concentration of celecoxib was selected because it is in the range of plasma concentrations required for anti-inflammatory effects in humans (McAdam et al., 1999), and is less than the concentration necessary for significant anti-inflammatory effects in the carrageenan-induced paw oedema model (Smith et al., 1998). Leukocyte adherence was quantified on video playback in a blind manner. A leukocyte was considered adherent to the endothelium if it remained stationary for 30 s or more.
Additional studies were performed to determine if hypertension altered the propensity of leukocytes to adhere the vascular endothelium in response to a different agonist (N-formyl-methionyl-leucine-phenylalanine, FMLP, 5 μM).
Effects of celecoxib on platelet aggregation
Normal rats and rats given L-NAME in the drinking water for 2 weeks were used. The rats were anaesthetized with Halothane and blood was drawn into a syringe from the inferior vena cava. Platelet-rich plasma (PRP) was prepared as described previously (Radomski & Moncada, 1983). Aliquots (0.4 ml) of the PRP were placed in the cuvette of a platelet aggregometer (Payton Instruments, Buffalo, NY, U.S.A.) and continuously stirred at 37°C. After 1 min, celecoxib (3 μM) or vehicle (10 μl) was added to the platelet suspension. One minute later, adenosine diphosphate (ADP; 2.5–40 μM) or thrombin (1–100 u ml−1) was added to the cuvette and aggregation was monitored for the following 5 min.
Statistical analysis
All data are shown as mean±s.e.mean. Differences among groups were analysed using one-way analysis of variance followed by the Student-Neuman-Keuls test for multiple comparisons or, where appropriate, with a paired t-test. Values of probability less than 5% (P<0.05) were considered significant.
Results
Selection of dose of celecoxib
Celecoxib did not significantly affect COX-1 activity, as measured by whole blood thromboxane synthesis, at any of the doses tested (1–45 mg kg−1). However, celecoxib dose-dependently inhibited COX-2 activity (inflammatory PGE2 synthesis). At a doses of 5 and 15 mg kg−1, celecoxib inhibited COX-2 activity by 64±10% and 88±3% (P<0.05), respectively. Thus, a dose of 10 mg kg−1 was selected for subsequent experiments.
Blood pressure studies in normotensive rats
Daily oral administration of celecoxib resulted in a significant and progressive increase in blood pressure in normal rats. The mean increases in blood pressure 1, 2 and 3 weeks after beginning daily treatment with celecoxib were 9, 14 and 30 mmHg, respectively (Figure 1). In contrast, blood pressure did not change significantly in the group treated with vehicle over the 3-week study period.
Figure 1.
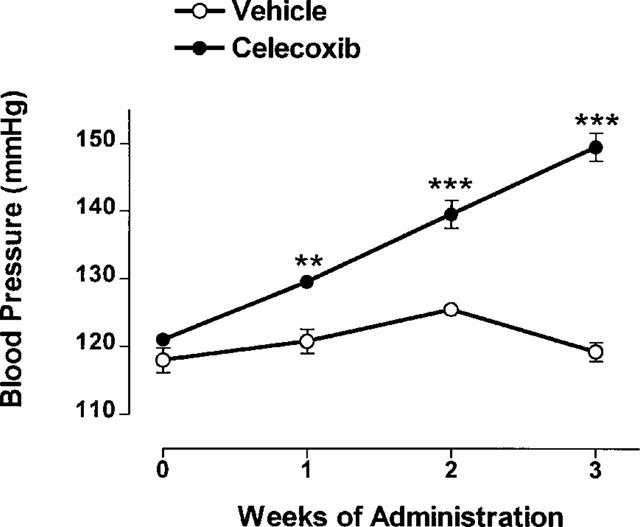
Systemic blood pressure in conscious rats over a 3-week period of daily oral treatment with celecoxib (10 mg kg−1) or vehicle (mean±s.e.mean, n=5 per group). **P<0.01, ***P<0.001 vs the corresponding vehicle-treated group.
Daily treatment with celecoxib for 3 weeks had no significant effect on serum thromboxane B2 or 6-keto PGF1α levels (Figure 2). Also, celecoxib treatment did not significantly affect serum nitrate (31.0±2.0 μM vs 27.7±2.1μM in vehicle-treated) or serum nitrite (1.78±0.07 μM vs 1.66±0.14 μM in vehicle-treated) levels.
