

Abstract
Cisapride is a prokinetic drug that is widely used to facilitate gastrointestinal tract motility.
Structurally, cisapride is a substituted piperidinyl benzamide that interacts with 5-hydroxytryptamine-4 receptors and which is largely without central depressant or antidopaminergic side-effects.
The aims of this study were to investigate the metabolism of cisapride in human liver microsomes and to determine which cytochrome P-450 (CYP) isoenzyme(s) are involved in cisapride biotransformation. Additionally, the effects of various drugs on the metabolism of cisapride were investigated.
The major in vitro metabolite of cisapride was formed by oxidative N-dealkylation at the piperidine nitrogen, leading to the production of norcisapride.
By using competitive inhibition data, correlation studies and heterologous expression systems, it was demonstrated that CYP3A4 was the major CYP involved. CYP2A6 also contributed to the metabolism of cisapride, albeit to a much lesser extent.
The mean apparent Km against cisapride was 8.6±3.5 μM (n=3). The peak plasma levels of cisapride under normal clinical practice are approximately 0.17 μM; therefore it is unlikely that cisapride would inhibit the metabolism of co-administered drugs.
In this in vitro study the inhibitory effects of 44 drugs were tested for any effect on cisapride biotransformation. In conclusion, 34 of the drugs are unlikely to have a clinically relevant interaction; however, the antidepressant nefazodone, the macrolide antibiotic troleandomycin, the HIV-1 protease inhibitors ritonavir and indinavir and the calcium channel blocker mibefradil inhibited the metabolism of cisapride and these interactions are likely to be of clinical relevance. Furthermore, the antimycotics ketoconazole, miconazole, hydroxy-itraconazole, itraconazole and fluconazole, when administered orally or intravenously, would inhibit cisapride metabolism.
Keywords: Cisapride, prokinetic, CYP3A4, CYP2A6, norcisapride, pharmacokinetics, drug–drug interactions, in vitro, human liver microsomes
Introduction
Cisapride ((±)-cis-4-amino-5-chloro-N-[1-[3-(4-fluorophen-oxy)propyl]-3-methoxy-4-piperidinyl]-2-methoxybenzamide) is a prokinetic agent used to treat the pathophysiologic abnormalities of gastrointestinal motility (Wiseman & Faulds, 1994; Verlinden et al., 1988). Without treatment this condition can lead to gastrointestinal reflux disease and also to a variety of gastrointestinal tract motility disorders including diabetic gastroparesis and irritable bowel syndrome (Wiseman & Faulds, 1994; Verlinden et al., 1988). Previously used prokinetic agents, such as bethanechol and metoclopramide, have been associated with central nervous system side-effects; however, these effects appear to be minimal when cisapride is administered instead (Horowitz et al., 1987; Ramirez & Richter, 1993; McCallum et al., 1988). The mechanism of action of cisapride is not completely understood, but it is believed to be caused by an increase of acetylcholine release from the myenteric plexus. This effect is likely to be mediated through stimulation of 5-hydroxytryptamine 4 (5-HT4) receptors (Buchheit & Buhl, 1991).
The metabolism and excretion of 14C-cisapride have been studied after oral dosing to healthy volunteers. Cisapride monohydrate is rapidly and almost completely absorbed after an oral dose, as demonstrated in a mass balance trial in humans with radiolabelled drug. However, the absolute bioavailability of an oral solution of cisapride monohydrate is approximately 40–50%, due to a significant first pass metabolism. Although intravenous clearance is 7.9 l/h, clearance after oral dosing is 20 l/h. Since oral clearance is higher than expected from hepatic blood flow, presystemic metabolism is not confined to the liver but also occurs in the gut wall. Cisapride is extensively metabolised, primarily by oxidative N-dealkylation to norcisapride (43% of the dose) and aromatic hydroxylation (16% of the dose) (Meuldermans et al., 1988). The contribution of the metabolites to the overall pharmacological activity of cisapride is reported to be negligible. In vitro experiments using human liver microsomes highly reflect the metabolic pattern observed in vivo. Cisapride is metabolised in vitro primarily to norcisapride via oxidative N-dealkylation at the piperidine nitrogen. In vivo, the major metabolic product is also norcisapride (Meuldermans et al., 1988). Generally, cisapride is a well-tolerated drug and the most commonly reported side-effects (loose stools, diarrhoea, borborygmi and abdominal cramps) are usually self-limiting and can be predicted from the pharmacological profile of the drug. Prepulsid® has been on the market since 1988 and is available in more than 90 countries world-wide. It is estimated that over 190 million patient treatments have been administered since the launch of cisapride. Some concerns have been raised about the cardiovascular safety of cisapride following postmarketing reports of cardiac arrhythmias and potentially fatal polymorphic ventricular arrhythmias (torsades de pointes). However, an analysis of these reports of serious ventricular arrhythmias (Serious VA) show that most of the reports described patients with labelled risk factors for these events such as significant underlying conditions, co-morbidities predisposing to arrhythmias, concomitant administration of medications which might result in QT prolongation or concomitant use of contraindicated CYP3A4 inhibitors (Wysowski & Bacsanyi, 1996). The Company Core Data Sheet (CCDS) of cisapride emphasizes appropriate contraindications and warnings/precautions for the use of cisapride in patients with conditions or concomitant medications that could predispose them to QT prolongation and/or cardiac arrhythmias. A review of all post-marketing reports received by Janssen world-wide since the first marketing of Prepulsid® confirms the infrequent world-wide occurrence of serious VA and sudden death in association with cisapride use. Several epidemiological trials have been performed and concluded that cisapride is not associated with an increased risk of serious VA or other cardiac events. A large epidemiological study was performed in more than 36,000 patients prescribed cisapride and shows an incidence of serious VA which is ‘consistent with an absence of any cisapride-induced increase in rates of arrhythmic events, at least under the conditions of cisapride usage that were prevalent in the first half of this decade' (Walker et al., 1999). The investigators concluded that ‘serious rhythm disorders were not associated with cisapride use, although the upper confidence bounds to not rule out an increase in risk'. Also, the use of H2 blockers, omeprazole and metoclopramide, was examined in this study. After adjustment for confounding factors, neither the use of cisapride nor any of these drugs was associated with an appreciable increase in the incidence of serious arrhythmias. As a rule, the patients had a significant underlying pathology, such as history of coronary disease and arrhythmia, renal insufficiency or failure, electrolyte imbalance or were using medications associated with arrhythmia or QT prolongation. These patients may be at higher risk of developing ECG changes. The CCDS was revised to emphasize that cisapride is contraindicated in patients with conditions leading to QT prolongation and/or cardiac arrhythmia. Also grapefruit juice was added as a potential interaction. From that moment on, the number and percentage of reports in which patients received contraindicated CYP3A4 inhibitors decreased. When used according to the recommended dosage, and taking into consideration the contraindications and risk factors that have been identified in the CCDS, there is little evidence to suggest that cisapride poses a significant risk of arrhythmias in the general patient population.
Bedford & Rowbotham (1996), published clinically significant drug–drug interactions with cisapride. Concomitant administration of cisapride with drugs that inhibit the cytochrome P450 3A4 enzyme (such as ketoconazole, itraconazole, fluconazole, miconazole, erythromycin, clarithromycin, troleandomycin, nefazodone, indinavir and ritonavir) increased the cisapride plasma concentrations and might result in a prolonged QT interval. Because of the importance of metabolic drug–drug interactions, the aim of the present study was the investigation of the cytochrome P-450 forms involved in the metabolism of cisapride. Using inhibition, correlation and heterologous expression experiments, the results detailed in this study indicate the major CYP isoenzyme involved in cisapride metabolism. In addition, in vitro drug–drug interaction studies were performed for a variety of compounds that are likely to be co-administered with cisapride.
