

Abstract
Nine bis-quinolinyl and bis-quinolinium compounds related to dequalinium, and previously shown to block apamin-sensitive small conductance Ca2+-activated K+ channels (SKCa), have been tested for their inhibitory effects on actions mediated by intermediate conductance Ca2+-activated K+ channels (IKCa) in rabbit blood cells.
In most experiments, a K+-sensitive electrode was employed to monitor the IKCa-mediated net loss of cell K+ that followed the addition of the Ca2+ ionophore A23187 (2 μM) to red cells suspended at an haematocrit of 1% in a low K+ (0.12–0.17 mM) solution. The remainder used an optical method based on measuring the reduction in light transmission that occurred on applying A23187 (0.4 or 2 μM) to a very dilute suspension of red cells (haematocrit 0.02%).
Of the compounds tested, the most potent IKCa blocker was 1,12 bis[(2-methylquinolin-4-yl)amino]dodecane (UCL 1407) which had an IC50 of 0.85±0.06 μM (mean±s.d.mean).
The inhibitory action of UCL 1407 and its three most active congeners was characterized by (i) a Hill slope greater than unity, (ii) sensitivity to an increase in external [K+], and (iii) a time course of onset that suggested use-dependence. Also, the potency of the nonquaternary compounds tested increased with their predicted lipophilicity. These findings suggested that the IKCa blocking action resembles that of cetiedil rather than of clotrimazole.
Some quaternized members of the series were also active. The most potent was the monoquaternary UCL 1440 ((1-[N-[1-(3,5-dimethoxybenzyl)-2-methylquinolinium-4-yl]amino]-10-[N′-(2-methylquinolinium-4yl)amino] decane (trifluoroacetate) which had an IC50 of 1.8±0.1 μM. The corresponding bisquaternary UCL 1438 (1,10-bis[N-[1-(3,5-dimethoxybenzyl)-2-methylquinolinium-4-yl]amino] decane bis(trifluoroacetate) was almost as active (IC50 2.7±0.3 μM).
A bis-aminoquinolium cyclophane (UCL 1684) had little IKCa blocking action despite its great potency at SKCa channels (IC50 4.1±0.2 nM).
The main outcome is the identification of new intermediate-conductance Ca2+-activated K+ channel blockers with a wide range of IKCa/SKCa selectivities.
Keywords: Ca2+-activated K+ channel, IKCa blocker, SKCa blocker, UCL 1684, UCL 1407, clotrimazole, rabbit blood cells, superior cervical ganglion, bisquinolinium compounds, intermediate conductance K+ channel
Introduction
Calcium-activated potassium channels are found in cells as diverse as central and peripheral neurones, the parenchymal cells of the liver, and erythrocytes (for reviews see Sarkadi & Gárdos, 1985; Haylett & Jenkinson, 1990; Sah, 1996; Vergara et al., 1998; Castle, 1999). Three main types (BKCa, IKCa and SKCa) have been identified, and each has now been cloned (Kohler et al., 1996; Ishii et al., 1997b; Joiner et al., 1997; Logsdon et al., 1997: see also Vergara et al., 1998; Jensen et al., 1998). Furthermore, earlier functional, binding and structural studies which suggested that there are several kinds of SKCa channel (Lancaster & Adams, 1986; Wadsworth et al., 1994; Dunn et al., 1996) have been confirmed by DNA cloning that has revealed the existence of SK1, SK2 and SK3 subtypes (reviewed by Vergara et al., 1998).
Despite close structural similarities, the IKCa and SKCa channels differ not only in their conductance and gating but also in pharmacology. For example, the bee venom toxin apamin has long been known (Burgess et al., 1981) to block the Ca2+-activated K+ permeability (PK(Ca)) found in parenchymal hepatocytes, and mediated by SKCa channels (Capiod & Ogden, 1989), but not the PK(Ca) that occurs in red cells (Gárdos, 1958) and which involves IKCa channels (Grygorczyk & Schwarz, 1983; Christophersen, 1991; Leinders et al., 1992a,1992b; Dunn, 1998). Accordingly, apamin has proved a valuable tool for the study of the properties and physiological role of SKCa channels. Tubocurarine (Jenkinson et al., 1983; Nohmi & Kuba, 1984; Cook & Haylett, 1985; Ishii et al., 1997a) and more recently dequalinium (Castle et al., 1993; Dunn, 1994) have also been used for this purpose. A detailed examination of the steric and electronic features that determine the SKCa blocking action of dequalinium and its congeners has led to the development of novel non-peptidic blockers, some of which are as active as apamin (see e.g. Galanakis et al., 1996; Campos Rosa et al., 1998; Dunn, 1999; Benton et al., 1999a). The present study is concerned with the SKCa/IKCa selectivity of these new blockers. We have measured the ability of a representative subset of them to inhibit the IKCa-mediated PK(Ca) of red blood cells, in order to compare this with their SKCa-blocking action which has already been reported (Galanakis et al., 1996; Campos Rosa et al., 1998). In that work, the event of SKCa block was assessed from the reduction in the amplitude of the apamin-sensitive after-hyperpolarization (AHP) that follows the action potential in rat superior cervical ganglion (SCG) neurones. This AHP is thought to involve the SK3 subtype of SKCa channel (Hosseini et al., 1999). In the present study, IKCa inhibition was evaluated from the ability of the compounds to reduce the net loss of K+ that follows PK(Ca) activation by the application of the Ca2+ ionophore (A23187) to red blood cells (see Benton et al., 1999b). Our results show that whereas some of the compounds resemble apamin in being highly SKCa/IKCa selective, others have the potentially useful property of blocking both types of channel.
Methods
Preparation of red blood cells
The procedure used to prepare rabbit blood cells has recently been described in detail (Benton et al., 1999b). Briefly, heparinized blood drawn from an ear vein of an adult New Zealand White rabbit was centrifuged at 1200 × g for 2–3 min and the supernatant and buffy coat aspirated and discarded. The packed cells were resuspended and washed three times in a solution containing (mM): NaCl 145, KCl 0.1, MgSO4 1, EDTA 1, TRIS HCl 10, inosine 10; pH adjusted to 7.4 with 1 M NaOH. They were stored as a pellet in this solution at 4°C for up to 3 days.