Figure 2.
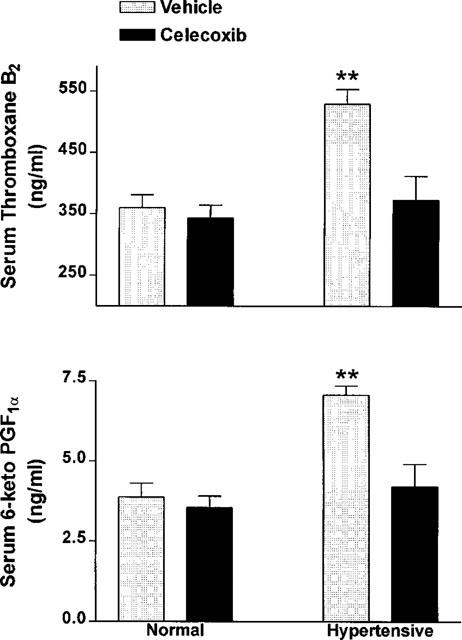
Serum levels of 6-keto prostaglandin F1α and thromboxane B2 in normal and hypertensive rats following 3 weeks of daily oral treatment with celecoxib (10 mg kg−1) or vehicle (mean±s.e.mean, n=5–7 per group). Hypertension was induced through addition of L-NAME to the drinking water. Blood was collected 2 h following the final dose of celecoxib or vehicle. **P<0.01 vs all other groups.
Rats treated daily with celecoxib gained weight at a significantly greater rate than the vehicle-treated controls. At the end of 3 weeks of treatment with celecoxib, the mean increase in body weight was 145.6±6.8 g, approximately 20% more than that in the vehicle-treated group (121.5±11.3 g; P<0.05). Plasma renin activity was significantly reduced in the celecoxib-treated rats (9.5±1.5 ng ml−1 h−1 vs 13.6±1.4 ng ml−1 h−1 in vehicle-treated; P<0.05). On the other hand, plasma levels of arginine-vasopressin were increased in the celecoxib-treated rats (24.1±5.9 pg ml−1 vs 9.7±1.4 pg ml−1 in vehicle-treated; P<0.05). Plasma endothelin-1 levels did not differ between the celecoxib- and vehicle-treated groups (19.7±8.6 pg ml−1 in celecoxib vs 11.4±1.2 pg ml−1 in vehicle). Plasma urea levels were significantly elevated in the celecoxib-treated rats (12.2±1.1 mg dl−1 vs 7.2±0.9 mg dl−1 in controls; P<0.05), while plasma creatinine levels were not different in the celecoxib group (197±86 mg dl−1) in comparison to controls (115±12 mg dl−1). None of the rats in this experiment exhibited gastric damage at the end of the 3-week period of treatment with either celecoxib or vehicle.
Blood pressure studies in hypertensive rats
Addition of L-NAME to the drinking water resulted in a progressive increase in blood pressure. By 2 weeks after beginning this treatment, the mean blood pressure had increased from 122±2 to 168±4 mmHg (Figure 3). In rats treated each day with vehicle for the subsequent 3 weeks, the blood pressure continued to increase, reaching a mean of 198±2 mmHg by the end of the fifth week. Daily treatment with celecoxib resulted in a significant increase in blood pressure over that in the vehicle-treated group at all three time-points, with the greatest increase being seen at the end of the fifth week of the study (mean increase of 33±2 mmHg; P<0.01).
Figure 3.
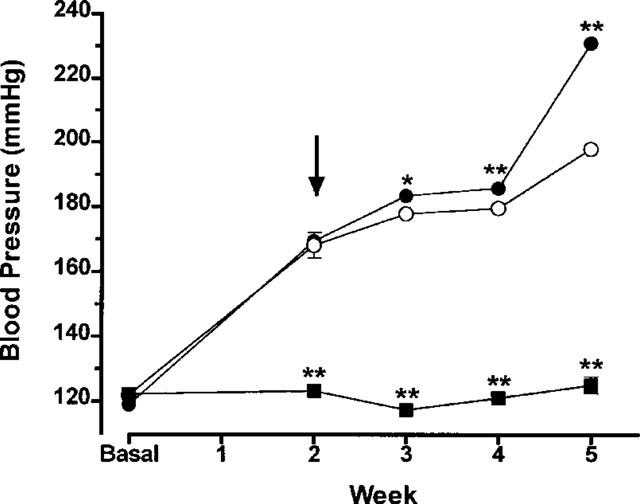
Systemic blood pressure in hypertensive rats over a 3-week period of daily oral treatment with celecoxib (10 mg kg−1, filled circles) or vehicle (open circles) (mean±s.e.mean, n=6–7 per group). Hypertension was induced through addition of L-NAME to the drinking water. Two weeks after beginning treatment with L-NAME (arrow), the rats started receiving celecoxib or vehicle. A control group received normal drinking water (no L-NAME; filled squares). *P<0.05, **P<0.01, vs the L-NAME+vehicle group.
