

Abstract
Previous studies described the metabolism-based discovery of a potent, selective inhibitor of intestinal absorption of cholesterol, SCH58235 (Ezetimibe). Here we demonstrate that the phenolic glucuronide (SCH60663) of SCH58235, was more potent at inhibiting cholesterol absorption in rats than SCH58235, when administered by the intraduodenal route.
To understand the increased potency of the glucuronide, the metabolism and distribution of SCH58235 and SCH60663 were studied in bile duct-cannulated rats.
One minute after intraduodenal delivery of SCH58235, significant levels of compound were detected in portal plasma; >95% was glucuronidated, indicating that the intestine was metabolizing SCH58235 to its glucuronide. When intraduodenally delivered as SCH58235, the compound was glucuronidated, moved through the intestinal wall, into portal plasma, through the liver, and into bile. However, when delivered as SCH60663, >95% of the compound remained in the intestinal lumen and wall, which may explain its increased potency. Significant inhibition of cholesterol absorption and glucuronidation of SCH58235 occurred when SCH58235 was intravenously injected into bile duct-cannulated rats. Autoradiographic analysis demonstrated that drug related material was located throughout the intestinal villi, but concentrated in the villus tip.
These data indicate that (a) SCH58235 is rapidly metabolized in the intestine to its glucuronide; (b) once glucuronidated, the dose is excreted in the bile, thereby delivering drug related material back to the site of action and (c) the glucuronide is more potent than the parent possibly because it localizes to the intestine. Taken together, these data may explain the potency of SCH58235 in the rat (ID50=0.0015 mg kg−1) and rhesus monkey (ID50=0.0005 mg kg−1).
Keywords: Cholesterol absorption, plasma cholesterol, intestine, inhibition, intestinal glucuronidation, enterohepatic circulation
Introduction
It has been well established that elevated plasma cholesterol is positively associated with the incidence of cardiovascular disease (National Research Council, 1989). Evidence is mounting from clinical trials that reducing plasma cholesterol by dietary and/or pharmacological means leads to reductions in the incidence of death from vascular events (4S. 1994; Shepherd et al., 1995; Sacks et al., 1996; LIPID, 1998). Plasma cholesterol is predominantly derived from biosynthesis and dietary intake of cholesterol. Present pharmacological interventions include the inhibition of cholesterol biosynthesis by inhibiting HMG CoA reductase (the statins) either alone or in combination with other agents such as cholestyramine, which sequesters bile acids. In contrast, research efforts have thus far failed to develop pharmacological agents that are selectively targeted towards inhibiting the absorption and traversal of cholesterol through the intestinal wall.
We have previously reported the discovery of a novel class of intestinal cholesterol absorption inhibitors, which have been shown to be selective and potent cholesterol-lowering agents in cholesterol-fed hamsters, rats, rabbits, dogs and cynomolgus and rhesus monkeys (Burnett et al., 1994; Salisbury et al., 1995; Sybertz et al., 1995; van Heek et al., 1997; Rosenblum et al., 1998). We have also shown that compounds of this series are synergistic with a number of the statins (Davis et al., 1994; 1995). The first compound of the cholesterol absorption inhibitors to go into clinical trials, SCH48461, reduced LDL by 15% in humans (Bergman et al., 1995). SCH58235 (Ezetimibe) was shown to be 400 times more potent than SCH48461 in the cholesterol-fed rhesus monkey and is rapidly progressing in clinical trials.
The cholesterol absorption inhibitors prevent the absorption of cholesterol by inhibiting the traversal of dietary and biliary cholesterol across the intestinal wall. The molecular mechanism by which this class of compounds inhibits cholesterol absorption, however, remains unknown and is being intensively investigated at Schering-Plough Research Institute. Previous experiments indicated that ACAT, pancreatic lipase, and HMG-CoA reductase are not potently inhibited by these compounds (Salisbury et al., 1995). The cholesterol absorption inhibitors do not sequester or precipitate cholesterol. These compounds also do not sequester bile acids as cholestyramine does. Other known pathways which play a role in cholesterol absorption are still under investigation. However, most of the data to date suggest that these compounds interfere with cholesterol uptake into the intestinal wall by an as yet undiscovered mechanism. The present experiments were undertaken to study the absorption, metabolism and trafficking of SCH58235, and to determine how these relate to the cholesterol absorption inhibition activity of this compound in vivo.
Methods
Schering-Plough compounds
The nonradiolabelled compounds used in the present experiments, SCH58235 and SCH60663 (the glucuronidated metabolite of SCH58235; see Figure 1 for structures), were synthesized by our Department of Chemical Research. 3H-SCH58235 (specific activity=68.3 mCi mg−1; purity=>99%) was radiolabelled by our Radiochemistry section. 3H-SCH60663 was not available as a tool at the time of these experiments, so it was necessary to generate radiolabelled material by dosing 3H-SCH58235 to bile duct-cannulated donor rats and collecting 3H-SCH60663 in the bile as described below.
Figure 1.