Measurement of K+ loss from red cells exposed to a Ca+ ionophore
In most experiments a valinomycin-based K+-sensitive electrode was used to monitor the rise in extracellular [K+] that resulted from the IKCa-mediated net loss of K+ which followed the application of the Ca2+ ionophore A23187 to a suspension of red cells. Details of the procedures, including the construction of the electrodes, have been given in Benton et al. (1999b). As in that study, most of the experiments were performed at 35–37°C in a low K+ solution containing (mM): NaCl 145, KCl 0.1, MgSO4 1, CaCl2 1, inosine 10, TRIS HCl 10. The pH was adjusted to 7.4 by addition of 1 M NaOH. There were two reasons for the choice of a low-K+ solution. First, as is shown, the blocking agents tested became more effective when the external K+ was reduced so that it was then possible to use lower concentrations of the compounds, some of which were available in limited quantity. Second, when using a K+-sensitive electrode, small increases in the K+ content of the external solution resulting from the loss of cell K+ are more easily detected when [K+]o is initially low.
A 2 ml sample of the standard solution was placed in the recording chamber containing the K+-sensitive and reference electrodes. Twenty μl of packed red cells was then added to give a final haematocrit of just under 1%. Because of the K+ content of the fluid adhering to the cells, their addition caused the concentration of K+ to rise to 0.12–0.17 mM, as indicated by the signal from the K+-sensitive electrode. When the reading from the electrode had become steady, K+ loss from the cells was initiated by the addition of a supramaximal concentration of A23187 (2 μM, added as 4 μl of a 1 mM solution in DMSO) to the cell suspension. This caused a rapid release of K+. Three minutes after the application of A23187, digitonin (100 μM) was added to lyse the cells and so allow the total available K+ to be estimated. The magnitude of the K+ loss initiated by A23187 could then be calculated as the increase in [K+]o 3 min after addition of A23187, expressed as a percentage of the total increase after addition of digitonin. This is equivalent to the quantity of K+ released by A23187 as a percentage of the total K+ content of the cells.
The inhibitory effects of IKCa-blocking drugs were tested by adding a small volume (usually <5 μl) of a concentrated stock solution in DMSO to the cell suspension for a preincubation period of 3 min before applying A23187. The amount of K+ lost in the presence of the drug was then compared with that in its absence, and the degree of inhibition was expressed as a percentage.
A limitation of the method was that the presence of quaternary ammonium ions can interfere with the function of valinomycin-based K+ sensitive electrodes. It was therefore necessary when testing IKCa blocking agents containing quaternized nitrogens to recalibrate the electrodes in solutions containing each concentration of the compound to be studied.
A second technique that did not suffer from this disadvantage, and which also made it easier to use solutions with the normal concentration of K+, was employed in the remaining experiments. It was based on the work of Alvarez et al. (1986; 1992) who showed that changes in the absorption of light by a suspension of red cells could provide a convenient, albeit indirect, measure of the IKCa-initiated loss of K+, Cl− and water, leading to cell shrinkage, that follows the application of a Ca2+ ionophore such as A23187. In our experiments, sufficient red cells to give a haematocrit of 0.02% were added to either 2 or 5 ml of the standard bathing solution in a glass test-tube. This was maintained at 35–37°C and mounted in the light path of a photometer (Vitatron UPS) to which a magnetic stirrer had been fitted. The photometer output was fed to a pen recorder to provide a continuous measure on a linear scale of the percentage of the incident light (692 nm) transmitted by the suspension. Initially this was set to 100%. Addition of the red cells caused a drop to 73–82%. The transmittance scale of the photometer was then reset to 100%. Transmission was monitored for 2–3 min before the addition of the compound to be tested, followed 3 min later by A23187 (at either 2 μM or, in later experiments, 0.4 μM added in 2 μl DMSO). The decrease in transmission 3 min after the application of A23187 in the presence of the blocker was expressed as a percentage of that in its absence and the degree of inhibition was calculated by subtracting this percentage from 100.
Inhibition of the after-hyperpolarization recorded from rat sympathetic neurones
In a few experiments, action potentials were recorded from isolated sympathetic neurones using the methods described in detail by Dunn (1994). Briefly, rat superior cervical ganglia from 17 day old rats were dissociated using collagenase followed by trypsin. The isolated neurones were maintained in tissue culture for 5–7 days. Intracellular microelectrodes filled with 1 M KCl, and with a tip resistance of 100–140 MΩ, were used to record action potentials elicited every 5 s by a 20–40 ms depolarizing current pulse passed through the electrode. The bathing fluid in these experiments contained (mM): NaCl 118, KCl 3.8, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, glucose 11, and was equilibrated with a 95% O2, 5% CO2 gas mixture. Drugs were applied in a continuously flowing solution, maintained at 30–31°C, as described by Dunn (1994). Their effect on the after-hyperpolarization (AHP) that followed the action potential was assessed by expressing the AHP in the presence of the drug as a percentage of the control value, measured at a time when the inhibition was maximal.
Data analysis
Values are given as the mean±s.e.mean except when the Hill equation was fitted to concentration-response data. This was done using a weighted least-squares minimization program, either CVFIT (written by Professor D. Colquhoun, Department of Pharmacology, University College London, and available from www.ucl.ac.uk/pharmacol/dc.html) or Origin 5.0 (Microcal, Northampton, MA, U.S.A.). Both programs provided estimates of the molar concentration (the IC50) of the compounds that caused 50% of the maximum attainable reduction in the measured response, and of the Hill coefficient (nH), ± an approximate standard deviation for each parameter (see Colquhoun et al., 1974, for details). The estimates of the parameters, and of their errors, given by the programs agreed closely provided that the ‘Use Chi-squared formula for errors' option in Origin 5.0 was not selected.
When comparing the effects of changes in external K+ on the potency of the compounds, the same programs were used to estimate the factors (equivalent to the equi-effective molar ratios) by which the concentration of the compound had to be altered to achieve the same inhibition under the new experimental conditions.