Addition of L-NAME to the drinking water resulted in significant increases in serum levels of both 6-keto PGF1α (∼82%) and thromboxane B2 (∼47%) compared to the group receiving normal drinking water Figure 4). Daily treatment of hypertensive rats with celecoxib resulted in a significant decrease in both 6-keto PGF1α and thromboxane B2 levels, such that they were not different from those in normotensive controls. Gastric damage was not observed in any of the hypertensive rats (vehicle- or celecoxib-treated).
Figure 4.
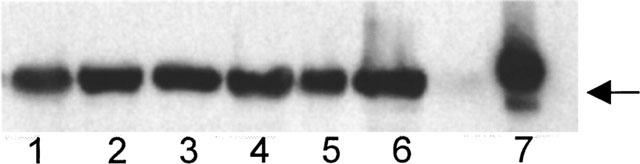
Western blot showing expression of COX-2 in dorsal aorta of normal rats (lanes 1–3) and rats that received L-NAME in the drinking water for 3 weeks (lanes 4–6). Lane 7 is a positive control (COX-2 from a mouse macrophage cell line; RAW 264.7). The monoclonal antibody used was a mouse anti-rat COX-2 from Transduction Labs (Lexington, KY, U.S.A.). Arrow indicates 70 kDa.
COX-2 mRNA and protein was constitutively expressed in the descending aorta and kidney of normal rats. Addition of L-NAME to the drinking water did not significantly change the expression of COX-2 mRNA or protein in these tissues relative to the control group (Figure 4).
In vitro contractility of descending aorta
Neither naproxen nor celecoxib (10−9–10−5 M) had any direct effect on the contractility of aortic rings prepared from either normotensive or hypertensive rats. The lack of effect of the two drugs was seen across the full range of pre-contraction with phenylephrine.
In vivo responses to endothelin and norepinephrine
Endothelin administration causes an early hypotensive response, followed by a more pronounced and long-lasting hypertensive response. Pretreatment with celecoxib resulted in a significant attenuationtion of the hypotensive response to endothelin. For example, in vehicle-treated rats, endothelin-1 at doses of 100 and 250 pmol kg−1 caused decreases in mean arterial pressure of 12±3 and 26±5%, respectively. In rats pretreated with celecoxib, these responses were significantly reduced to 5±4 and 14±4%, respectively (P<0.01). Pretreatment with celecoxib also significantly amplified the hypertensive response to endothelin. For example, at a dose of 50 pmol kg−1, mean arterial pressure increased by 28±4% in vehicle-treated rats, significantly (P<0.05) greater than the 11±3% increase in celecoxib-treated rats.
In contrast to the altered responsiveness to endothelin, pretreatment with celecoxib did not significantly affect the hypertensive responses observed following administration of norepinephrine. For example, the increases of mean arterial pressure in vehicle-treated rats produced with 0.25 and 2.5 μg kg−1 doses of norepinephrine were 42±6 and 69±9%, respectively. In celecoxib-treated rats, the responses to these doses of norepinephrine were 34±5 and 73±13%, respectively.
Leukocyte adherence
In normal rats under basal conditions, there were typically two to four adherent leukocytes per 100 μm length of mesenteric venule (Figure 5). During a 60-min superfusion with celecoxib (3 μM), there was a significant increase in the number of adherent leukocytes. The peak level of leukocyte adherence was observed after 45 min of superfusion with celecoxib, with a mean increase of approximately 6 fold over basal levels.
Figure 5.