Methods
Materials
14C-Cisapride was labelled in the amide group at Janssen Pharmaceutica, Beerse, Belgium (Figure 1) (Janssen et al., 1987). The compound was dissolved in ethanol, and its radiochemical purity was determined to be approximately 99% (radio-HPLC analysis). After evaporation of the solvent under a stream of nitrogen, the 14C-cisapride was dissolved in 0.5 M lactic acid in an appropriate range of concentrations for later kinetic studies.
Figure 1.
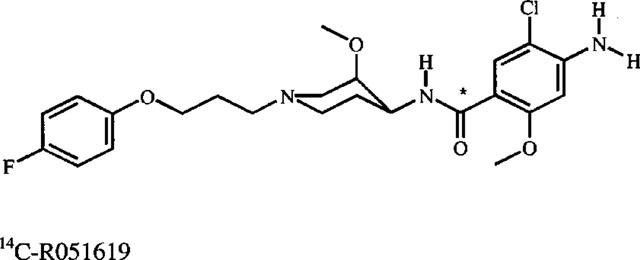
Structure of 14C-cisapride. The asterisk denotes the position of radiolabel.
The compounds used in this study were purchased from: Upjohn (alprazolam, clindamycin) Janssen Chimica (erythromycin), Sigma (azithromycin, diltiazem, nifedipine, ofloxacin, quinidine), Pliva (azithromycin), Smithkline Beecham (cimetidine, paroxetine), Abbot Laboratories (clarithromycin, ritonavir), Hoffman-Laroche (diazepam, midazolam, saquinavir), Pfizer (fluconazole, sertraline), Lily Research Laboratories (fluoxetine, norfluoxetine), SOL (fluvoxamine), Hoechst (furosemide), Merck Sharp & Dohme (indinavir), WAKO pure chemicals (josamycin), ICN biochemicals (lincomycin), Rhone-Poulenc-Rorer (metronidazole), Roche (mibefradil), Bristol-Myers Squibb (nefazodone), AB Astra (omeprazole), Glaxo (ranitidine), Abbott Laboratories (ritonavir), Roussel-UCLAF (roxithromycine), TCI America (terbinafine), Marion Merrell Dow (Terfenadine), Endo Laboratories (warfarin) or were synthesized at Janssen Pharmaceutica (astemizole, desmethylastemizole, itraconazole, hydroxy-itraconazole, ketoconazole, miconazole). A liquid scintillation cocktail (Picofluor 30™) was purchased from Packard and all other reagents were obtained from commercial sources, and were of the highest analytical grade. The heterologous expression systems were purchased from Gentest Corporation, MA, U.S.A.
Hepatic microsomes
Liver pieces were obtained from kidney transplant donors. The liver pieces were homogenized at 4°C with three volumes of a homogenization buffer (1.15% KCl-0.01 M phosphate buffer, pH 7.4), using a Potter-Elvehjem homogenizer with seven vertical strokes and rapid pestle rotation. The crude homogenate was centrifuged (12,000×g, 20 min, 4°C), and the supernatant was further centrifuged (110,000×g, 1 h, 4°C) to sediment the microsomes. The microsomes were washed by resuspending the pellets in an equal volume of homogenization buffer and then 1.0 or 2.0 ml aliquots were immediately frozen in liquid nitrogen prior to storage at ⩽−75°C. Protein content was measured by the method of Lowry et al. (1951), as modified by Miller (1959), using bovine serum albumin (Fluka, Germany, 98% pure) as a standard. The human liver microsomes were always characterized for their CYP content (Omura & Sato, 1964) and their enzymatic activity towards known CYP substrates. The CYP enzymes analysed were: caffeine N3-demethylase (CYP1A2, Berthou et al., 1989), coumarin 7-hydroxylase (CYP2A6, Miles et al., 1990), phenytoin hydroxylase (CYP2C9, Riley et al., 1990), tolbutamide hydroxylase (CYP2C8/9/10, Miners et al., 1988), dextromethorphan O-demethylase (CYP2D6, Dayer et al., 1989), chlorzoxazone 6-hydroxylase (CYP2E1, Peter et al., 1990), erythromycin N-demethylase (CYP3A4, Brian et al., 1990), cyclosporin A-oxidase (CYP3A4, Pichard et al., 1990).
14C-Cisapride incubation conditions
Aliquots of microsomal suspensions, containing 0.5 mg of protein, were pipetted into 10-ml glass tubes which were immersed in ice. Twenty-five μl of 14C-cisapride solution was then included to give a final concentration of 5 μM. To determine the kinetic parameters, the concentration of 14C-cisapride was varied from 1.1–30 μM. After adding 500 μl of a co-factor mixture containing, 0.5 mg glucose-6-phosphate, 0.5 mg MgCl2.6H2O and 0.25 units of glucose 6-phosphate dehydrogenase in 0.5 M phosphate buffer (pH 7.4), homogenization buffer was added to give a final volume of 1.0 ml. The incubations (30 min, 37°C) were initiated by adding 100 μl of a solution of NADP (0.125 mg) in homogenization buffer. After a pre-incubation (5 min at 37°C) the tubes were continuously shaken at 100 oscillations/min in an Heto® shaking water bath. For the determination of the kinetic parameters, reactions were initiated by adding of 14C-cisapride. Reactions were terminated by immersing the tubes in dry ice. Control incubates contained the whole incubation mixture except that drug vehicle was substituted for the drug solution. Blank incubates containing boiled microsomes were incubated under the same conditions as the drug incubates. The samples were stored at ⩽−18°C until analysis by high performance liquid chromatography with on-line radioactivity detection (radio-HPLC).
Diagnostic inhibition experiments
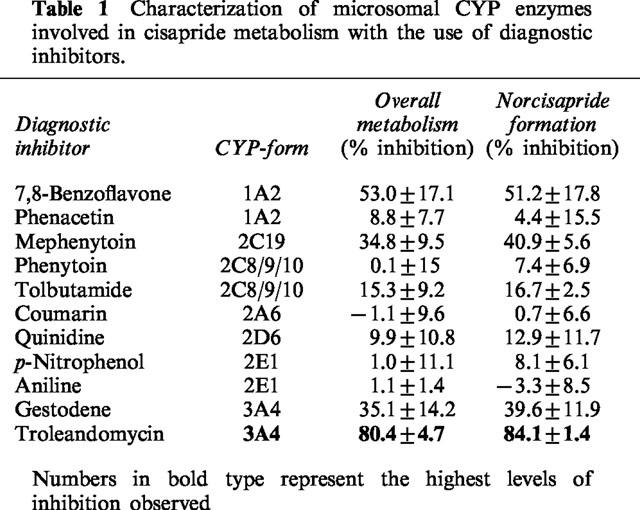
For the inhibition experiments, incubations were carried out in the presence of diagnostic inhibitors, in order to reveal specific CYP isoenzyme activities which might be involved in cisapride metabolism (Table 1). The following diagnostic inhibitors were used: 7,8-benzoflavone (10 μM, CYP1A2, Lee et al., 1994), phenacetin (50 μM, CYP1A2, Tassaneeyakul et al., 1993), coumarin (25 μM, CYP2A6, Miles et al., 1990), phenytoin (50 μM, CYP2C8/9/10, Riley et al., 1990), tolbutamide (50 μM, CYP2C8/9/10, Miners et al., 1988), mephenytoin (500 μM, CYP2C19, Goldstein et al., 1994), quinidine (10 μM, CYP2D6, Ching et al., 1995), p-nitrophenol (50 μM, CYP2E1, Duescher & Elfarra, 1993), aniline (50 μM, CYP2E1, Ono et al., 1996), gestodene (40 μM, CYP3A4, Ward & Back, 1993), troleandomycin (20 μM, CYP3A4, Periti et al., 1992).