Drugs and reagents
The UCL-prefixed compounds were synthesized in the Department of Chemistry at University College London by methods described elsewhere: for UCL 1407 (1,12 bis[(2-methylquinolin-4-yl)amino]dodecane), see compound 2 in Galanakis et al. (1996); for UCL 1144 (4,4′-[decane-1,10-diyl-bis(oxy)]bis[quinoline] see compound 4 loc. cit.; for UCL 1440 (1-[N-[ 1-(3, 5-dimethoxybenzyl) - 2 - methylquinolinium - 4 - yl] amino]-10-[N′-(2-methylquinolinium-4yl)amino] decane bis (tri-fluoroacetate) see compound 19 loc. cit.; for UCL1417 (1,10-bis[(2-methylquinolin-4-yl)amino]decane) see compound 1 loc. cit.; for UCL1438 (1,10-bis[N-[1-(3,5-dimethoxybenzyl)-2-methylquinolinium-4-yl]amino] decane bis(trifluoroacetate) see compound 18 loc. cit.; for UCL 1091 (1,10-bis(N-quinolin-4-ylamino)decane) see compound 3 loc. cit.; for UCL 1156 (4,4′-[decane-1,10-diylbis(oxy)]bis[1-methylquinolium] diiodide) see compound 9 loc. cit.; for UCL 1118 (1,10-bis[N-(1-methylquinolinium-4-yl)amino]decane diiodide) see compound 8 loc. cit.; for UCL 1684 (6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane) see Campos Rosa et al. (1998).
Dequalinium (decamethylene bis-4-aminoquinaldinium) and high molecular weight polyvinyl chloride (PVC) were generous gifts from the Laboratories for Applied Biology (London U.K.) and ICI respectively. Dibutyl sebacate, dimethyl sulphoxide (DMSO), inosine, valinomycin, clotrimazole, A23187 (calcimycin), EGTA (1,2-di(2-aminoethoxy)ethane-tetra-acetic acid) and heparin were purchased from Sigma, TRIS (tris (hydroxymethyl)aminomethane) from Calbiochem and sodium tetraphenyl borate from Koch-Light. Standard salts were of Analar quality and were obtained from BDH, who also provided EDTA (ethylene diamine tetra-acetic acid), digitonin and tetrahydrofuran.
Results
Figure 1 compares the SKCa and IKCa blocking action of the bisquaternary compounds dequalinum and UCL 1684 (6,10-diaza-3 (1,3), 8(1,4)-dibenzena-1, 5 (1, 4) -diquinolinacyclodecaphane). Confirming earlier work (Campos Rosa et al., 1998; see also Dunn, 1999), low nanomolar concentrations of UCL 1684 inhibited the SKCa-mediated afterhyperpolarization (AHP) recorded from rat sympathetic ganglion cells. Dequalinium was also effective (Dunn, 1994; Galanakis et al., 1996) though ∼150 times less active. The potency order was strikingly different when the same two compounds were tested for their effect on the IKCa-mediated loss of K+ that follows the application of a Ca2+ ionophore to red cells. UCL 1684 now caused little inhibition (<20% at 10 μM). Like apamin, it is evidently highly selective for SKCa over IKCa channels.
Figure 1.
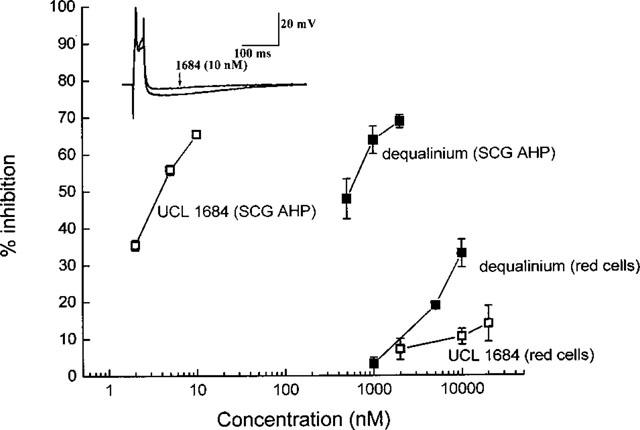
Comparison of the blocking action of dequalinium and UCL1684 on the SKCa-mediated afterhyperpolarization (AHP) in rat superior cervical ganglion (SCG) cells and on the IKCa-mediated loss of K+ from rabbit red cells exposed to a Ca2+ ionophore. Each point is the mean of 3–5 observations and the error bars show the s.e.means. Inset. Superimposed traces showing actions potentials and AHPs recorded from an individual SCG neurone before, at the end of, and 5.5 min after a 100 s application of UCL 1684 at 10 nM. Each trace is the average of three responses. The membrane potential of the cell was −66 mV and was unaffected by the drug. Note the reduction in the AHP recorded in the presence of the drug and the rapid and complete reversal afterwards (the control and recovery traces superimpose almost exactly).
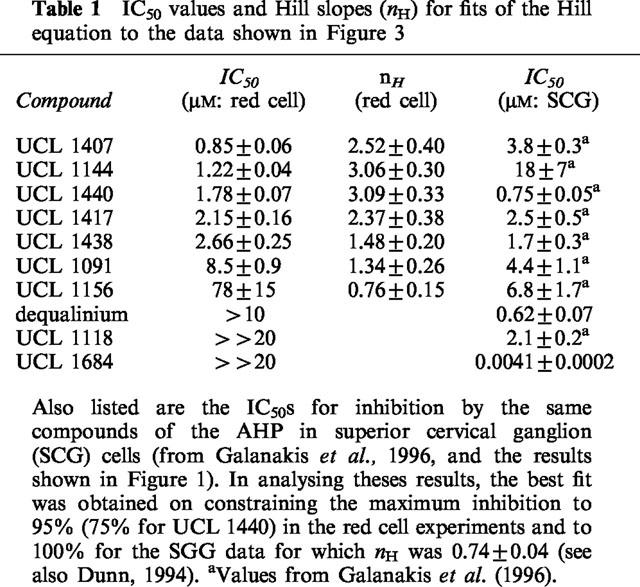
Though the IKCa blocking action of dequalinium was modest (Figure 1), it was also unexpected. This prompted us to examine other compounds with the same general structure in which two aminoquinolinium or quinolinyl moieties are linked by a hydrophobic chain (see Figure 2). Each of the substances selected had already been tested for its SKCa blocking action on rat SCG neurones (Galanakis et al., 1996). The results obtained have been combined in Figure 3, and Table 1 lists the IC50 and Hill coefficient values obtained on fitting the Hill equation to the concentration-inhibition data for the more potent compounds. The IC50 values for the inhibition of the SKCa-mediated AHP in rat SCG neurones are also included, for comparison.