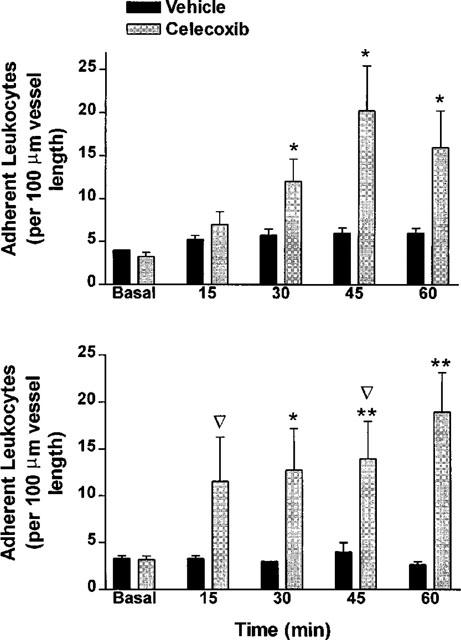
Leukocyte adherence to post-capillary mesenteric venules before and during superfusion of the vessels with celecoxib (3 μM) or vehicle in normotensive (top panel) and L-NAME-induced hypertensive (bottom panel) rats. The data are expressed as mean±s.e.mean (n=5–6 per group). *P<0.05, **P<0.01 vs the corresponding vehicle-treated group. ▿ At the time point indicated by this symbol, blood flow through the vessel stopped in one of the rats in the group treated with celecoxib.
In hypertensive rats, basal levels of leukocyte adherence were similar to those observed in the normal rats. Also in similarity to the studies of normal rats, superfusion with celecoxib resulted in a marked increase in leukocyte adherence (Figure 5). A significant increase in leukocyte adherence was detected as early as 15 min after exposure to celecoxib. In hypertensive rats, flow through the superfused vessel ceased in two of the five rats prior to completion of the experiment (Figure 5). This contrasted to the situation in normal rats, where cessation of blood flow was not observed in any of the five rats.
The diameter of the vessels examined by intravital microscopy did not change in normotensive or hypertensive rats during the superfusion with buffer or celecoxib. For example, in normal rats in which celecoxib was superfused over the mesenteric venules, the diameter at the end of the 60-min superfusion was 106±10% of the initial diameter. In the case of hypertensive rats, the final diameter in the three rats in which flow did not stop was 102±2% of the initial diameter. Even in the two rats in which flow stopped during the celecoxib superfusion, there was no significant change in vessel diameter.
Superfusion of mesenteric venules with FLMP also caused a significant increase in leukocyte adherence (Figure 6). The magnitude of the increase in leukocyte adherence in hypertensive rats was significantly greater than that observed in normotensive rats. Blood flow through the mesenteric venules did not stop in any of the normotensive or hypertensive rats during the 60-min superfusion with FMLP.
Figure 6.
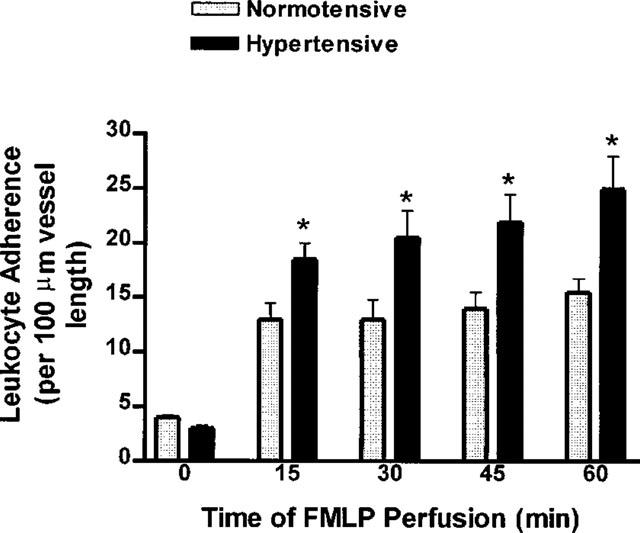
Leukocyte adherence to post-capillary mesenteric venules of normal and L-NAME-induced hypertensive rats before and during superfusion of the vessels with N-formyl-methionine-leucine-phenylalanine (FMLP; 5 μM) or vehicle. In both groups, the levels of leukocyte adherence were significantly elevated (P<0.05) over basal levels at all times after starting the superfusion with FMLP. Results are expressed as the mean±s.e.mean (n=5–6 per group). *P<0.05 vs the corresponding data from normotensive rats.