Table 1.
Characterization of microsomal CYP enzymes involved in cisapride metabolism with the use of diagnostic inhibitors
Correlation experiments
The rates of 14C-cisapride metabolism and the formation of the major metabolite were correlated with specific CYP enzyme activities in a bank of 16 batches of human liver microsomes. The microsomes were characterized previously by their CYP content and by the level of CYP isoenzyme activities (data not shown). The same incubation conditions as previously described were used. The rate of cisapride metabolism and formation of norcisapride varied approximately 5 fold (data not shown). The metabolism rates of cisapride or the norcisapride formation rates in these batches of liver microsomes were correlated with the caffeine N3-demethylation, coumarin 7-hydroxylation, phenytoin hydroxylation, tolbutamide hydroxylation, dextromethorphan O-demethylation, chlorzoxazone 6-hydroxylation, erythromycin N-demethylation and cyclosporin A-oxidation by linear regression analysis.
Co-medication studies
The incubations were carried out exactly as described in the section 14C-cisapride incubation conditions. Depending on the solubility of the drug, drugs were preferentially dissolved in water and if necessary in methanol or dimethyl sulfoxide (DMSO). Solvent concentrations did not exceed 0.5%. Prior to addition of 14C-cisapride, troleandomycin and diltiazem was metabolized for 10 or 20 min, with the microsomes and an NADPH-generating system.
Heterologous expression systems
The metabolism of 5 μM 14C-cisapride was studied in microsomes prepared from human lymphoblastoid or insect cells expressing human CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 or CYP3A4, in combination with human reductase. A microsomal suspension containing 50 pmol P-450, microsomes containing CYP1A2, CYP2C19, CYP2D6, CYP2E1 or CYP3A4 were diluted in 50 mM Tris-HCl buffer pH 7.5; microsomes containing CYP2A6 were diluted in a 50 mM Tris-HCl buffer pH 7.5 and microsomes containing CYP2C9 were diluted in a 100 mM Tris-HCl buffer pH 7.5. 25 μl of a 14C-cisapride solution was added to give a final cisapride concentration of 5 μM. After adding 500 μl of a co-factor mixture containing 3.3 mM glucose-6-phosphate, 3.3 mM MgCl2·6H2O and 0.4 units of glucose-6-phosphate dehydrogenase (or 1 unit in case of CYP2E1) dissolved in the corresponding buffer, homogenization buffer was added to give a final volume of 1.0 ml. After a pre-incubation of 5 min at 37°C, the incubations were started by adding 100 μl of a solution of NADP (final concentration of 1.3 mM) in the corresponding buffer. After 30 min the reactions were stopped by freezing the incubates in dry ice and the samples were stored at ⩽−18°C, until analysis by radio-HPLC. Control incubates were performed with blank human lymphoblastoid or insect cell microsomes.
High performance liquid chromatography
1.0-ml samples were thawed and diluted with 200 μl of methanol. The samples were sonicated for 10 min and then centrifuged (10 min). Suitable aliquots of the supernatant were analysed in duplicate by liquid scintillation counting to ensure quantitative recovery of radioactivity and then the remaining supernatant was injected directly onto the HPLC-column. The metabolism of 14C-cisapride was monitored using on-line radioactivity detection with a Berthold Radioactivity Monitor LB 507 A equipped with a flow-through cell of 1000 μl. The eluate was mixed with Picofluor 30 (Packard) scintillation cocktail delivered by a FMI LB 5031 pump at a flow rate of 4 ml/min. The HPLC-system was composed of a Waters 600/616 MS pump system and the samples were automatically injected by using a Wisp 717 plus automatic injector. A stainless steel column (30 cm×4.6 mm i.d.) was packed with ODS Hypersil C-18 (5 μm) bound phase by a balanced density slurry procedure (Haskel DSTV 122-C pump, 107 Pa). UV-detection at 230 nm was performed using a Waters 996 Diode Array Detector.
In order to make up mass balances of cisapride and its major metabolites, linear gradient elution was applied, starting from 100% solvent A (distilled water containing 0.2% diethylamine) to a mixture of 73% of solvent A and 27% of solvent B (distilled water containing 2% diethylamine/acetonitrile/methanol/tetrahydrofuran (10 : 20 : 20 : 50, v/v/v/v)) over a 1-min period at a flow rate of 1.0 ml/min. These conditions were maintained for 7 min. Subsequently, another linear gradient elution was applied from the previous conditions to 60% solvent B and 40% solvent system A over a 4-min period. These conditions were maintained for 5 min. Finally, a third linear gradient step was applied elution from the previous conditions to 100% solvent B over a 1-min period. These conditions were maintained for 2 min before returning to the start elution conditions.
 |
The amount of unchanged 14C-cisapride and of its major metabolites were calculated from the peaks of radioactivity. The conversion of the peak areas into d.p.m. values was performed by a Nelson 3000 or Millennium version 2 data system, based on a calibration curve of 14C-cisapride. The calibration curve was plotted after injection of known amounts of 14C-cisapride and linear regression analysis of the corresponding radioactive peak areas from the radio-HPLC chromatogram. At regular times, known amounts of 14C-cisapride were injected to check that the detector's output was still quantitative.
Metabolite identification
In order to characterize cisapride and its metabolites, a mixture of the parent compound and a number of reference compounds, postulated as metabolites, were co-injected with the radioactive samples (Figure 2). The reference compounds were monitored by UV-detection, and the radiolabelled drug and metabolites in the incubates were monitored by radioactivity detection. The structures of the possible reference compounds are shown in Figure 3. Mass spectrometry also confirmed the identity of the metabolites.
Figure 2.
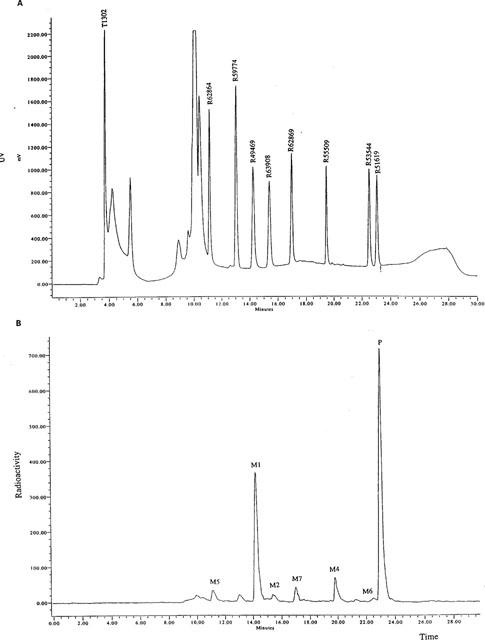
Metabolism of 14C-cisapride by human liver microsomes. (A) illustrates the HPLC separation profile of the cisapride reference compounds. (B) is representative of a typical HPLC metabolic profile for 14C-cisapride in the presence of human liver microsomes (n=4). The metabolites, M1, M2, M5, M6 and M7, were identified by co-chromatography and mass spectrometry. M4 was identified by mass spectrometry. P=parent compound.
Figure 3.
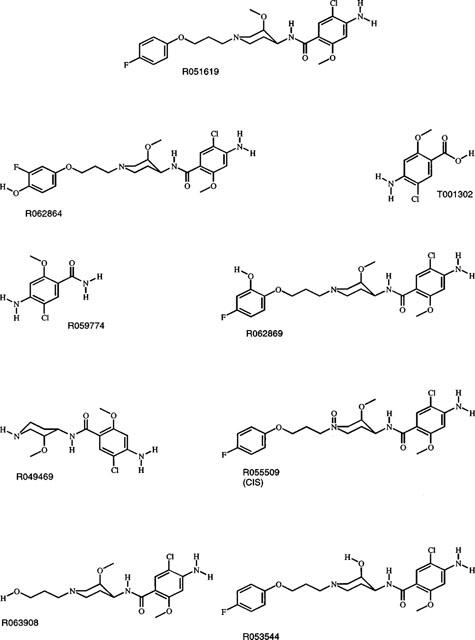
Chemical structures of the reference compounds. The reference compound and the corresponding compound number are indicated in the figure.