Figure 2.
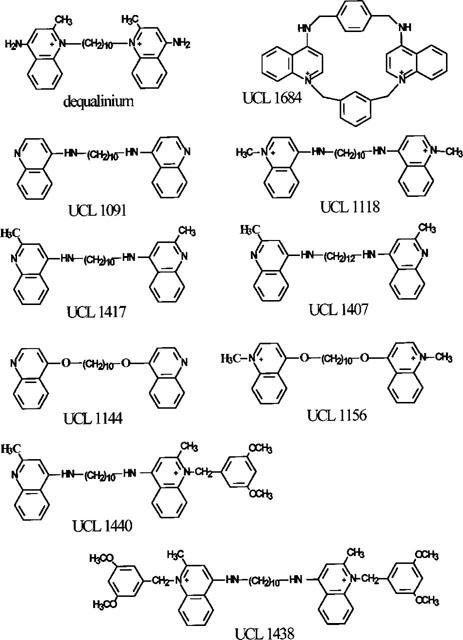
Structures of the compounds.
Figure 3.
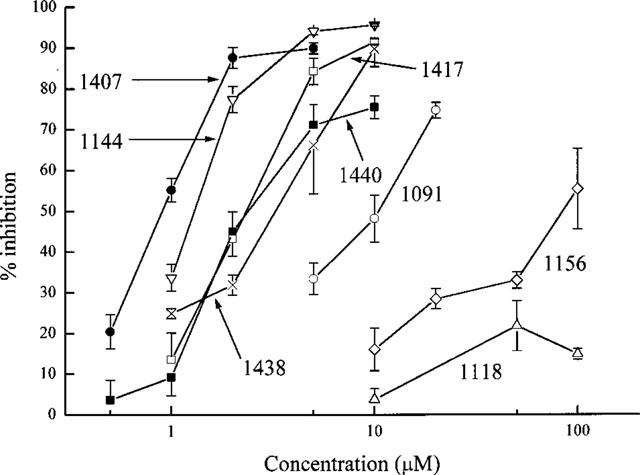
The relationship between the concentration of the compounds tested and inhibition of K+ loss from a suspension of rabbit red cells exposed to the Ca2+ ionophore A23187. The ionophore was applied 3 min after the drug was added to the cell suspension. Each point is the mean of 3–5 observations and the error bars show the s.e.means. The UCL prefixes of the compound identifiers have been omitted for clarity.
Table 1.
IC50 values and Hill slopes (nH) for fits of the Hill equation to the data shown in Figure 3
Characterization of the blocking action: K+ dependence
The results in Figure 3 and Table 1 show that several of the compounds tested are effective IKCa inhibitors. The most potent of them (UCL 1407, 1144, 1438, 1417 and 1440) are 10–30 times more active than quinine and cetiedil, examined under the same conditions (Benton et al., 1999b). In that study, it was found that IKCa blockers acting on red cells could be placed in two categories. For one set of compounds, exemplified by quinine and by cetiedil and its congeners, the Hill slopes of the concentration-inhibition relationships were much greater than unity (ranging from 2.7 to 3.6). In contrast, for a second group of inhibitors that included clotrimazole, nitrendipine and charybdotoxin, nH values close to or a little less than unity were found. The Hill slopes of the four most active compounds (UCL 1407, 1144, 1440 and 1417) listed in Table 1 were all significantly greater than one, and comparable to those observed with quinine and cetiedil (Benton et al., 1999b). This suggests the possibility of a common mechanism of action. Were this to be so, the blocking action would be expected to be reduced on increasing the concentration of K+ in the bathing solution, as has been shown for quinine (Armando-Hardy et al., 1975; Reichstein & Rothstein, 1981; Benton et al., 1999b) and for cetiedil and its congeners (Benton et al., 1999b). This was tested by examining the effect of raising [K+]o on the blocking action of UCL 1438 and dequalinium. As before, A23187 was used to activate the IKCa channels by raising cytosolic [Ca]2+ but the response was now measured by means of an optical method described by Alvarez et al. (1986; 1992). In this technique, changes in the absorption of light by a suspension of red cells provide an index of cell shrinkage consequent to the loss of K+, Cl− and water that follows the application of a calcium ionophore and the subsequent opening of IKCa channels. The method has the advantage over the K+-sensitive electrode technique that it allows external [K+]o to be changed without affecting the sensitivity of the detection method (see Methods). Figure 4 illustrates the effect of raising [K+]o from 0.12–0.17 mM to 5.4 mM on the IKCa blocking action of UCL 1438. As with quinine and cetiedil, the inhibition is reduced and it is necessary to raise the concentration of the blocker by a factor of 1.90±0.29 to achieve the same amount of inhibition. In similar experiments with dequalinium the factor was 3.1±1.1.
Figure 4.
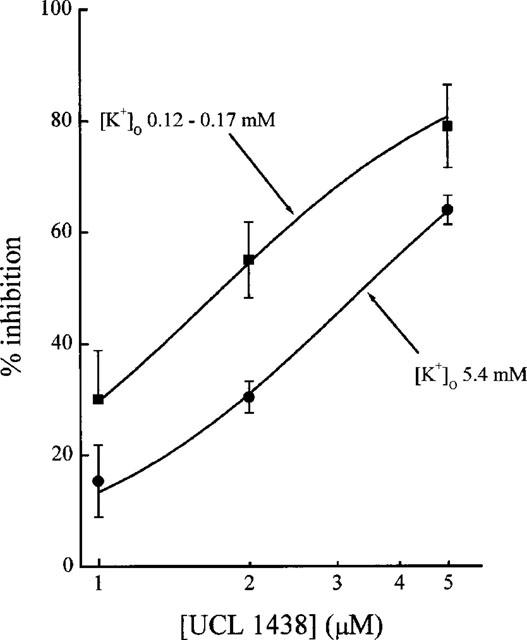
Influence of external K+ concentration on the IKCa blocking action of UCL 1438. The measurements were made using an optical method (see Text and also Figure 5) based on observing the changes in light absorption by a suspension of rabbit blood cells (haematocrit 0.02%) exposed to A23287 (0.4 μM). Each point is the mean of 3–4 observations and the error bars show the s.e.means.