Platelet aggregation
ADP and thrombin caused concentration-dependent aggregation of rat platelets in vitro. The responses of platelets from rats that had received L-NAME in the drinking water for 2 weeks did not differ from the responses observed with platelets from normal rats. Pre-incubation of the platelets with celecoxib (3 μM) for 1 min prior to stimulation with ADP or thrombin did not significantly affect the magnitude of aggregatory responses.
Discussion
Selective inhibition of COX-2 has been shown to result in markedly less gastrointestinal ulceration than is seen with conventional NSAIDs (Boyce et al., 1994; Vane, 1994; Simon et al., 1998). Originally, selective COX-2 inhibition was also proposed to represent a renal-sparing approach to anti-inflammatory therapy (Vane, 1994). However, a number of studies have now documented constitutive expression of COX-2 in the kidney of humans and several other species, as well as the importance of COX-2 in regulating renal function (Harris et al., 1994; Harding et al., 1997; Komhoff et al., 1997). Moreover, two recent reports highlighted the possibility that selective inhibition of COX-2 may, through inhibition of prostacyclin synthesis, promote thrombogenesis (McAdam et al., 1999; Catella-Lawson et al., 1999). In the present study, celecoxib at an anti-inflammatory dose that is selective for COX-2 significantly increased systemic blood pressure in healthy rats and in rats with pre-existing hypertension. This was not a transient effect of celecoxib, since blood pressure measurements were performed 16–20 h after the final dose of the drug. Celecoxib also significantly increased leukocyte adherence to post-capillary mesenteric venules when superfused over those vessels at a clinically relevant concentration. Thus, the results of the present study suggest that COX-2 can play an important role in terms of modulating blood pressure and adhesive interactions between the vascular endothelium and circulating leukocytes.
Our initial hypothesis was that celecoxib would increase blood pressure by virtue of suppression of vascular prostacyclin synthesis. McAdam et al. (1999) found that doses of celecoxib in the range used for treatment of rheumatoid arthritis were capable of suppressing urinary excretion of prostacyclin metabolites in healthy volunteers by more than 80%. In the present study, however, celecoxib did not significantly affect serum levels of 6-keto PGF1α yet did markedly elevate systemic blood pressure. In rats with L-NAME-induced hypertension, serum levels of 6-keto PGF1α were significantly elevated above those of controls. The finding that celecoxib treatment reduced serum 6-keto PGF1α levels to those of normotensive controls indicates that the increased production of this mediator occurred via COX-2. We could not detect a significant change in COX-2 mRNA or protein expression in descending aorta or kidney following administration of L-NAME for 3 weeks, but we cannot rule out the possibility that such an induction occurred in the smaller, resistance vessels. On the other hand, the elevated production of prostacyclin could have occurred as a consequence of increased COX-2 activity or increased substrate availability, rather than being due to a change in COX-2 expression. Suppression of prostacyclin synthesis by celecoxib may have contributed to the increased blood pressure in the L-NAME-treated rats. We have previously found that a conventional NSAID, naproxen, could significantly elevate blood pressure in normal rats and in rats receiving L-NAME in the drinking water, and in both cases, was associated with significantly reduced prostacyclin synthesis (Muscará et al., 1998). Thus, chronic suppression of nitric oxide synthesis may result in a greater dependence on prostacyclin synthesis in terms of maintenance of vascular tone.
While we did not detect any effect on celecoxib on serum levels of 6-keto PGF1α, celecoxib did alter vascular reactivity. Endothelin-1 administration results in an early hypotensive response, which is mediated via the release of prostacyclin (De Nucci et al. 1988), followed by a pronounced pressor response. Acute celecoxib administration significantly attenuated the hypotensive response to endothelin-1, while significantly enhancing the pressor response. A similar effect can be produced through acute administration of a conventional NSAID, naproxen (unpublished observation). Thus, it is likely that suppression of endothelial prostacyclin synthesis by celecoxib accounts for the effects of this drug on vascular responiveness to endothelin-1. Celecoxib did not directly cause vascular contractile responses in vitro, nor did it alter the pressor response to norepinephrine in vivo.