Mass spectrometry
For mass spectrometric analysis, uncharacterized metabolites were isolated from a 150-ml incubate of 30 μM 14C-cisapride with human liver microsomes (37°C, 120 min). Briefly, the supernatant from the incubation was concentrated using a Sep-pak cartridge (Waters) and proteins were precipitated using acetonitrile. The sample was injected directly onto the HPLC-column and eluted exactly as described above. The radioactive eluate fractions corresponding to cisapride metabolites were then collected for mass spectrometry. The mass spectra were recorded on a Fisons Autospec Q mass spectrometer coupled to an Opus data system. Approximately 1 μg of sample was dissolved in methanol/dichloromethane (1/1 v v−1) and transferred into the sample cup which was heated until all the solvent was removed. The residue was introduced into the mass spectrometer by means of the direct inlet probe. A programmed evaporation (50–350°C, in about 5 min) of this residue yielded the electron impact (EI) spectra. A comparison of the EI mass spectra of the metabolites with respect to their reference compounds could then be made (data not shown). The mass spectrometric conditions were: electron energy–70 eV; emission current–1.4 mA; ion source temperature–200°C and ion source pressure–10−6 Torr.
Fast Atom Bombardment (FAB) mass spectrometry for M4 was used to gain molecular weight information, since the identity of M4 could not be identified by EI mass spectrometry (data not shown). Approximately 1 μg of sample was dissolved in methanol, deposited on the FAB probe target and mixed with the glycerol matrix. The caesium ion gun produced positively charged caesium ions which bombarded the sample with 20 keV. The anode current was 2 μA.
Data analysis
The relative amounts of unchanged cisapride and of its major metabolites were calculated as the percentage of the amount of injected radioactivity. The apparent Km and Vmax values were determined graphically using Lineweaver-Burke plots.
The following calculation was used to determine the per cent inhibition for the incubations with cisapride and increasing inhibitor concentration:
where C(+ inhibitor) and C(control) represent the relative amounts of overall metabolism of cisapride or its major metabolite in the presence and absence of inhibitor, respectively.
The IC50 values, inhibition concentration resulting in 50% inhibition of the metabolism, were determined from a plot of the per cent inhibition versus the logarithm of the drug concentration. The value was obtained by regression analysis of the linear part of the curve.
Results
14C-cisapride (Figure 1) was incubated with human liver microsomes and any resulting metabolites were identified by HPLC co-chromatography with authentic reference compounds. A UV-chromatogram of the parent and reference compounds, and a typical radio-HPLC chromatogram, are displayed in Figure 2. Up to six metabolite peaks were observed in the radio-HPLC chromatogram, five of these co-chromatographed with authentic reference compounds; the structures of which are depicted in Figure 3. No metabolism of 14C-cisapride was observed in incubates with boiled microsomes (data not shown).
The major metabolite, M1, co-chromatographed with norcisapride (R049469). M2 co-chromatographed with R063908, M5 co-chromatographed with R062864, M6 co-chromatographed with R053544 and M7 co-chromatographed with R062869. Metabolite identity was confirmed by comparing mass spectrometra of the purified metabolites and the available reference compounds.
M4 did not correspond to any of the available reference compounds, however, this metabolite could be identified as the N-oxide of cisapride on the basis of its FAB mass spectrographic characteristics, in addition to its behaviour after pH-dependent HPLC-chromatography and its UV spectrum when compared with another N-oxide of cisapride (R055509). Since it did not co-chromatograph with R055509, M4 most likely represents the trans-isomer of R055509. The metabolic pathways deduced are shown in Figure 4.
Figure 4.
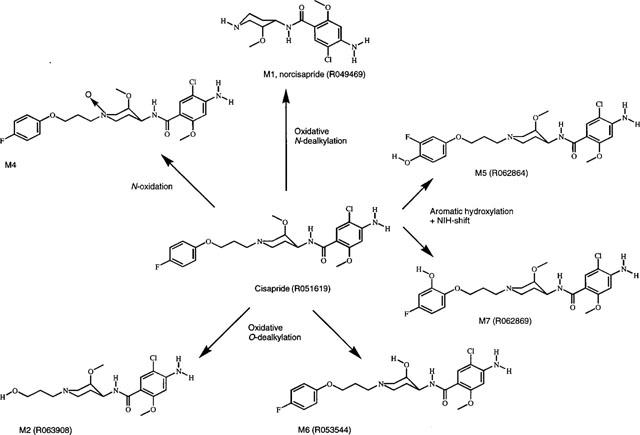
Metabolic profile for the in vitro metabolism of cisapride in human liver microsomes.
The kinetics of cisapride metabolism were investigated using a cisapride concentration range from 1.1–30 μM and an incubation time of 30 min. Unmetabolized cisapride was determined by radio-HPLC. Lineweaver-Burke plots were then plotted in order to ascertain the kinetic parameters for cisapride metabolism (Figure 5). The Vmax value was 523±330 pmol mg−1 min−1 (s.d., n=3) and the apparent Km was 8.6±3.5 μM (s.d., n=3).
Figure 5.
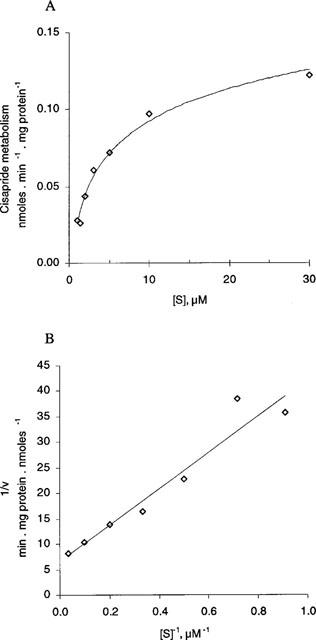
Michaelis-Menten (Figure 5A) and Lineweaver-Burke (Figure 5B) plot for the overall metabolism rate of cisapride in human liver microsomes. Incubations were carried out as described in the Methods section, with the exception of 14C-cisapride concentration which varied between 1.1–30 μM and the incubations which were terminated after 30 min. A representative graph using one batch of human liver microsomes is shown in this figure. Similar results were obtained with a further two batches of human liver microsomes.
To determine which CYP isoenzymes were involved in cisapride elimination, the effects of CYP diagnostic enzyme inhibitors were studied. To achieve this, the inhibitory effect on the overall metabolism of cisapride, and the effect on the formation of its major metabolites, by various CYP inhibitors was investigated (Table 1). Three batches of human liver microsomes were used and the mean±standard deviation values for the per cent inhibition are displayed in Table 1. The relative amounts of cisapride were determined by radio-HPLC. The three batches of microsomes were also analysed for their total CYP content which was as follows: H2–0.309 nmol mg−1, H13–0.262 nmol mg−1 and 9284–0.464 nmol mg−1. Troleandomycin was the most potent inhibitor in the three batches of human liver microsomes, indicating the involvement of CYP3A4 as a major enzyme in the overall metabolism of cisapride and the formation of its major metabolites (Periti et al., 1992). Marked inhibition was also observed with 7,8-benzoflavone (CYP1A2), mephenytoin (CYP2C19) and gestodene (CYP3A4).
The metabolic rates of various probes, specific for various human CYP forms, in 16 batches of human liver microsomes, were then correlated by linear regression with the rates of cisapride metabolism or the rate of norcisapride formation. Values shown are the square of the correlation coefficient (r2) obtained by comparing 16 different batches of human liver microsomes. The correlation coefficients in Table 2 indicate that the major CYP form involved in cisapride metabolism is CYP3A4. In addition, CYP2A6 was also implicated in the metabolism of cisapride and the formation of norcisapride.