Although the influence of raising [K+]o from 0.1 to 5.4 mM on the transmission of light through the resting cell suspension was not systematically examined, no substantial changes were apparent. In keeping with this, the response to the standard application of A23187 in the absence of blockers was little affected by the change in external [K+]o. For example, in the experiment of Figure 4, the ratio of the average decrease in light transmission produced by A23187 in 0.1 mM K+ (ten observations) to that in 5.4 mM K+ (eight observations) was 1.02 (95% confidence limits 0.83 to 1.21).
Effects of the inhibitors on the time course of A23187-induced K+ loss
Further evidence that the new compounds tested resemble cetiedil and quinine rather than clotrimazole in their blocking actions came from their effects on the time course of the responses to A23187. The was examined in two ways. Preliminary experiments, illustrated by records a and b in Figure 5, showed that when the most potent compound of the present series, UCL 1407 (0.3 μM), was applied during the course of the K+ loss induced by A23187, the associated decrease in light transmission slowed almost immediately, ceasing after ∼60 s. Clearly the inhibition occurred rapidly. Nevertheless, for consistency with the procedure used in the measurements summarized in Figure 3, in the remainder of the experiments the blocking agents were added ∼3 min before A23187 was applied. A typical record is shown in Figure 5d which illustrates that when UCL 1438 is present the reduction in transmission initiated by A23187 slows and then stops, as with UCL 1407. Similar results were obtained with the other compounds tested in the present work. In contrast, the response in the presence of clotrimazole, though reduced in comparison with the control, continued throughout the 3 min exposure to A23187 (Figure 5f). This difference was equally evident when K+-sensitive electrodes were used to measure K+ loss, as previously described and quantified for cetiedil and clotrimazole by Benton et al. (1999b) who have suggested that it may reflect a use-dependent component to the blocking action of cetiedil but not clotrimazole.
Figure 5.
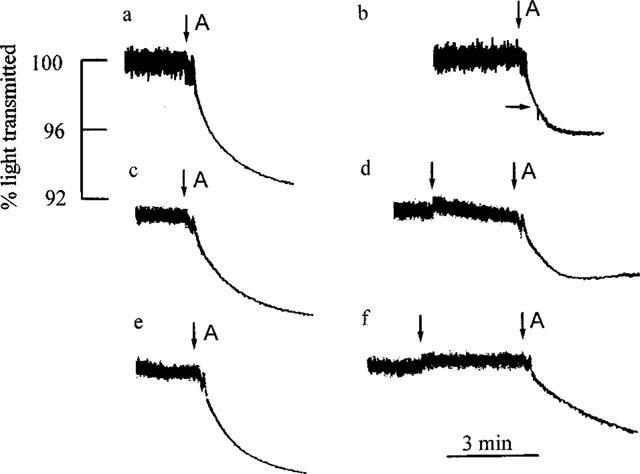
Effects of IKCa blockers on the time course of the response to A23187. The records show the decreases in the transmission of light by a suspension of rabbit red cells exposed to A23187 (0.4 μM, applied at A in each record). (a), (c) and (e) are control responses for the tests shown in (b), (d) and (f) respectively. A fresh sample of cells was used on each occasion. In (b), UCL 1407 (0.3 μM) was added at the horizontal arrow, 30 s after A23187. Note the rapid and complete inhibition of the change in light transmission. In record (d), UCL 1438 (0.5 μM) was introduced at the first vertical arrow. 2.7 min later, A23187 was applied in the presence of UCL 1438. Though the initial response to A23187 was little reduced, the fall in light transmission progressively slowed and ceased after ∼1.5 min. Record (f) illustrates the inhibitory action of clotrimazole (0.2 μM, added at the first vertical arrow). In contrast to the findings with UCL 1407 and 1438, the fall in light transmission induced by A23187 (second arrow) continued throughout the recording, albeit at a reduced rate. The time and transmission calibrations apply to all the traces.
Two incidental aspects of the use of the light absorption method merit comment. First, it allowed much lower haematocrits to be employed (0.02% as compared with 1% when K+ electrodes were used in the present work). Comparing the results of a large number of experiments with each technique showed that the IC50s for highly lipophilic substances were lower by a factor of up to 3 when the optical method was employed with a haematocrit of 0.02% (M. Malik-Hall & D.H. Jenkinson, unpublished observations). The difference is substantial even though exact correspondence is not to be expected in view of the indirectness of both methods. A possible contributory factor is that when a very lipophilic substance is added to a relatively dense suspension of red cells, an appreciable quantity may enter the lipid component of the cell, and perhaps also the cytosol, resulting in a reduction in the concentration in the suspension medium. In keeping with this, when the observations were repeated, but now with denser cell suspensions (e.g. haematocrits of 1 and 3%) and with blocking agents more lipophilic than the present compounds, the expected dependence of the IC50 on cell density was confirmed (M. Malik-Hall & D.H. Jenkinson, unpublished observations). A similar situation has been described and analysed by Boer et al. (1989) in binding studies with lipophilic Ca2+ channel blockers. This factor is likely to account at least partly for the greater potency of UCL 1407 and UCL 1438 in the experiments illustrated in Figure 5 (records b and d respectively) as compared with those of Figure 3. The puzzling and as yet unexplained variation in the potencies of IKCa blockers over long periods of time (Benton et al., 1999b) could also contribute.
The second incidental observation was that some of the compounds tested themselves caused changes in the absorption of light by the suspension (see e.g. record d in Figure 5), thereby complicating the measurement of the response. Though not examined further, it was noted that the subsequent application of A23187 to the suspension seemed to partly reverse this action. The susceptibility of the light absorption method to changes in the optical properties of the red cells unrelated to K+ loss, and its indirectness, have to be set against the advantages of allowing the use of much lower haematocrits than practicable with the K+ electrode technique and also the greater ease with which the effects of changes in external K+ can be studied. In our hands, the K+ electrode procedure proved more reliable and reproducible when large numbers of measurements were to be made, as in the experiments of Figure 3.
Discussion
The results summarized in Table 1 show that it has been possible to identify compounds with a wide range of SKCa/IKCa selectivities. At one extreme, the bisquaternary quinolinium cyclophane UCL 1684 is much less active (by a factor of >5000) as a blocker of actions mediated by IKCa as compared with SKCa channels (Figure 1). This selectivity is comparable to that of apamin (Burgess et al., 1981) and of UCL 1848 (Benton et al., 1999a). In contrast, UCL 1144 (Figure 2) is 15 times more effective at the IKCa channel. In the midrange are UCL 1417 and UCL 1438.