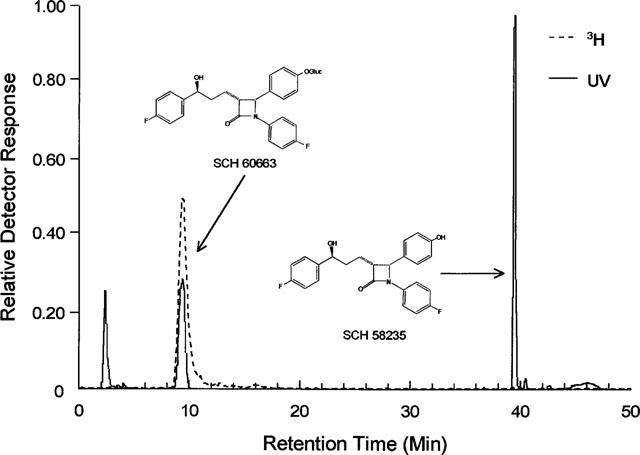
HPLC chromatogram and chemical structures. HPLC chromatogram of Schering-Plough reference compounds SCH58235 and its phenolic glucuronide, SCH60663 (UV detector). The 3H-metabolite from an extract of bile from rats dosed with 3H-SCH58235 (radiometric detector) reveals a single peak which co-elutes with SCH60663.
Identification of the 3H-metabolite (SCH60663) of SCH58235 by reversed-phase HPLC
To generate bile containing metabolites of SCH58235 for reversed-phase HPLC analysis, identification, and for other experiments described later, male Sprague Dawley rats (300–500 g; Charles River Laboratories, Wilmington, MA, U.S.A.) were intraduodenally and bile duct-cannulated as previously described (van Heek et al., 1997). Rats were intraduodenally dosed with 3H-SCH58235 (0.01 mg kg−1; 5 μCi per rat;) and bile was collected for 3–5 h and pooled. Metabolites were extracted from the bile sample with 1.5 times the volume of acetonitrile. RP-HPLC profiling was performed using two Waters M6000 pumps (Waters Corp., Milford, MA, U.S.A.), a Waters Model 480 UV (245 nm) detector and a 5 μM MetaChem Inertsil C-8 reversed-phase column (4.6×150 mm). Using a constant flow rate (1.0 ml min−1), a concave gradient (Waters Expert-Ease Curve #10) was programmed initially at 30% acetonitrile/70% 0.1 M ammonium acetate (pH 6) and then changed to 100% acetonitrile over 40 min. Flo-One for Windows (Packard Instrument Co., Meriden, CT, U.S.A.) was used for acquisition of data from the radiometric (Packard Flo-One) and/or UV detectors. Packard Flo-Scint II (Packard Instrument Co., Meriden, CT, U.S.A.), the scintillation cocktail used with the in-line detector, was mixed post-column at a 3 : 1 ratio (v v−1) with mobile phase. A fraction of the sample was subjected to deglucuronidation utilizing Glucurase (Sigma, St. Louis, MO, U.S.A.).
Determination of cholesterol absorption inhibitory activity of SCH58235 vs SCH60663 in the bile duct-cannulated rat
Male Sprague Dawley rats were surgically prepared as previously described (van Heek et al., 1997). Briefly, fasted rats (18 h) which had been on normal chow diet were anaesthetized with Inactin (100 mg kg−1 i.p.: Abbott Labs, Chicago, IL, U.S.A.). All rats were fitted with tracheal tubes, were intraduodenally cannulated, and were bile duct-cannulated. Rats (n=5 per group) were intraduodenally dosed either with vehicle only (1 ml control bile from donor rats), or with bile containing SCH58235 or SCH60663 (0.01 and 0.003 mg kg−1). Five minutes after drug dosing, 3 ml of an emulsion containing triolein (35.4 mg per dose), L-α-phosphatidylcholine (6.69 mg per dose), sodium taurocholate (3 ml per dose; 19 mM in DPBS, pH 6.4; Sigma Chemical Company, St Louis, MO, U.S.A.), unlabelled cholesterol (3.02 mg per dose), and 14C-cholesterol (1 μCi per dose; NEN, Boston, MA, U.S.A.) was delivered directly into the intestine. At t=1.5 h, rats were sacrificed, plasma was isolated and analysed for radioactivity in triplicate. We had previously determined that an absorption period of 1.5 h is the shortest timeframe in which we can detect a significant window of 14C-cholesterol in plasma. Data are expressed as total 14C d.p.m. per total plasma volume which was calculated by multiplying 0.04 by the body weight. Four per cent of body weight is a general conversion factor for plasma volume (Hawk & Leary, 1995).
Timecourse and identification of 3H compounds in portal plasma and bile
Twelve fasted rats were bile duct-cannulated and intestinally cannulated as described above. At t=0, rats were given an intraduodenal dose of 3H-SCH58235 (0.01 mg kg−1; 5 μCi per rat) using bile as a vehicle. Portal plasma and bile were collected from four rats at each timepoint (1, 7 and 15 min post-dosing). Aliquots of portal plasma and bile were directly analysed for radioactivity. 3H-compound was extracted from portal plasma and bile with 1.5 volumes of acetonitrile; the extract was dried down, resolubilized in methanol, and an aliquot was applied to thin layer chromatography (TLC) plates. The solvent system used to separate 3H-SCH58235 from its glucuronidated metabolite was methylene chloride:methanol:acetic acid (75 : 20 : 5). In this system, the parent (SCH58235) moves with the solvent front while the glucuronide (SCH60663) migrates a small distance from the origin. The TLC bands containing the parent and glucuronide were scraped and analysed for radioactivity. The portal plasma data are expressed as d.p.m. ml−1 while the bile data is expressed as total d.p.m. in the cumulative sample collected.