Table 2.
Correlation table for CYP isoenzyme activities and the metabolism of 14C-cisapride in human liver microsomes
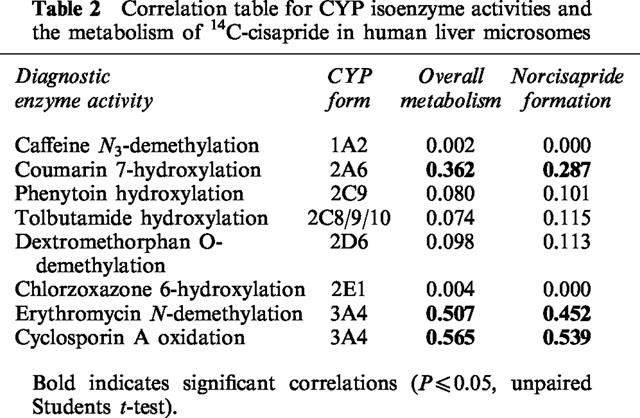
Figure 6 illustrates that the overall metabolism of 14C-cisapride and the formation of norcisapride significantly correlated with erythromycin N-demethylation and cyclosporin A oxidation activity (P⩽0.05). These two metabolic pathways are markers for the involvement of CYP3A4 (Brian et al., 1990; Pichard et al., 1990). The correlation results support the hypothesis that CYP3A4 plays a major role in cisapride metabolism. More direct evidence was obtained from the heterologous expression systems. 14C-cisapride was metabolized by CYP3A4 at a rate of 1.38 pmol min−1 pmol P450−1. For the other CYP's the activity was ⩽0.14 pmol min−1 pmol P450−1.
Figure 6.

Correlation between the overall metabolism rate of cisapride, or the formation of norcisapride, and the metabolism of CYP substrates. The metabolism rates of two specific probes for CYP3A4 were correlated with the metabolism of 14C-cisapride, and norcisapride formation, in the presence of 16 different batches of human liver microsomes. The correlation with erythromycin N-demethylation or cyclosporin A-oxidase was determined by linear regression analysis. The correlation between the CYP3A4 substrate metabolism and the metabolism of cisapride (B,D) and formation of norcisapride (A,C) was significant (P⩽0.05).
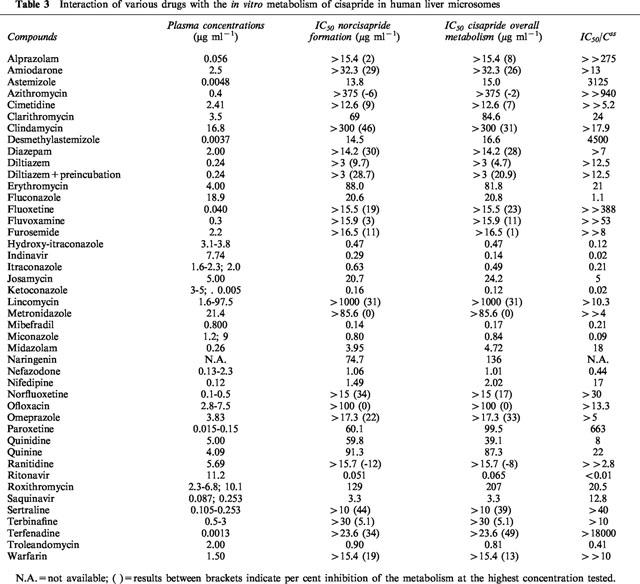
The issue of potential drug–drug interactions was addressed by investigating the effect of primarily CYP3A4-interacting drugs on the in vitro biotransformation of cisapride. Dose response curves were constructed for the various compounds tested and the IC50 values for inhibition of cisapride overall metabolism and for the inhibition of norcisapride formation were calculated. IC50/Css ratio were shown in Table 3, representing the ratio between the IC50 value for the metabolism of cisapride and the therapeutic plasma concentration of the inhibitor at steady state. The results in Table 3 clearly show that the HIV protease inhibitors ritonavir and indinavir; the antifungals ketoconazole, miconazole, hydroxy-itraconazole, itraconazole and to a lesser extent, fluconazole; the macrolide antibiotic troleandomycin, the antidepressant nefadozone and the calcium channel blocker mibefradil were the most potent inhibitors of cisapride metabolism, displaying IC50/Css ratios of ⩽1 μg/ml.
Table 3.
Interaction of various drugs with the in vitro metabolism of cisapride in human liver microsomes
Discussion and conclusion
Cisapride is a registered gastro-intestinal prokinetic agent used for the treatment of motility-related gastro-intestinal disorders (Wiseman & Faulds, 1994). The drug is generally well tolerated (McCallum et al., 1988) but rare cases of cardiac arrhythmias and QT prolongation have been reported. The number of reports of serious VA remains very low considering the extensive patient exposure over the last 11 years. As a rule, these patients had significant underlying pathology, such as histories of coronary disease and arrhythmia, electrolyte imbalance and were using CYP3A4 inhibiting drugs and/or medications associated with arrhythmia or QT prolongation.
The cytochrome P-450 form involved in the metabolism of cisapride was investigated in vitro. The in vitro results described in this study demonstrate that cisapride is metabolized principally via oxidative metabolism by CYP3A4 (Table 1, Table 2, Figure 6). The major metabolite formed is norcisapride (Figure 2). In accordance with the in vitro results, in vivo studies have demonstrated also that cisapride is primarily metabolized to norcisapride (Meuldermans et al., 1988). Other metabolic pathways detected in this study were N-oxidation at the piperidine nitrogen forming the N-oxide of cisapride (M4), O-dealkylation leading to M2 formation and M6 formation (low levels) and aromatic hydroxylation at the fluorophenoxy moiety, leading to the formation of 2-hydroxy-cisapride (M7) and 3-fluoro-4-hydroxy-cisapride (M5), respectively.
Over the concentration range 1.1–30 μM, the metabolism of 14C-cisapride was linear, consistent with a single CYP enzyme being responsible; the apparent Km was 8.6±3.5 μM (n=3, s.d.). Since the peak plasma level of cisapride at steady-state under normal clinical practice is 0.17 μM (Wiseman & Faulds, 1994), it is clear that cisapride is unlikely to inhibit the metabolism of co-administered drugs itself. Thus, the presently available in vitro data do not indicate any relevant interaction by cisapride on the metabolism of other drugs. However, it is possible that cisapride itself may influence the pharmacokinetics of co-administered agents through its pharmacological effect on accelerated gastric-emptying or increased absorption in the small intestine. No clinically related problems have been encountered during the therapeutic trials with cisapride. These effects are generally indicated by increased peak plasma concentration and a shortened time to attain the peak level (Greiff & Rowbotham, 1994). In former pharmacokinetic studies, cisapride was shown to increase the absorption rate of concomitantly given H2-antagonists (cimetidine (Kirch et al., 1989), ranitidine (Rowbotham et al., 1991), diazepam (Bateman, 1986) and ethanol. Accelerated absorption, for example explains transiently enhanced sedative effects of diazepam and ethanol (Bedford & Rowbotham, 1996) in those studies.