The hypertensive effects of celecoxib may have been due to effects of this drug on the kidney. COX-2 is constitutively expressed in the macula densa and thick ascending limb of the rat (Wang et al., 1999), and also constitutively expressed in the human kidney (Komhoff et al., 1997). COX-2 has been suggested to play a role in regulating renin release (Harris et al., 1994; Harding et al., 1997) and, as confirmed in the present study, selective inhibition of COX-2 in the rat results in a reduction in plasma renin activity (Wang et al., 1999). It is interesting that the rats treated once-daily with celecoxib for 3 weeks exhibited significantly increased weight gain, consistent with fluid retention. Moreover, celecoxib treatment caused a 2.5 fold increase in plasma levels of arginine-vasopressin, which would contribute to fluid retention. The significant elevation of plasma urea together with unchanged plasma levels of creatinine suggest the celecoxib interfered with the normal excretion of urea, independent of generalized effects on renal blood flow or glomerular filtration rate.
Leukocyte adhesion is normally down-regulated by the release from the endothelium of nitric oxide and prostaglandins; thus, inhibition of nitric oxide synthesis or prostaglandin synthesis results in a significant increase in leukocyte adherence (Kubes et al., 1991; Wallace et al., 1993). In the present study, superfusion of mesenteric venules with celecoxib resulted in a marked increase in the numbers of leukocytes adhering to the vascular endothelium, similar to what has previously been observed with conventional NSAIDs (Asako et al., 1992; Wallace et al., 1993). The increase in leukocyte adherence observed following NSAID administration has been suggested to be due, at least in part, to a rapid up-regulation of ICAM-1 expression on the vascular endothelium (Wallace et al., 1993). Interestingly, COX-2 has been shown to down-regulate ICAM-1 (and VCAM-1) expression on cultured human vascular smooth muscle cells, and has been suggested to play ‘a protective role in cardiovascular and inflammatory processes' (Bishop-Bailey et al., 1998). In rats that had received L-NAME in the drinking water for the previous 2 weeks, the increase in leukocyte adherence during superfusion with celecoxib occurred more rapidly than in normal rats, and was accompanied in some rats by cessation of flow through the vessel being studied. Enhanced leukocyte adherence in the L-NAME treated rats was also seen when FMLP was used as an agonist, rather than celecoxib. However, cessation of blood flow was not observed when FMLP was used, even though a greater level of leukocyte adherence was observed than in the celecoxib experiments. Despite causing cessation of blood flow in some rats, celecoxib did not affect vessel diameter. Thus, it would appear that in a setting of diminished nitric oxide synthesis, celecoxib produced changes in the vasculature, independent of alterations in vessel diameter that led to impairment of flow. One possibility was that celecoxib increased the propensity of platelets to aggregate. However, the in vitro studies of platelet aggregation suggested that platelet reactivity was not significantly affected by prior exposure to celecoxib, consistent with previous reports that this drug does not alter platelet aggregation (McAdam et al., 1999).
In summary, a major limitation to the use of conventional NSAIDs is their interference with the effectiveness of anti-hypertensive therapy. NSAIDs are used primarily by the elderly, the same group that is most likely to be using anti-hypertensive therapy. Selective COX-2 inhibitors are an attractive alternative to conventional NSAIDs because of their reduced toxicity in the gastrointestinal tract. However, the results of the present study suggest that effects of selective COX-2 inhibitors on prostacyclin synthesis and on renal function could result in detrimental effects on blood pressure regulation similar to what is seen with conventional NSAIDs. Moreover, selective COX-2 inhibitors, while not affecting platelet aggregation, can promote the adherence of leukocytes to the vascular endothelium. Taken together, these results suggest that studies of these potential adverse effects of selective COX-2 inhibitors in humans are warranted and that, until such studies are performed, caution should be exercised in using these drugs in patients pre-disposed to hypertension and thrombosis.
Acknowledgments
This work was supported by a grants from the Heart and Stroke Foundation of Canada and the MRC of Canada. Dr M.N. Muscará is supported by a Merck Pharmacology Fellowship. Dr J.K. Wallace is supported by a MRC Senior Scientist award, and by an Alberta Heritage Foundation for Medical Research Senior Scientist award.