Timecourse of tissue distribution of 3H-SCH58235 vs 3H-SCH58235-glucuronide (3H-SCH60663) in bile duct-cannulated rats
Fasted rats which had been on normal chow diet were anaesthetized with Inactin (100 mg kg−1 i.p.). All rats were fitted with tracheal tubes, intraduodenally cannulated, and bile duct-cannulated. Radiolabelled glucuronidated SCH58235 was not available as a tool, so it was necessary to generate radiolabelled material using bile duct-cannulated donor rats. 3H-SCH60663, the glucuronide of SCH58235, was generated by dosing bile duct-cannulated rats with 3H-SCH58235 (50 μCi per rat) of known specific activity and unlabelled SCH58235 (0.01 mg kg−1) and collecting the resulting labelled glucuronide in the bile. Using the specific activity and the dose given, the measured radioactivity in the bile, and the molecular weights of SCH58235 (409.0 g mol−1) and SCH60663 (585.6 g mol−1), the molar concentration in the bile was calculated to be 0.0025 μmole ml−1. 3H-SCH58235 and unlabelled SCH58235 were added to control bile to equal the radioactivity and molar concentration of the naturally generated SCH60663-containing bile. Rats (six groups of n=4 per group) were bile duct-cannulated and fitted with duodenal catheters as described above. Groups were dosed with 0.0025 μmoles of the labelled SCH58235 or SCH60663 in bile and sacrificed at 7, 60 and 120 min post-dosing. During the time course, bile was collected. Immediately prior to sacrifice, a large portal plasma sample was taken from each rat. Postmortem, livers were collected, and small intestines were collected after luminal contents were obtained by rinsing intestines with 50 ml of cold saline. Aliquots of intestinal luminal contents, intestinal walls, portal plasma and bile were taken directly for determination of radioactivity. The portal plasma data is expressed as d.p.m. ml−1 while all other data are expressed as total d.p.m. per total volume or per total tissue. Overall, recoveries of radioactivity were >85% of dose and did not differ between treatment groups.
Effect on cholesterol absorption of intravenously administered SCH58235 in intact vs bile duct-cannulated rats
To prepare SCH58235 for intravenous drug delivery, donor plasma was collected using citrate (final concentration 0.016 mM sodium citrate, 0.004 mM citric acid) as an anti-coagulant. SCH58235 was solubilized in ethanol and added to the citrated plasma. Twenty fasted rats were anaesthetized, fitted with a tracheal tube and an intraduodenal catheter, and 10 of these animals were bile duct-cannulated. The other 10 rats were left bile duct intact. At t=0, rats were dosed with 1 ml of citrated plasma only (control intact, n=5; control bile duct-cannulated, n=5) or 0.3 mg kg−1 SCH58235 in citrated plasma (3H-SCH58235 intact, n=5; 3H-SCH58235 bile duct-cannulated, n=5) via the jugular vein. Three millilitres of the cholesterol-containing emulsion described above was delivered to the duodenum of all rats immediately thereafter. At t=3 h, rats were sacrificed, plasma was isolated and analysed for radioactivity in triplicate. Data are expressed as total 14C d.p.m. per plasma volume which was calculated by multiplying 0.04 by the body weight. Livers were also taken, weighed and lipids were extracted by the method of Folch (Folch et al., 1957). Aliquots of the resulting organic layer were dried under N2 and analysed for radioactivity in triplicate. Data are expressed as total 14C d.p.m. per liver. The sum of the radioactivity in plasma and liver was approximately 10% of the dose in the control groups and <2% of the dose in the groups treated with SCH58235.
Distribution and identification of 3H compound in the intestinal wall after intravenously administered SCH58235 in bile duct-cannulated rats
Five bile duct-cannulated rats were administered intravenously 2 μCi 3H-SCH58235 and 0.3 mg kg−1 SCH58235 in citrated plasma. Bile was cumulatively collected throughout the experimental period. Five additional rats were left bile duct intact, but otherwise were dosed in an identical manner to the duct-cannulated rats. After 3 h, all rats were sacrificed, the intestines were rinsed copiously with ice cold saline, and the intestinal mucosa was quickly removed by scraping with a glass slide. Intestinal luminal contents and intestinal mucosa were immediately extracted with two volumes of acetonitrile; the extract was dried down, was resolubilized in methanol, and an aliquot was applied to TLC plates to separate the parent compound from the glucuronide as described above. After separation, the TLC bands were analysed for radioactivity. Data are expressed as total d.p.m. in the intestinal lumen, intestinal wall, plasma, whole liver and the cumulative bile sample (in the case of the bile duct-cannulated rats). Recoveries of radioactivity were >85% of dose and did not differ between the intact and bile duct-cannulated groups.