In order to make a prediction towards possible competitive inhibition between the metabolism of two drugs, the cytochrome P-450 forms involved in the metabolism of cisapride were investigated. In this in vitro study which was performed by the use of diagnostic inhibitors, correlation studies and heterologous expression systems, the metabolism of cisapride was demonstrated to primarily involve CYP3A4, however, CYP2A6 may be implicated in the formation of norcisapride (Table 2). It is likely, though, that CYP3A4 plays the predominant role in cisapride metabolism since this CYP enzyme accounts for approximately 35% of the total CYP in human liver (Guengerich & Shimada, 1991; Shaw et al., 1989). In contrast, CYP2A6 is only a minor form in human liver, accounting for less than 1% of the total CYP content (Yun et al., 1991). This probably explains why the potent inhibitors of other CYP2 enzymes, coumarin (Miles et al., 1990), quinidine (Ching et al., 1995), and fluoxetine (Otton et al., 1993), did not significantly affect cisapride metabolism (Tables 1 and 3). Two other compounds, 7,8-benzoflavone and mephenytoin, did exhibit inhibitory properties against cisapride metabolism (Table 1). Previous publications have demonstrated that 7,8-benzoflavone can stimulate, inhibit or be without effect on CYP3A4-catalysed metabolism (Guengerich & Shimada, 1991), and mephenytoin has a Ki value of approximately 160 μM for typical CYP2C19 reactions (Chiba et al., 1993). This would suggest that cisapride metabolism should have been inhibited by more than 50% where an inhibitor concentration of 500 μM if CYP2C19 was involved, since this was not the case (Table 1), involvement of CYP2C19 in cisapride metabolism does not appear to catalyse a major metabolic route.
Identification of the major CYP forms involved in the metabolism of a potential drug is of extreme importance to allow for future predictions for potential drug–drug interactions. The finding that cisapride is predominantly metabolized by CYP3A4 led to the testing of compounds which are known to interact with CYP3A4, and thus potentially affect cisapride metabolism. The results demonstrate that this is indeed the case for a number of the drugs tested (Table 3). The following rank order of potency for the inhibition of the overall metabolism of cisapride and the inhibition of the formation of its major metabolite, norcisapride, was found for the antimycotics: ketoconazole>miconazole>hydroxy-itraconazole>itraconazole>fluconazole. For these compounds, inhibition occurred at IC50 values that were lower or similar to their clinically relevant plasma levels after oral or intravenous administration. Consequently, the inhibition observed in this in vitro study is most probably of clinical relevance. However, after topical application of miconazole (as a cream) or ketoconazole (as a cream, ovule or shampoo) the plasma concentrations are 100–1000 times lower than after oral or intravenous administration, therefore no clinically relevant inhibition of cisapride metabolism is expected in these applications (Daneshmend & Warnock, 1988; Blatchford, 1988). Unpublished data on file at JRF demonstrated that co-administration of cisapride with ketoconazole or fluconazole resulted in an increased area under the concentration-time curve (AUC) of cisapride (personal communication). Ketoconazole resulted in an 8 fold increase and fluconazole more than doubled the AUC of cisapride. The in vitro data on the effect of itraconazole on the metabolism of cisapride were confirmed by Shulman (1996). The antimycotic terbinafine did not affect the metabolism of cisapride at an in vitro concentration of 30 μg ml−1. Therefore, no interaction of terbinafine with the cisapride metabolism at clinically relevant concentrations is to be expected since therapeutical levels of terbinafine are in the range of 0.5–3 μg ml−1. These results are in agreement with a former in vitro study (Back et al., 1989) in which it was demonstrated that a high terbinafine concentration (14.6 μg ml−1 or 50 μM) had no or very low inhibitory effect on CYP3A4 mediated activities such as cyclosporin hydroxylation and ethinylestradiol 2-hydroxylation.
Of the other drugs tested in this study, the antidepressant nefazodone (IC50 1.01 μg ml−1, plasma level 2.3 μg ml−1), the macrolide antibiotic troleandomycin (IC50 0.81 μg ml−1, plasma level 2.0 μg ml−1), the calcium channel blocker mibefradil (IC50 0.17 μg ml−1, plasma level 0.8 μg ml−1) and the HIV-1 protease inhibitors ritonavir (IC50 0.065 μg ml−1, plasma level 11.2 μg ml−1) and indinavir (IC50 0.14 μg ml−1, plasma level 7.74 μg ml−1) will probably show clinically relevant interactions if co-administered with cisapride. For these compounds, the IC50 values were lower than their therapeutic plasma levels (Table 3), which would enable an inhibition of cisapride metabolism.
Interestingly, the in vitro results demonstrated no metabolic interaction between the macrolides clarithromycin and erythromycin and the metabolism of cisapride. In vivo studies, however, did show interaction between the macrolides and cisapride. Van Haarst et al. (1998) reported the results of a study with 12 healthy volunteers who were randomized in an open two-way crossover study with washout periods of 1 week. The subjects received cisapride (4×10 mg daily) for 10 days with concomitant clarithromycin (500 mg bid) from day 6 through 10, or clarithromycin (500 mg bid) for 10 days combined with cisapride (4×10 mg daily) from day 6 through 10. No serious adverse experiences occurred. Clarithromycin alone was associated with a minimal increase in QTc intervals. Monotherapy with cisapride or clarithromycin showed QTc elevations within the normal ranges of diurnal variation. Combination of cisapride and clarithromycin caused an average QT-increase of 25 msec above pre-treatment values and 3 fold increases in cisapride concentrations. Unpublished data on file at Janssen Research Foundation showed that chronic administration of erythromycin doubled the mean cisapride steady-state plasma levels and peak concentrations. This underprediction of the interaction of the macrolides with the metabolism of cisapride can be explained by the following mechanism. The macrolides clarithromycin and erythromycin first need to be converted to a metabolite by cytochrome P-450 3A4, this metabolite binds to cytochrome P-450 resulting in an inactive cytochrome P-450 metabolite complex (Periti, 1992). The severity of the interaction of macrolides with the metabolism of co-administered drugs will depend on the duration of the clinical treatment with macrolides and is therefore difficult to predict in vitro.
The interaction potential of the antibiotics is based on structural factors such as the accessibility of the nitrogen atom and the hydrophobic character of the antibiotic studied (Periti et al., 1992; Delaforge et al., 1983). In case of lincomycin and its synthetically derived analogue clindamycin, interaction is rather unlikely to occur since the nitrogen atom is inserted in a cyclic structure making it unable to produce a complex with the cytochrome P-450 enzyme. This was confirmed in the present in vitro interaction study showing no effect of lincomycin and clindamycin on the metabolism of cisapride. Roxithromycin, however, possesses a freely accessible N-dimethylamino group. The interaction caused by roxithromycin seems of minor importance since the roxithromycin metabolite-cytochrome P-450 complexes are formed to a lesser extent resulting in a rare occurrence of clinically relevant interactions (Periti et al., 1992), as confirmed in the present in vitro study.
A clinically relevant interaction between the protease inhibitors indinavir and ritonavir and the cisapride metabolism is to be expected. Both indinavir and ritonavir showed to be strong inhibitors of CYP3A4. Indinavir strongly inhibited the testosterone 6-β hydroxylation (specifically CYP3A4 mediated). Kinetic analysis showed a Ki value of 0.5 μM (0.31 μg ml−1) (Chiba et al., 1996; Lin et al., 1996). Since the Ki value of indinavir is lower than the plasma level of indinavir observed in normal clinical practice, interaction of indinavir with the metabolism of other CYP-3A4-mediated drugs is to be expected, as has been shown in the present study for cisapride. Ritonavir was also found to be a potent inhibitor of CYP3A4 mediated biotransformation such as nifedipine oxidation, 17-α-ethynylestradiol 2-hydroxylation and terfenadine hydroxylation (Kumar et al., 1996). Based on the present in vitro study, interaction between ritonavir and cisapride is to be expected at clinically relevant concentrations. The protease inhibitor saquinavir will most likely not affect the metabolism of cisapride. At a concentration of 0.3 μg ml−1 saquinavir, a concentration approximating the plasma level of saquinavir, approximately 10% inhibition of the cisapride metabolism could be observed. The quinolone antibacterial agent ofloxacin did not inhibit the cisapride metabolism, these findings are in accordance with a previous study showing that ofloxacin has a low inhibitory effect on the cytochrome P-450 system (Brouwers, 1992).