Although the mechanisms of the IKCa blocking action of these compounds were not examined directly in the present work, some inferences are possible. As already mentioned, the previously known IKCa inhibitors can be placed in two categories with different mechanisms of action (Benton et al., 1999b). For one set (e.g., cetiedil, quinine) the block in red cells is strongly dependent on the K+ content of the bathing fluid, the concentration-inhibition curve is steep with a Hill slope (nH) of 2–3, and the rate of K+ loss initiated by A23187 progressively declines in their continued presence, suggesting use-dependence. For the others (e.g., clotrimazole), nH is close to unity, the inhibition is little affected by moderate changes in external K+, and K+ loss continues in the presence of a submaximal concentration of the blocker, albeit at a lower rate (Benton et al., 1999b). The present findings suggest that at least the five most active IKCa blockers shown in Figure 2 belong to the first category. The Hill slopes are greater than 1 (Table 1), the inhibition is reduced on increasing external K+ (Figure 4) and the time course of the response to A23187 is changed in a use-dependent manner (Figure 5).
An additional characteristic of the block produced by compounds of the first type is that activity increases with the lipophilicity of the molecule (Benton et al., 1994; 1999b). This has been studied in detail by Benton (1995) who examined the relation between the potency of 35 cetiedil-related compounds and their lipophilicity, as predicted from the hydrophobic fragmental constant approach introduced by Nys & Rekker (1973) and recently refined (see Mannhold et al., 1998). Benton found that the relationship between the IC50 and the sum, Σf, of the fragmental constants for each compound could be described by the expression:
This has been plotted in Figure 6, together with the values for the four nonquaternary compounds examined in the present work. That the points lie around the line adds to the similarities in the action of the cetiedil derivatives and of the compounds we have now tested, suggesting that the latter act in the same manner as cetiedil, and perhaps at a common site.
Figure 6.
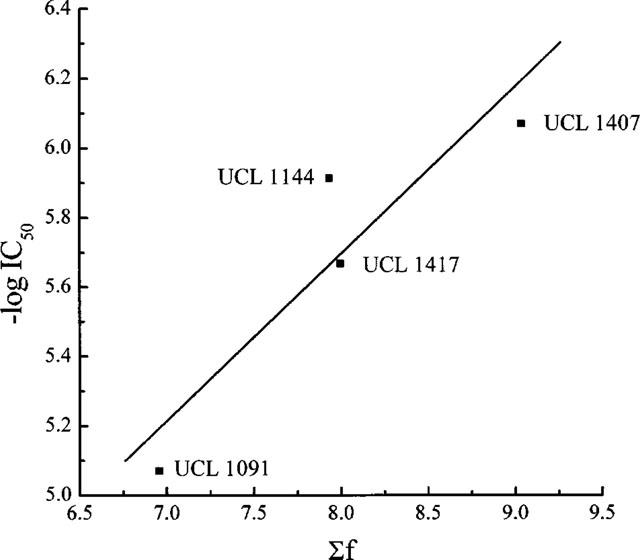
A plot of −log IC50 against Σf, a predictor of hydrophobicity, for four nonquaternary bisquinolinyl compounds (see Figure 2 for structures). The line shows the relationship observed in identical experiments with 35 derivatives of the mono-tertiary base cetiedil (from Benton, 1995).
However, if this is so, it is at first sight surprising that the monoquaternary compound UCL 1440 and the bisquaternaries UCL 1438, 1156 and dequalinium should also be active, although to very different degrees. Earlier work (Benton et al., 1994) had shown that the quaternary derivative formed by N-methylation of the tertiary base cetiedil had little activity (though the low potency of cetiedil itself limited the range of concentrations that could be tested). Three factors at least may underlie the activity of the quaternized compounds studied in the present work. First, the positive charges carried by these molecules are extensively delocalized, as discussed by Galanakis et al. (1995; 1996). Second, the relative size of the group used to form the quaternary derivative will influence the extent to which the charge is screened, allowing the molecule access to relatively lipophilic regions. A third possibility is raised by the ability of quaternized aminoquinoline (quinolinium) compounds to lose a proton to give an electrically neutral, and lipophilic, imino-base (see Galanakis et al., 1996). The presence of even a small proportion of the unionized form, provided that it is sufficiently lipophilic, could allow it to reach the site of its blocking action which, if the same as for cetiedil, probably lies within the channel (Benton et al., 1999b).
Here it is of interest to compare the activities of the compound pairs UCL 1091 and 1118, 1417 and 1438, and 1144 and 1156. Each pair comprises a bisquinolinyl base and its bisquaternary derivative. UCL 1091 is the least potent of the nonquaternary members of the sets and, accordingly, its quaternized derivative (UCL 1118), formed by methylation, is relatively inactive. However, the base UCL 1417 is substantially more lipophilic, and correspondingly more active, than UCL 1091. Moreover, its bisquaternary derivative, UCL 1438, was formed by quaternization with the relatively large and lipophilic dimethoxy benzyl moiety, rather than a methyl, and although a large proportion of the 1438 molecules will be charged (>99.9%), the charge will be relatively screened. The finding that the bisquaternary UCL 1438, the monoquaternary 1440 and the parent bisquinolinyl 1417 have similar activities (Table 1) suggests that these factors are almost in balance. With UCL 1144 and UCL 1156, an oxygen atom replaces the amino nitrogen so that deprotonation to form an imino-base cannot occur. Also, since the dication UCL 1156 is quaternized by the relatively small methyl group, it would be predicted to be much less active than 1144, as is indeed observed (Figure 3, Table 1).
These results show that it is possible to identify structural and electronic features that influence the IKCa blocking activity of compounds in which quinolinyl or quinolinium groups are linked by a single alkyl chain connected via heteroatoms (N or O). The explanation for the lack of IKCa blocking action by the more complex cyclophanes UCL 1684 (Figure 1) and UCL 1848 (see Benton et al., 1999a) remains to be established. Possible factors include the lower lipophilicity, greater rigidity and larger molecular diameter of these substances, features that may preclude access to the putative site of action in the pore region of the IKCa channel.