Autoradiographic analysis of intestinal wall after intravenous administration of 3H-SCH58235 in bile duct-cannulated rats
Five bile duct-cannulated rats were intravenously administered 2 μCi 3H-SCH58235 and 0.3 mg kg−1 SCH58235 in citrated plasma. After 3 h, rats were sacrificed and intestinal sections approximately 15 cm distal to the stomach were taken without rinsing. Sections were immersion fixed in 10% neutral buffered formalin. Cryostat sections were cut at 10 μm, mounted on glass slides and subsequently immersed in Kodak NTB2 emulsion. The slides were allowed to dry completely and placed in air and light-tight boxes and exposed for 3 weeks. The slides were then developed using Kodak D-19 developer, counterstained with haematoxylin and coverslipped with an aqueous mounting media.
AAALAC statement
All studies were conducted in an AAALAC accredited facility following protocols approved by the Schering-Plough Research Institute's Animal Care and Use Committee. The procedures were performed in accordance with the principles and guidelines established by the NIH for the care and use of laboratory animals.
Results
Identification of the metabolite (SCH60663) of SCH58235 by reverse phase HPLC
HPLC analysis of the extract of bile (extraction efficiency=90%) from rats after intraduodenal dosing of 3H-SCH58235 demonstrated that no parent compound was excreted into the bile and that a single metabolite was present (Figure 1). β-glucuronidase (Glucurase) treatment of this metabolite regenerated the parent compound, indicating that the metabolite was a glucuronide (data not shown). Comparison to related compounds of this series indicated that the metabolite was the phenolic glucuronide of SCH58235 thenceforth referred to as SCH60663). Structures of SCH58235 and SCH60663 are shown in Figure 1. Extensive work in other labs at Schering-Plough occurring concomitantly corroborated these results, and showed that phenolic glucuronidation was also the only metabolic pathway in humans (Iannucci et al., 1999).
Determination of cholesterol absorption inhibitory activity of SCH58235 vs SCH60663 in the bile duct-cannulated rat
Figure 2 shows that the glucuronide, SCH60663, is more potent than the parent compound, SCH58235, in inhibiting 14C-cholesterol appearance in plasma when delivered into the duodenum of bile duct-cannulated rats. Note that these doses were given in mass equivalents, not molar equivalents, suggesting that potency would be greater for SCH60663 if the doses had been given in equimolar doses, since the molecular weight of SCH60663 (585.6 g mol−1) is greater than SCH58235 (409.0 g mol−1).
Figure 2.
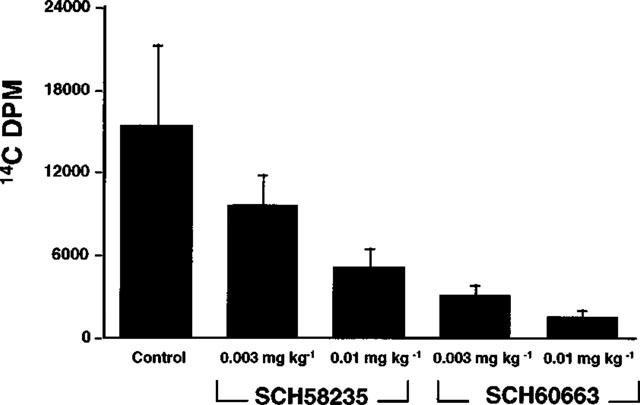
Determination of cholesterol absorption inhibitory activity of SCH58235 vs SCH60663 in the bile duct-cannulated rat. Inhibition of 14C-cholesterol appearance in plasma of bile duct-cannulated rats after intraduodenal delivery of control bile (control) or SCH58235 or SCH60663 in bile at the doses indicated, followed by an intraduodenal bolus delivery of an emulsion containing 14C-cholesterol. Values are mean±s.e.mean, n=5 per group.
Timecourse and identification of 3H compounds in portal plasma and bile
Previous ex vivo experiments with SCH58235 indicated that the compound underwent extensive Phase II metabolism primarily in the liver and to a lesser extent in the kidney (Zhaida et al., 1998). However, these experiments did not address whether the compound was metabolized in the intestine; this possibility was of interest because it was thought that the primary mechanism of action of cholesterol absorption inhibition occurred in the intestine. A timecourse of appearance of 3H-compound in portal plasma and bile 1, 7 and 15 min after intraduodenal dosing of 3H-SCH58235 is shown in Figure 3. After separation by TLC, it was determined that, in portal plasma and in bile, >95% and >99%, respectively, of the radiolabelled compound was glucuronidated at all timepoints. TLC identification of the radiolabelled compounds was verified by reversed-phase HPLC (data not shown, see Figure 1 for example chromatogram). These results indicate that SCH58235 is very rapidly absorbed, is extensively glucuronidated by the intestine, and appears in portal plasma within 1 min of intraduodenal dosing.
Figure 3.