The in vitro findings show that paroxetine will have no relevant inhibitory effect on the biotransformation of cisapride. The IC50-value for the inhibition of the overall biotransformation of cisapride was much higher than the plasma levels observed after clinical trials. The present findings were in agreement with previous findings showing that paroxetine was a relatively weak inhibitor of the CYP3A4 mediated hydroxylation of alprazolam (von Moltke et al., 1995). Thomas et al. (1998) described a 45-year-old female patient who was reported with near syncope and QT-interval prolongation. Multiple drug interactions were suspected in this patient since she was taking cisapride in combination with diltiazem, albuterol inhaler, doxepin, fluoxetine, furosemide, glyburide, ipratropium inhaler, isosorbide mononitrate, omeprazole, potassium chloride and prednisone. After discontinuing cisapride, the QT-interval returned to normal and symptoms did not recur. According to the authors, diltiazem might act as an inhibitor, resulting in elevated levels of cisapride and in possible prolongation of the QT interval. In the current in vitro study, the antihypertensive diltiazem did only slightly inhibit the cisapride overall metabolism and the formation of norcisapride at a concentration of 3 μg ml−1, a concentration which was more than ten times above the plasma level of diltiazem of 0.24 μg ml−1 observed during normal clinical practice (Uges, 1996). The effect of diltiazem on the cisapride metabolism was investigated with and without preincubation, as a previous study (Sutton et al., 1997) indicated that the N-demethylated metabolites of diltiazem selectively inhibited the CYP3A4 activity. After preincubation with diltiazem, a slightly higher inhibition of the cisapride overall metabolism and norcisapride formation was observed, however still being of no clinical relevance.
Based on the in vitro data, no clinically significant interaction between cimetidine and cisapride is to be expected. A pharmacokinetic study in eight healthy subjects demonstrated that cimetidine slightly but significantly increased the cisapride peak plasma concentration and the AUC (Kirch et al., 1989 and McCallum et al., 1988). The interaction with cimetidine is very limited and according to the authors probably not of clinical significance. Grapefruit juice has been shown to increase the bioavailability of several drugs metabolized by the CYP3A enzymes. The inhibitory effects of a grapefruit bioflavonoid, naringenin, was investigated, showing 50% inhibition of the metabolism of cisapride at an in vitro concentration of 136 μg ml−1. In vivo data show that grapefruit juice increases the oral bioavailability of cisapride in 14 subjects (Gross et al., 1999). The results of this study indicate that both Cmax and AUC values increased when taking cisapride with grapefruit juice. The terminal half-life of cisapride was not affected by co-administration of grapefruit juice, thereby confirming published data that grapefruit juice inhibits intestinal, rather than hepatic, CYP3A4.
In conclusion, it was shown that cisapride is mainly metabolized by CYP3A4 but CYP2A6 also contributes to the metabolism of cisapride. Based on Km determinations it was shown that it is unlikely that cisapride would inhibit the metabolism of co-administered drugs. Potential inhibitors of cisapride metabolism at in vitro concentrations which were clinically relevant were nefazodone, troleandomycin, mibefradil, ritonavir, indinavir and the antifungals ketoconazole, miconazole, hydroxy-itraconazole, itraconazole, fluconazole.
Acknowledgments
We would like to thank the synthesis group headed by Dr C. Janssen for delivering of the radiolabelled cisapride, in particular H. Lenoir, J. Thijssen and A. Knaeps. In addition we are extremely grateful to C. Zwijsen, M. Bockx, L. Le Jeune and D. Deleersnijder for their expert technical assistance. Finally, we would like to thank Dr Gerben van ‘t Klooster, Dr Erik Mannaert, Marc Denayer and Dr Dirk Reyn for their interesting comments.
Abbreviations
- AUC
area under the concentration-time curve
- CCDS
Company Core Data Sheet
- CYP
cytochrome P-450
- DMSO
dimethyl sulphoxide
- EI
electron impact
- FAB
Fast Atom Bombardment
- IC50
inhibition concentration resulting in 50% inhibition of the metabolism
- Radio-HPLC
high performance liquid chromatography with on-line radioactivity detection
- VA
ventricular arrythmias
References
- BACK D.J., STEVENSON P., TJIA J.F. Comparative effects of two antimycotic agents, ketoconazole and terbinafine on the metabolism of tolbutamide, ethinylestradiol, cyclosporin and ethoxycoumarin by human liver microsomes in vitro. Br. J. Clin. Pharmacol. 1989;28:166–170. doi: 10.1111/j.1365-2125.1989.tb05410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATEMAN D.N. The action of cisapride on gastric emptying and the pharmacodynamics and pharmacokinetics of oral diazepam. Eur. J. Clin. Pharmacol. 1986;30:205–208. doi: 10.1007/BF00614304. [DOI] [PubMed] [Google Scholar]
- BEDFORD T.A., ROWBOTHAM D.J. Cisapride drug interactions of clinical significance. Drug Safety. 1996;15:167–175. doi: 10.2165/00002018-199615030-00002. [DOI] [PubMed] [Google Scholar]
- BERTHOU F., RATANASAVANH D., RICE C., PICARD D., VOIRIN T., GUILLOUZO A. Comparison of caffeine metabolism by slices, microsomes and hepatocyte cultures from adult human liver. Xenobiotica. 1989;19:401–417. doi: 10.3109/00498258909042282. [DOI] [PubMed] [Google Scholar]
- BLATCHFORD N.R.The pharmacokinetics of oral and topical ketoconazole Seborrhoeic dermatitis and dandruff a fungal disease Royal Society of Medicine Services International Congress and Symposium Series 198813229–34.Shuster, S and Blatchford, N. (eds)Shuster S, Blatchford, N. (eds). 1988 [Google Scholar]
- BRIAN W.R., SARI M.A., IWASAKI M., SHIMADA T., KAMINSKY L.S., GUENGERICH F.P. Catalytic activities of human liver cytochrome P-450IIIA4 expressed in Saccharomyces cerevisiae. Biochemistry. 1990;29:11280–11292. doi: 10.1021/bi00503a018. [DOI] [PubMed] [Google Scholar]
- BROUWERS J.R. Drug interactions with quinolone antibacterials. Drug Safety. 1992;7:268–281. doi: 10.2165/00002018-199207040-00003. [DOI] [PubMed] [Google Scholar]
- BUCHHEIT K.H., BUHL T. Prokinetic benzamides stimulate peristaltic activity in the isolated guinea pig ileum by activation of 5-HT4 receptors. Eur. J. Pharmacol. 1991;205:203–208. doi: 10.1016/0014-2999(91)90821-7. [DOI] [PubMed] [Google Scholar]