Abbreviations
- COX
cyclo-oxygenase
- L-NAME
Nω-nitro-L-arginine methylester
- mRNA
messenger ribonucleic acid
- NSAID
nonsteroidal anti-inflammatory drug
- PG
prostaglandin
- RT–PCR
reverse transcriptase polymerase chain reaction
References
- ASAKO H., KUBES P., WALLACE J.L., WOLF R.E., GRANGER D.N. Modulation of leukocyte adherence in rat mesenteric venules by aspirin and salicylate. Gastroenterology. 1992;103:146–152. doi: 10.1016/0016-5085(92)91107-f. [DOI] [PubMed] [Google Scholar]
- BISHOP-BAILEY D., BURKE-GAFFNEY A., HELLEWELL P.G., PEPPER J.R., MITCHELL J.A. Cyclo-oxygenase-2 regulates inducible ICAM-1 and VCAM-1 expression in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998;249:44–47. doi: 10.1006/bbrc.1998.8966. [DOI] [PubMed] [Google Scholar]
- BOYCE S., CHAN C.C., GORDON R., LI C.-S., RODGER I.W., WEBB J.K., RUPNIAK N.M.J., HILL R.G. L-745,337: A selective inhibitor of cyclooxygenase-2 elicits antinociception but not gastric ulceration in rats. Neuropharmacology. 1994;33:1609–1611. doi: 10.1016/0028-3908(94)90137-6. [DOI] [PubMed] [Google Scholar]
- BUNTING S., MONCADA S., VANE J.R. The prostacyclin–thromboxane A2 balance: pathophysiological and therapeutic implications. Br. Med. Bull. 1983;39:271–276. doi: 10.1093/oxfordjournals.bmb.a071832. [DOI] [PubMed] [Google Scholar]
- CATELLA-LAWSON F., MCADAM B., MORRISON B.W., KAPOOR S., KUJUBU D., ANTES L., LASSETER K.C., QUAN H., GERTZ B.J., FITZGERALD G.A. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharmacol. Exp. Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- DE NUCCI G., THOMAS R., D'ORLEANS-JUSTE P., ANTUNES E., WALDER C., WARNER T.D., VANE J.R. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin. Proc. Natl. Acad. Sci. U.S.A. 1988;85:9797–9800. doi: 10.1073/pnas.85.24.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS J.C.W., SEDGWICK A.D., WILLOUGHBY D.A. The formation of a structure with features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J. Pathol. 1981;134:147–156. doi: 10.1002/path.1711340205. [DOI] [PubMed] [Google Scholar]
- FLOWER R.J., VANE J.R. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamide-phenol) Nature New Biol. 1972;240:410–411. doi: 10.1038/240410a0. [DOI] [PubMed] [Google Scholar]
- HARDING P., SIGMON D.H., ALFIE M.E., HUANG P.L., FISHMAN M.C., BEIERWALTES W.H., CARRETERO O.A. Cyclooxygenase-2 mediates increased renal content induced by low-sodium diet. Hypertension. 1997;29:297–302. doi: 10.1161/01.hyp.29.1.297. [DOI] [PubMed] [Google Scholar]
- HARRIS R.C., MCKANNA J.A., AKAI Y., JACOBSON H.R., DUBOIS R.N. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J. Clin. Invest. 1994;94:2504–2510. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMHOFF M., GRONE H.J., KLEIN T., SEYBERTH H.W., NUSING R.M. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am. J. Physiol. 1997;272:F460–F468. doi: 10.1152/ajprenal.1997.272.4.F460. [DOI] [PubMed] [Google Scholar]
- KUBES P., SUZUKI M., GRANGER D.N. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUJUBU D.A., FLETCHER B.S., VARNUM B.C., LIM R.W., HERSCHMAN H.R. TIS10, a phorbol ester tumor promoter-inducible mRNA from swiss eTe cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- MCADAM B.F., CATELLA-LAWSON F., MARDINI I.A., KAPOOR S., LAWSON J.A., FITZGERALD G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: The human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. U.S.A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZUNO H., SAKAMOTO C., MATSUDA K., WADA K., UCHIDA T., NOGUCHI H., AKAMATSU T., KASUGA M. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112:387–397. doi: 10.1053/gast.1997.v112.pm9024292. [DOI] [PubMed] [Google Scholar]