To summarize, our work has identified a group of simple nonquaternary and bisquaternary derivatives of dequalinium that are able to block IKCa as well as SKCa calcium-activated potassium channels. Compounds of this kind are likely to be of value for the study of physiological mechanisms that are thought to involve both kinds of channel.
Acknowledgments
We are grateful to the Wellcome and Leverhulme Trusts for support and to Dr D.C.H. Benton and Dr D.G. Haylett for many helpful discussions.
Abbreviations
- AHP
afterhyperpolarization
- IKCa
intermediate conductance Ca2+-activated K+ channel
- SCG
superior cervical ganglion
- SKCa
small conductance Ca2+-activated K+ channel
References
- ALVAREZ J., GARCIA SANCHO J., HERREROS B. The role of calmodulin on Ca2+-dependent K+-transport regulation in the human red-cell. Biochim. Biophys. Acta. 1986;860:25–34. doi: 10.1016/0005-2736(86)90494-3. [DOI] [PubMed] [Google Scholar]
- ALVAREZ J., MONTERO M., GARCIA SANCHO J. High-affinity inhibition of Ca2+-dependent K+ channels by cytochrome- p-450 inhibitors. J. Biol. Chem. 1992;267:11789–11793. [PubMed] [Google Scholar]
- ARMANDO-HARDY M., ELLORY J.C., FERREIRA H.G., FLEMINGER S., LEW V.L. Inhibition of the calcium-induced increase in the potassium permeability of human red blood cells by quinine. J. Physiol. 1975;250:32P. [PubMed] [Google Scholar]
- BENTON D.C.H.The pharmacology of calcium and ATP-dependent potassium channels in erythrocytes and in smooth and skeletal muscle 1995University of London; Ph.D Thesis [Google Scholar]
- BENTON D.C.H., ATHMANI S., ROXBURGH C.J., SHINER M.A.R., HAYLETT D.G., GANELLIN C.R., JENKINSON D.H. Effects of cetiedil and its analogues on the Ca2+-activated K+ permeability of rabbit erythrocytes and on levcromakalim-stimulated 86Rb+ efflux from rat aorta. Br. J. Pharmacol. 1994;112:466P. [Google Scholar]
- BENTON D.C.H., DUNN P.M., CHEN J.Q., GALANAKIS D., GANELLIN C.R., MALIK-HALL M., SHAH M., HAYLETT D.G., JENKINSON D.H. UCL 1848: a novel bis-quinolinium cyclophane which blocks apamin-sensitive K+ channels with nanomolar affinity. Br. J. Pharmacol. 1999a;128:39. [Google Scholar]
- BENTON D.C.H., ROXBURGH C.J., GANELLIN C.R., SHINER M.A.R., JENKINSON D.H. Differences in the actions of some blockers of the calcium-activated potassium permeability in mammalian red cells. Br. J. Pharmacol. 1999b;126:169–178. doi: 10.1038/sj.bjp.0702292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOER R., GRASSEGGER A., SCHUDT C., GLOSSMANN H. (+)-Niguldipine binds with very high-affinity to Ca2+ channels and to a subtype of alpha1-adrenoceptors. Eur. J. Pharmacol. 1989;172:131–145. doi: 10.1016/0922-4106(89)90004-7. [DOI] [PubMed] [Google Scholar]
- BURGESS G.M., CLARET M., JENKINSON D.H. Effects of quinine and apamin on the calcium-dependent potassium permeability of mammalian hepatocytes and red cells. J. Physiol. 1981;317:67–90. doi: 10.1113/jphysiol.1981.sp013814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPOS ROSA J., GALANAKIS D., GANELLIN C.R., DUNN P.M., JENKINSON D.H. Bis-quinolinium cyclophanes: 6,10-diaza - 3(1,3),8 (1,4) - dibenzena - 1,5 (1,4) - diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca2+-activated K+ channel. J. Med. Chem. 1998;41:2–5. doi: 10.1021/jm970571a. [DOI] [PubMed] [Google Scholar]
- CAPIOD T., OGDEN D.C. The properties of calcium-activated potassium ion channels in guinea-pig isolated hepatocytes. J. Physiol. 1989;409:285–295. doi: 10.1113/jphysiol.1989.sp017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASTLE N.A. Recent advances in the biology of small conductance calcium-activated potassium channels. Perspectives in drug discovery and design. 1999;15/16:131–154. [Google Scholar]
- CASTLE N.A., HAYLETT D.G., MORGAN J.M., JENKINSON D.H. Dequalinium: a potent inhibitor of apamin-sensitive K+ channels in hepatocytes and of nicotinic responses in skeletal muscle. Eur. J. Pharmacol. 1993;236:201–207. doi: 10.1016/0014-2999(93)90590-e. [DOI] [PubMed] [Google Scholar]
- CHRISTOPHERSEN P. Ca2+-activated K+ channel from human erythrocyte membranes–single channel rectification and selectivity. J. Mem. Biol. 1991;119:75–83. doi: 10.1007/BF01868542. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D., RANG H.P., RITCHIE J.M. The binding of tetrodotoxin and α-bungarotoxin to normal and denervated mammalian muscle. J. Physiol. 1974;240:199–226. doi: 10.1113/jphysiol.1974.sp010607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK N.S., HAYLETT D.G. Effects of apamin, quinine and neuromuscular blockers on calcium-activated potassium channels in guinea-pig hepatocytes. J. Physiol. 1985;358:373–394. doi: 10.1113/jphysiol.1985.sp015556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNN P.M. Dequalinium, a selective blocker of the slow afterhyperpolarization in rat sympathetic neurons in culture. Eur. J. Pharmacol. 1994;252:189–194. doi: 10.1016/0014-2999(94)90596-7. [DOI] [PubMed] [Google Scholar]
- DUNN P.M. The action of blocking agents applied to the inner face of Ca2+-activated K+ channels from human erythrocytes. J. Mem. Biol. 1998;165:133–143. doi: 10.1007/s002329900427. [DOI] [PubMed] [Google Scholar]
- DUNN P.M. UCL1684: a potent blocker of Ca2+-activated K+ channels in rat adrenal chromaffin cells in culture. Eur. J. Pharmacol. 1999;368:119–123. doi: 10.1016/s0014-2999(99)00029-1. [DOI] [PubMed] [Google Scholar]