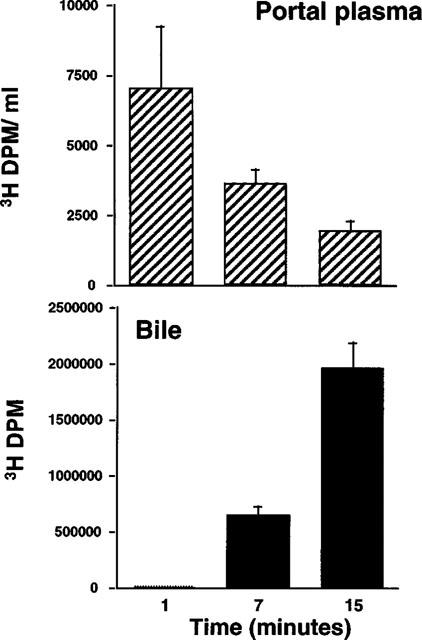
Time-course of 3H compounds in portal plasma and bile after intraduodenal delivery of 3H-SCH58235. 3H-SCH58235 was delivered directly into the intestines of bile duct-cannulated rats and portal plasma was taken at the indicated time after dosing. Each time-point constitutes a separate group of animals. Values are mean±s.e.mean, n=4 per group.
Timecourse of tissue distribution of 3H-SCH58235 vs 3H-SCH58235-glucuronide (3H-SCH60663) in bile duct-cannulated rats
The experiments described above presented an apparent paradox: SCH58235 was primarily metabolized to its glucuronide (SCH60663) in the intestine (Figure 3), yet when SCH60663 was delivered intraduodenally, it was more potent in preventing cholesterol absorption than the parent compound (Figure 2). One possibility was that intraduodenal delivery of SCH58235 vs SCH60663 would lead to different tissue distributions, despite the fact that SCH58235 was converted to the glucuronide in the intestine. A timecourse of the tissue distribution of 3H-compound after intraduodenal dosing of 3H-SCH58235 or 3H-SCH60663 is shown in Figure 4. Seven minutes after dosing, the majority of the labelled compound remained in the intestinal lumen or was associated with the intestinal wall in both treatment groups. In the rats dosed with 3H-SCH58235, significantly more 3H-compound was found in the portal plasma, liver and bile compared to SCH60663. By 60 and 120 min, the tissue distribution differed greatly between treatments; the large majority of the dose was found in the bile at these timepoints after SCH58235 dosing, while most of the dose remained in the intestinal lumen or was associated with the intestinal wall after dosing SCH60663, indicating that the rates at which these two compounds moved through the body were very different. At these timepoints with both treatments, very little radiolabelled compound could be found in portal plasma or liver.
Figure 4.
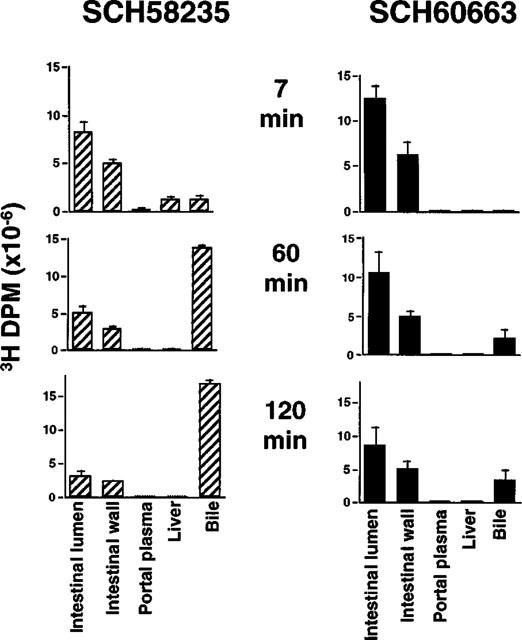
Time course of tissue distribution of 3H-SCH58235 vs 3H-SCH58235-glucuronide (3H-SCH60663) in bile duct-cannulated rats. Distribution of 3H-compound after intraduodenal dosing of 3H-SCH58235 (left panels) or 3H-SCH60663 (right panels) after 7 min (top panels), 60 min (middle panels) or 120 min (bottom panels). Each treatment and time-point constitutes one group of rats. Values are mean±s.e.mean, n=4 per group.
Effect on cholesterol absorption, and distribution and metabolism of intravenously administered SCH58235 in intact vs bile duct-cannulated rats
Previous experiments which assessed cholesterol absorption inhibitory activity of SCH58235 always included either oral or intraduodenal delivery of the drug. In the present study, we assessed whether an intravenous delivery of SCH58235 to the bile duct-cannulated rat would still inhibit the absorption of cholesterol. Groups of rats with bile ducts intact were used for cholesterol absorption inhibition comparison. An additional group of bile duct-cannulated rats received 3H-SCH58235 alone to determine the tissue distribution, as well as the identity, of drug related material after intravenous injection. Intravenous injection of 0.3 mg kg−1 SCH58235 into intact and bile duct-cannulated rats decreased the absorption of cholesterol by 90 and 59%, respectively (Figure 5A). Accumulation of absorbed 14C-cholesterol in livers was decreased 88 and 69% in intact and bile duct-cannulated rats, respectively (Figure 5B). In the intact rats, the dose would reach the luminal side of the intestinal wall via secretion into the biliary circulation. However, in the bile duct-cannulated rat, most of the injected dose would be diverted out of the body via the bile, and any remaining dose would have to reach the intestine from the serosal side. In fact, in the group of bile duct-cannulated rats which received intravenous 3H-SCH58235 alone, 0.63% (equivalent to 0.0019 mg kg−1) and 0.53% (equivalent to 0.0016 mg kg−1) of the radioactive dose was found in the intestinal lumen and wall, respectively; the majority of the dose was found in the bile (Figure 6). TLC separation of 3H-compound indicated that >80% of the compound in the intestinal wall and lumen was glucuronidated in both the intact and bile duct-cannulated rats after intravenous dosing (data not shown).