- MUSCARÁ M.N., DE NUCCI G. Simultaneous determination of nitrite and nitrate anions in plasma, urine and cell culture supernatants by high-performance liquid chromatography with post column reactions. J. Chromatogr. B Biomed. Appl. 1996;686:157–164. doi: 10.1016/s0378-4347(96)00229-0. [DOI] [PubMed] [Google Scholar]
- MUSCARÁ M.N., MCKNIGHT W., DEL SOLDATO P., WALLACE J.L. Effect of a nitric oxide-releasing naproxen derivative on hypertension and gastric damage induced by chronic nitric oxide inhibition in the rat. Life Sci. 1998;62:PL235–PL240. doi: 10.1016/s0024-3205(98)00072-1. [DOI] [PubMed] [Google Scholar]
- RADOMSKI M., MONCADA S. An improved method for washing of human platelets with prostacyclin. Thromb. Res. 1983;30:383–389. doi: 10.1016/0049-3848(83)90230-x. [DOI] [PubMed] [Google Scholar]
- REUTER B.K., ASFAHA S., BURET A., SHARKEY K.A., WALLACE J.L. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J. Clin. Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEDGWICK A.D., MOORE A.R., AL-DUAIJ A.Y., EDWARDS J.C., WILLOUGHBY D.A. Studies into the influence of carrageenan-induced inflammation on articular cartilage degradation using implantation into air pouches. Br. J. Exp. Pathol. 1985;66:445–453. [PMC free article] [PubMed] [Google Scholar]
- SEIBERT K., ZHANG Y., LEAHY K., HAUSER S., MASFERRER J., ISAKSON P. Distribution of COX-1 and COX-2 in normal and inflamed tissues. Adv. Exp. Med. Biol. 1997;400:167–170. doi: 10.1007/978-1-4615-5325-0_24. [DOI] [PubMed] [Google Scholar]
- SEIBERT K., ZHANG Y., LEAHY K., HAUSER S., MASFERRER J., PERKINS W., LEE L., ISAKSON P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMON L.S., LANZA F.L., LIPSKY P.E., HUBBARD R.C., TALWALKER S., SCHWARTZ B.D., ISAKSON P.C., GEIS G.S. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor - Efficacy and safety in two placebo-controlled trials in osteoarthritis and rheumatoid arthritis, and studies of gastrointestinal and platelet effects. Arthritis Rheum. 1998;41:1591–1602. doi: 10.1002/1529-0131(199809)41:9<1591::AID-ART9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- SMITH C.J., ZHANG Y., KOBOLDT C.M., MUHAMMAD J., ZWEIFEL B.S., SHAFFER A., TALLEY J.J., MASFERRER J.L., SEIBERT K., ISAKSON P.C. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANE J.R. Towards a better aspirin. Nature. 1994;367:215–216. doi: 10.1038/367215a0. [DOI] [PubMed] [Google Scholar]
- VANE J.R., MITCHELL J.A., APPLETON I., TOMLINSON A., BISHOP-BAILEY D., CROXTALL J., WILLOUGHBY D.A. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc. Natl. Acad. Sci. U.S.A. 1994;91:2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACE J.L. Selective COX-2 inhibitors: is the water becoming muddy. Trends Pharmacol. Sci. 1999;20:4–6. doi: 10.1016/s0165-6147(98)01283-8. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., CHAPMAN K., MCKNIGHT W. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. Br. J. Pharmacol. 1999;126:1200–1204. doi: 10.1038/sj.bjp.0702420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALLACE J.L., MCKNIGHT W., MIYASAKA M., TAMATANI T., PAULSON J., ANDERSON D.C., GRANGER D.N., KUBES P. Role of endothelial adhesion molecules in NSAID-induced gastric mucosal injury. Am. J. Physiol. 1993;265:G993–G998. doi: 10.1152/ajpgi.1993.265.5.G993. [DOI] [PubMed] [Google Scholar]
- WANG J.L., CHENG H.R., HARRIS R.C. Cyclooxygenase-2 inhibition decreases renin content and lowers blood pressure in a model of renovascular hypertension. Hypertension. 1999;34:96–101. doi: 10.1161/01.hyp.34.1.96. [DOI] [PubMed] [Google Scholar]
- XIE W., CHIPMAN J.G., ROBERTSON D.L., ERIKSON R.L., SIMMONS D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE W., ROBERTSON D.L., SIMMONS D.L. Mitogen-inducible prostaglandin G/H synthase: A new target for nonsteroidal antiinflammatory drugs. Drug Dev. Res. 1992;25:249–265. [Google Scholar]