- DUNN P.M., BENTON D.C.H., CAMPOS ROSA J., GANELLIN C.R., JENKINSON D.H. Discrimination between subtypes of apamin-sensitive Ca2+-activated K+ channels by gallamine and a novel bis-quaternary quinolinium cyclophane, UCL 1530. Br. J. Pharmacol. 1996;117:35–42. doi: 10.1111/j.1476-5381.1996.tb15151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALANAKIS D., DAVIS C.A., DEL REY HERRERO B., GANELLIN C.R., DUNN P.M., JENKINSON D.H. Synthesis and structure-activity relationships of dequalinium analogues of K+ channel blockers. Investigations on the role of the charged heterocycle. J. Med. Chem. 1995;38:595–606. doi: 10.1021/jm00004a005. [DOI] [PubMed] [Google Scholar]
- GALANAKIS D., DAVIS C.A., GANELLIN C.R., DUNN P.M. Synthesis and quantitative structure-activity relationship of a novel series of small-conductance Ca2+-activated K+ channel blockers related to dequalinium. J. Med. Chem. 1996;39:359–370. doi: 10.1021/jm950520i. [DOI] [PubMed] [Google Scholar]
- GÁRDOS G. The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta. 1958;30:653–654. doi: 10.1016/0006-3002(58)90124-0. [DOI] [PubMed] [Google Scholar]
- GRYGORCZYK R., SCHWARZ W. Properties of the Ca2+-activated K+ conductance of human red-cells as revealed by the patch-clamp technique. Cell Calcium. 1983;4:499–510. doi: 10.1016/0143-4160(83)90025-8. [DOI] [PubMed] [Google Scholar]
- HAYLETT D.G., JENKINSON D.H.Calcium-activated potassium channels Potassium Channels 1990Ellis Horwood: Chichester; 70–95.In: Cook, N.S. (ed) [Google Scholar]
- HOSSEINI R., BENTON D.C.H., HAYLETT D.G., MOSS G.W.J. Cloning of an SK channel from rat sympathetic neurones. J. Physiol. 1999;518:133P. [Google Scholar]
- ISHII T.M., MAYLIE J., ADELMAN J.P. Determinants of apamin and d-tubocurarine block in SK potassium channels. J. Biol. Chem. 1997a;272:23195–23200. doi: 10.1074/jbc.272.37.23195. [DOI] [PubMed] [Google Scholar]
- ISHII T.M., SILVIA C., HIRSCHBERG B., BOND C.T., ADELMAN J.P., MAYLIE J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. U.S.A. 1997b;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENKINSON D.H., HAYLETT D.G., COOK N.S. Calcium-activated potassium channels in liver cells. Cell Calcium. 1983;4:429–437. doi: 10.1016/0143-4160(83)90019-2. [DOI] [PubMed] [Google Scholar]
- JENSEN B.S., STROBAEK D., CHRISTOPHERSEN P., JORGENSEN T.D., HANSEN C., SILAHTAROGLU A., OLESEN S.P., AHRING P.K. Characterization of the cloned human intermediate-conductance Ca2+-activated K+ channel. Am. J. Physiol. 1998;275:C848–C856. doi: 10.1152/ajpcell.1998.275.3.C848. [DOI] [PubMed] [Google Scholar]
- JOINER W.J., WANG L.Y., TANG M.D., KACZMAREK L.K. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11013–11018. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOHLER M., HIRSCHBERG B., BOND C.T., KINZIE J.M., MARRION N.V., MAYLIE J., ADELMAN J.P. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- LANCASTER B., ADAMS P.R. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J. Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- LEINDERS T., VANKLEEF R.M., VIJVERBERG H.M. Distinct metal-ion binding-sites on Ca2+-activated K+ channels in inside-out patches of human erythrocytes. Biochim. Biophys. Acta. 1992a;1112:75–82. doi: 10.1016/0005-2736(92)90256-l. [DOI] [PubMed] [Google Scholar]
- LEINDERS T., VANKLEEF R.M., VIJVERBERG H.M. Single Ca2+-activated K+ channels in human erythrocytes–Ca2+ dependence of opening frequency but not of open lifetimes. Biochim. Biophys. Acta. 1992b;1112:67–74. doi: 10.1016/0005-2736(92)90255-k. [DOI] [PubMed] [Google Scholar]
- LOGSDON N.J., KANG J., TOGO J.A., CHRISTIAN E.P., AIYAR J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 1997;272:32723–32726. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- MANNHOLD R., REKKER R.F., DROSS K., BIJLOO G., DE VRIES G. The lipophilic behaviour of organic compounds: 1. An updating of the hydrophobic fragmental constant approach. Quant. Struct.-Act. Relat. 1998;17:517–536. [Google Scholar]
- NOHMI M., KUBA K. (+)-Tubocurarine blocks the Ca2+-dependent K+-channel of the bullfrog sympathetic-ganglion cell. Brain Research. 1984;301:146–148. doi: 10.1016/0006-8993(84)90412-8. [DOI] [PubMed] [Google Scholar]
- NYS G.G., REKKER R.F. Statistical analysis of a series of partition coefficients with special reference to the predictability of folding of drug molecules. The introduction of the hydrophobic fragmental constant (f-value) Chim. Ther. 1973;5:521–535. [Google Scholar]
- REICHSTEIN E., ROTHSTEIN A. Effects of quinine on Ca++-induced K+ efflux from human red cells. J. Mem. Biol. 1981;59:57–63. doi: 10.1007/BF01870821. [DOI] [PubMed] [Google Scholar]
- SAH P. Ca2+-activated K+ currents in neurones: Types, physiological roles and modulation. Trends In Neurosciences. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- SARKADI B., GÁRDOS G.Calcium-induced potassium transport in cell membranes The Enzymes of Biological Membranes 1985Vol 3Plenum Press: New York; 193–234.In: Martonosi, A.N. (ed) [Google Scholar]
- VERGARA C., LATORRE R., MARRION N.V., ADELMAN J.P. Calcium-activated potassium channels. Cur. Opin. Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- WADSWORTH J.D.F., DOORTY K.B., STRONG P.N. Comparable 30 kDa apamin binding polypeptides may fulfil equivalent roles within putative subtypes of small-conductance Ca2+-activated K+ channels. J. Biol. Chem. 1994;269:18053–18061. [PubMed] [Google Scholar]