Figure 5.
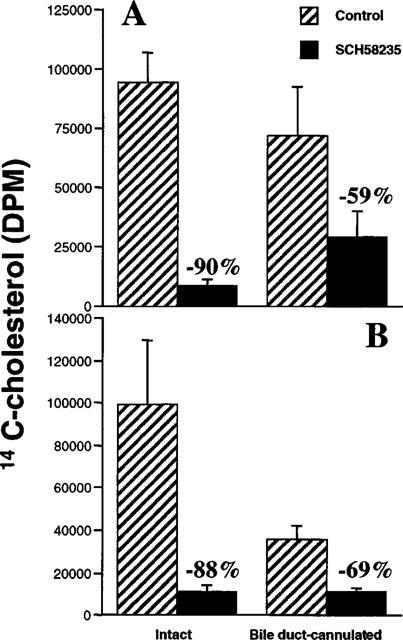
Effect on cholesterol absorption of intravenously administered SCH58235 in intact vs bile duct-cannulated rats. Inhibition of 14C-cholesterol appearance in plasma (A) or liver (B) of intact or bile duct-cannulated rats after intravenous delivery of control plasma (control) or SCH58235 in plasma at 0.3 mg kg−1, followed by an intraduodenal bolus delivery of an emulsion containing 14C-cholesterol. Cholesterol absorption was allowed to take place for 3 h after dosing. Percentages refer to the per cent change from the appropriate control group. Values are mean±s.e.mean, n=5 per group.
Figure 6.
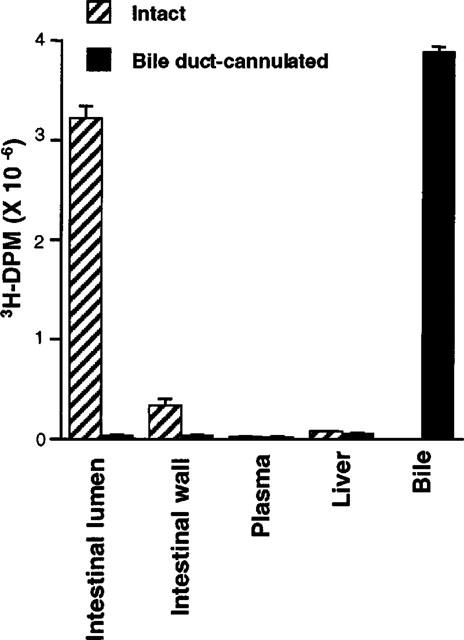
Distribution of 3H compound in the intestinal wall after intravenously administered SCH58235 in intact and bile duct-cannulated rats. Distribution of 3H compound in intestinal lumen, intestinal wall, plasma, liver, and bile 3 h after intravenous dosing of 3H-SCH58235 in intact or bile duct-cannulated rats. Values are mean±s.e.mean, n=5 per group.
Autoradiographic analysis of intestinal wall after intravenous administration of 3H-SCH58235
Autoradiographic analysis (Figure 7) of cross sections of intestines of rats dosed intravenously with 3H-SCH58235 indicated that compound was localized throughout the intestinal villi, but was concentrated in the villus tip where most of the absorptive cells are located.
Figure 7.
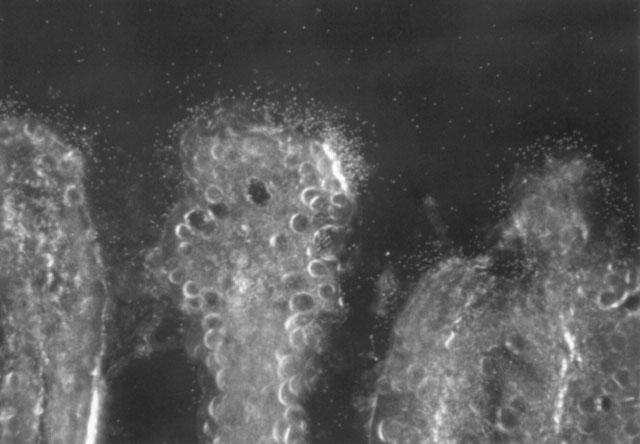
Autoradiographic analysis of intestinal wall after intravenous administration of 3H-SCH58235 in bile duct-cannulated rats. Localization of 3H compound in the intestinal villi 3 h after an intravenous dose of 3H-SCH58235. Three distinct villi can be seen. The dark background is the intestinal lumen. The emulsion autoradiogram demonstrates that the 3H compound (white grains) concentrates on the surface of the enterocytes at the tips of the villi (dark field microscopy, original magnification 250×).
Discussion
We have demonstrated that the novel cholesterol absorption inhibitor, SCH58235, undergoes rapid and extensive glucuronidation in the intestine after intraduodenal delivery. We have also shown that SCH58235 is effective at inhibiting cholesterol absorption even when delivered intravenously to the bile duct-cannulated rat; a small fraction of the dose does reach the intestine, presumably from the serosal side. Thirdly, we have found that the glucuronidated metabolite, SCH60663, is more potent than SCH58235 itself in inhibiting cholesterol absorption after intraduodenal administration. A timecourse of tissue distribution of the radiolabelled compounds in the bile duct-cannulated rat indicated that, after intestinal delivery of SCH60663, the large majority of compound is always associated with the intestinal lumen or wall. In contrast, when SCH58235 is delivered, compound moves quickly from the intestinal lumen, through the intestine wall where it is glucuronidated, into the portal plasma and through the liver into the bile. In an intact animal, the glucuronide would be delivered via the bile back to the intestinal wall which is the site of action. Taken together, the localization of drug in the intestinal wall and the return of the drug to the site of action may explain the potency of SCH58235 in inhibiting cholesterol absorption in the rat (ID50=0.0015 mg kg−1) and preventing the induction of hypercholesterolemia caused by a high fat/cholesterol-containing diet in the rhesus monkey (ID50=0.0005 mg kg−1) (van Heek et al., 1997).
Glucuronidation is generally considered one of the major detoxification processes in xenobiotic metabolism. Making a drug more polar, and thus more water soluble, improves its ability to be excreted into the bile and/or urine thereby eliminating it from the body. Glucuronidation is also generally regarded as a process which inactivates the drug. Neither of these generalizations holds true for this class of compounds. Due to the unknown molecular mechanism of these cholesterol absorption inhibitors, and therefore the lack of an in vitro assay, it is not presently known whether SCH58235 itself, or the glucuronidated form, or both are the active moieties. However, glucuronidation of SCH58235 appears to improve its activity in at least two ways: (1) the drug is repeatedly delivered back to the site of action via enterohepatic circulation and (2) glucuronidation appears to increase its residence time in the gut. In addition, once SCH58235 is glucuronidated, >95% of the compound is either in the intestinal lumen or wall, indicating that systemic exposure of this compound will likely be very low. Significant glucuronidation of compounds by the intestine has been reported (Back & Rogers, 1987, Krieter et al., 1995), although glucuronidation by the liver is generally considered the major route of this metabolism for compounds. Enhanced potency of compounds after glucuronidation has also been reported only recently, perhaps most extensively for the 6-glucuronide of morphine (Milne et al., 1996). Only when the molecular mechanism of action is discovered for this novel class of cholesterol absorption inhibitors, will we be able to discern whether the glucuronide of SCH58235 has inherent activity per se or whether the glucuronide simply indirectly enhances the activity by delivering parent compound back to the site of action after enzymatic hydrolysis of the glucuronide moiety.
From a clinical perspective, this novel class of potent, selective cholesterol absorption inhibitors may present a new approach to the pharmacological treatment of hypercholesterolemia by preventing the absorption of cholesterol from the intestine (Dawson & Rudel, 1999). Currently, the most widely used pharmacological therapy affects plasma cholesterol by decreasing cholesterol biosynthesis. The combination of SCH58235 and the HMG CoA reductase inhibitors has shown synergy in cholesterol-lowering in preclinical trials (Davis et al., 1995). Thus, the development of SCH58235 may provide the clinician with the first opportunity to treat the two primary contributors of plasma cholesterol, biosynthesis and dietary intake.
Abbreviations
- AAALAC
American Association for Accreditation of Laboratory Animal Care
- ACAT
acyl-CoA:cholesterol acyltransferase
- HMG-CoA
hydroxymethylglutaryl coenzyme A
- ID50
dose at which 50% inhibition occurs
- LDL
low density lipoprotein, MW, molecular weight
- RP-HPLC: reversed phase-high performance liquid chromatography; SCH58235 (Ezetimibe) ((1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone); TLC
thin-layer chromatography
References
- BACK D.J., ROGERS S.M. Review: first-pass metabolism by the gastrointestinal mucosa. Aliment. Pharmacol. Ther. 1987;5:339–357. doi: 10.1111/j.1365-2036.1987.tb00634.x. [DOI] [PubMed] [Google Scholar]
- BERGMAN M., MORALES H., MELLARS L., KOSOGLOU T., BURRIER R., DAVIS H.R., Jr, SYBERTZ E.J., POLLARE T.The clinical development of a novel cholesterol absorption inhibitor XII International Symposium on Drugs Affecting Lipid Metabolism 1995(abstract)
- BURNETT D.A., CAPLEN M.A., DAVIS H.R., BURRIER R.E., CLADER J.W. 2-Azetidinones as inhibitors of cholesterol absorption. J. Med. Chem. 1994;37:1733–1736. doi: 10.1021/jm00038a001. [DOI] [PubMed] [Google Scholar]
- DAVIS H.R., WATKINS R.W., SALISBURY B.G., COMPTON D.S., SYBERTZ E.J., BURRIER R.E. Effect of the cholesterol absorption inhibitor SCH48461 in combination with the HMG-CoA reductase inhibitor lovastatin in rabbits, dogs, and rhesus monkeys. Atherosclerosis. 1994;109:162. [Google Scholar]
- DAVIS H.R., VAN HEEK M., WATKINS R.W., ROSENBLUM S.B., COMPTON D.S., HOOS L., MCGREGOR D.G., PULA K., SYBERTZ E.J. The hypocholesterolemic activity of the potent cholesterol absorption inhibitor SCH58235 alone and in combination with HMG CoA reductase inhibitors (abstract) XII International Symposium on Drugs Affecting Lipid Metabolism (DALM) 1995.
- DAWSON P.A., RUDEL L.L. Intestinal cholesterol absorption. Curr. Opin Lipidol. 1999;10:315–320. doi: 10.1097/00041433-199908000-00005. [DOI] [PubMed] [Google Scholar]
- FOLCH J., LEES M., SLOAN-STANLEY G.H. A simple method for isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- HAWK C.T., LEARY S.L. Formulary for Laboratory Animals 1995American College of Laboratory Animal Medicine; p. 74 [Google Scholar]
- IANNUCCI R.M., KACZYNSKI E.J., ACHANFUO-YEBOAH J., ALVAREZ M., BLUMENKRANTZ N.B., CHOWDHURY S.K., ALTON K.B., PATRICK J.E., CAYEN M.N. Metabolism of SCH58235 in the human, rat and dog (abstract) 47th American Society for Mass Spectrometry. 1999.
- KRIETER P.A., COLLETTI A.E., MILLER R.R., STEARNS R.A. Absorption and glucuronidation of the angiotensin II receptor antagonist losartan by the rat intestine. J. Pharm. Exp. Ther. 1995;273:816–822. [PubMed] [Google Scholar]
- LIPID The Long-Term Intervention with Pravastatin in Ischaemic Disease. (1998). Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Eng. J. Med. 1998;339:1349–1357. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- MILNE R.W., NATION R.L., SOMOGYI A.A. The disposition of morphine and its 3- and 6-glucuronide metabolites in humans and animals, and the importance of the metabolites to the pharmacological effects of morphine. Drug Metab. Rev. 1996;28:345–472. doi: 10.3109/03602539608994011. [DOI] [PubMed] [Google Scholar]
- NATIONAL RESEARCH COUNCIL Diet and Health . National Academy Press, Washington, DC; 1989. Implications for Reducing Chronic Disease Risk; pp. 529–547. [PubMed] [Google Scholar]
- ROSENBLUM S.B., HUYNH T., AFONSO A., DAVIS H.R., YUMIBE N., CLADER J.W., BURNETT D.A. Discovery of 1-(4-fluorophenyl) - (3R) - [3 - (4 - fluorophenyl) -(3S)-hydroxypropyl]-(4S)-(4-hydroxyphenyl)-2-azetidinone ( SCH58235): a designed, potent, orally active inhibitor of cholesterol absorption. J. Med. Chem. 1998;41:973–980. doi: 10.1021/jm970701f. [DOI] [PubMed] [Google Scholar]
- SACKS F.M., PFEFFER M.A., MOYE L.A., ROULEAU J.L., RUTHERFORD J.D., COLE T.G., BROWN L., WARNICA J.W., ARNOLD J.M., WUN C.C., DAVIS B.R., BRAUNWALD E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- SALISBURY B.G., DAVIS H.R., BURRIER R.E., BURNETT D.A., BOYKOW G., CAPLEN M.A., CLEMMONS A.L., COMPTON D.S., HOOS L.M., MCGREGOR D.G., SCHNITZER-POLOKOFF R., SMITH A.A., WEIG B.C., ZILLI D.L., CLADER J.W., SYBERTZ E.J. Hypocholesterolemic activity of a novel inhibitor of cholesterol absorption, SCH48461. Atherosclerosis. 1995;115:45–63. doi: 10.1016/0021-9150(94)05499-9. [DOI] [PubMed] [Google Scholar]
- 4S Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- SHEPHERD J., COBBE S.M., FORD I., ISLES C.G., LORIMER A.R., MACFARLANE P.W., MCKILLOP J.H., PACKARD C.J. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N. Engl. J. Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- SYBERTZ E.J., DAVIS H.R., VAN HEEK M., BURRIER R.E., SALISBURY B.G., ROMANO M.T., CLADER J.W., BURNETT D.A.SCH48461, a novel inhibitor of cholesterol absorption Atherosclerosis X 1995Pub. Elsevier; 311–315.ed. Woodford, F.P., Davignon, J. & Sniderman, A. pp [Google Scholar]
- VAN HEEK M., FRANCE C.F., COMPTON D.S., MCLEOD R.L., YUMIBE N.P., ALTON K.B., SYBERTZ E.J., DAVIS H.R. In vivo metabolism-based discovery of a potent cholesterol absorption inhibitor, SCH58235, in the rat and rhesus monkey through the identification of the active metabolites of SCH48461. J. Pharm. Exp. Ther. 1997;283:157–163. [PubMed] [Google Scholar]
- ZBAIDA S., SHANNON D., DU Y., NG K., BLUMENKRANTZ N., YUMIBE N., PATRICK J., CAYEN M.In vitro metabolism of SCH58235, 1-(4-fluorophenyl)-(3R)-[3-(4-fluorophenyl)-(3S)-hydroxypropyl] - (4S) -(4-hydroxyphenyl)-2-azetidinone, by liver and kidney slices 12th International Symposium of Microsomes and Drug Oxidations, Montpellier, France 1998(abstract)