- CHIBA K., KOBAYASHI K., MANABE K., TANI M., KAMATAKI T., ISHIZAKI T. Oxidative metabolism of omeprazole in human liver microsomes. Co-segregation with S-mephenytoin. J. Pharmacol. Exp. Ther. 1993;266:52–59. [PubMed] [Google Scholar]
- CHIBA M., HENSLEIGH M., NISHIME J.A., BALANI S.K., LIN J.H. Role of cytochrome P-450 3A4 in human metabolism of MK-639, a potent HIV protease inhibitor. Drug Metab. Dispos. 1996;24:307–314. [PubMed] [Google Scholar]
- CHING M.S., BLAKE C.L., GHABRIAL H., ELLIS S.W., LENNARD M.S., TUCKER G.T., SMALLWOOD R.A. Potent inhibition of yeast-expressed CYP2D6 by dihydroquinidine, quinidine, and its metabolites. Biochem. Pharmacol. 1995;50:833–837. doi: 10.1016/0006-2952(95)00207-g. [DOI] [PubMed] [Google Scholar]
- DANESHMEND T.K., WARNOCK D.W. Clinical pharmacokinetics of ketoconazole. Clin. Pharmacokinet. 1988;14:13–34. doi: 10.2165/00003088-198814010-00002. [DOI] [PubMed] [Google Scholar]
- DAYER P., LEEMANN T., STRIBERNI R. Dextromethorphan O-demethylation in liver microsomes as a prototype reaction to monitor P-450db1 activity. Clin. Pharmacol. Ther. 1989;45:34–40. doi: 10.1038/clpt.1989.6. [DOI] [PubMed] [Google Scholar]
- DELAFORGE M., JAOUEN M., MANSUY D. Dual effects of macrolide antibiotics on rat liver cytochrome P-450. Biochem. Pharmacol. 1983;32:2309–2318. doi: 10.1016/0006-2952(83)90178-8. [DOI] [PubMed] [Google Scholar]
- DUESCHER R.J., ELFARRA A.A. Determination of p-nitrophenol hydroxylase activity of rat liver microsomes by high pressure liquid chromatography. Anal. Biochem. 1993;212:311–314. doi: 10.1006/abio.1993.1335. [DOI] [PubMed] [Google Scholar]
- GOLDSTEIN J.A., FALETTO M.B., ROMKES-SPARKS M. Evidence that CYP2C19 is the major (S)-mephenytoin 4′-hydroxylase in humans. Biochemistry. 1994;33:1743–1752. doi: 10.1021/bi00173a017. [DOI] [PubMed] [Google Scholar]
- GREIFF J.M., ROWBOTHAM D. Pharmacokinetic drug interactions with gastrointestinal motility modifying agents. Clin. Pharmacokin. 1994;27:447–461. doi: 10.2165/00003088-199427060-00004. [DOI] [PubMed] [Google Scholar]
- GROSS A.J., GOH Y.D., ADDISON R.S., SHENFIELD C.M. Influence of grapefruit juice on cisapride pharmacokinetics. Clin. Pharmacol. Ther. 1999;64:395–401. doi: 10.1016/S0009-9236(99)70133-5. [DOI] [PubMed] [Google Scholar]
- GUENGERICH P., SHIMADA T. Oxidation of toxic and carcinogenic chemicals by human cytochrome P-450 enzymes. Chem. Res. Toxicol. 1991;4:391–407. doi: 10.1021/tx00022a001. [DOI] [PubMed] [Google Scholar]
- HOROWITZ M., MADDOX A., HARDING P.E., MADDERN G.J., CHATTERTON B.E., WISHART J., SHEARMAN D.J. Effect of cisapride on gastric and oesophageal emptying in insulin-dependent diabetes mellitus. Gastroenterology. 1987;93:1899–1907. doi: 10.1016/0016-5085(87)90622-6. [DOI] [PubMed] [Google Scholar]
- JANSSEN C.G.M., LENOIR H.A.C., THIJSSEN J.B.A., KNAEPS A.G., HEYKANTS J.J.P. The synthesis of 3H- and 14C-cisapride. J. Labelled. Compd. Radiopharm. 1987;24:1493–1501. [Google Scholar]
- KIRCH W., JANISCH H.D., OHNHAUS E.E., VAN PEER A. Cisapride-cimetidine interaction: enhanced cisapride bioavailability and accelerated cimetidine absorption. Ther. Drug Monit. 1989;11:411–414. [PubMed] [Google Scholar]
- KUMAR G.N., RODRIGUES A.D., BUKO A.M., DENISSEN J.F. Cytochrome P-450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J. Pharmacol. Exp. Ther. 1996;277:423–431. [PubMed] [Google Scholar]
- LEE H.S., JIN C., PARK J., KIM D.H. Modulation of cytochrome P450 activities by 7,8-benzoflavone and its metabolites. Biochem. Molec. Biol. Int. 1994;34:483–491. [PubMed] [Google Scholar]
- LIN J.H., CHIBA M., BALANI S.K., CHEN I.-W., KWEI G.Y.-S., VASTAG K.J., NISHIME J.A. Species differences in the pharmacokinetics and metabolism of indinavir, a potent human immunodeficiency virus protease inhibitor. Drug Metab. Dispos. 1996;24:1111–1120. [PubMed] [Google Scholar]
- LOWRY O.H., ROSENBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MCCALLUM R.W., PRAKASH C., CAMPOLI-RICHARDS D.M., GOA K.L. Cisapride. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use as a prokinetic agent in gastrointestinal motility disorders. Drugs. 1988;36:652–681. doi: 10.2165/00003495-198836060-00002. [DOI] [PubMed] [Google Scholar]
- MEULDERMANS W., VAN PEER A., HENDRICKX J., LAUWERS W., SWYSEN E., BOCKX M., WOESTENBORGHS R., HEYKANTS J. Excretion and biotransformation of cisapride in dogs and humans after oral administration. Drug. Metab. Dispos. 1988;16:403–409. [PubMed] [Google Scholar]
- MILES J.S., MCLAREN A.W., FORRESTER L.M., GLANCEY M.J., LANG M.A., WOLF C.R. Identification of the human liver cytochrome P-450 responsible for coumarin 7-hydroxylase activity. Biochem. J. 1990;267:365–371. doi: 10.1042/bj2670365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER G.L. Protein determination for large numbers of samples. Anal. Chem. 1959;31:964–971. [Google Scholar]
- MINERS J.O., SMITH K.J., ROBSON R.A., MCMANUS M.E., VERONESE M.E., BIRKETT D.J. Tolbutamide hydroxylation by human liver microsomes. Kinetic characterization and relationship to other cytochrome P-450 dependent xenobiotic oxidations. Biochem. Pharmacol. 1988;37:1137–1144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- OMURA T., SATO R. The carbon monoxide binding pigment of liver microsomes. II Solubilisation, purification and properties. J. Biol. Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- ONO S., HATANAKA T., HOTTA H., SATOH T., GONZALEZ F.J., TSUTSUI M. Specificity of substrate and inhibitor probes for cytochrome P450s: evaluation of in vitro metabolism using cDNA-expressed human P450s and human liver microsomes. Xenobiotica. 1996;26:681–693. doi: 10.3109/00498259609046742. [DOI] [PubMed] [Google Scholar]
- OTTON S.V., WU D., JOFFE R.T., CHEUNG S.W., SELLERS E.M. Inhibition by fluoxetine of cytochrome P450 2D6 activity. Clin. Pharmacol. Ther. 1993;53:401–409. doi: 10.1038/clpt.1993.43. [DOI] [PubMed] [Google Scholar]
- PERITI P., MAZZEI T., MINI E., NOVELLI A. Pharmacokinetic drug interactions of macrolides. Clin. Pharmacokin. 1992;23:106–131. doi: 10.2165/00003088-199223020-00004. [DOI] [PubMed] [Google Scholar]
- PETER R., BÖCKER R., BEAUNE P.H., IWASAKI M., GUENGERICH F.P., YANG C.S. Hydroxylation of chlorzoxazone as a specific probe for human liver cytochrome P-450IIE1. Chem. Res. Toxicol. 1990;3:566–573. doi: 10.1021/tx00018a012. [DOI] [PubMed] [Google Scholar]
- PICHARD L., FABRE I., FABRE G., DOMERGUE J., SAINT AUBERT B., MOURAD G., MAUREL P. Screening for inducers and inhibitors of cytochrome P-450 (cyclosporin A oxidase) in primary cultures of human hepatocytes and in liver microsomes. Drug. Metab. Dispos. 1990;18:595–606. [PubMed] [Google Scholar]
- RAMIREZ B., RICHTER J.E. Promotility drugs in the treatment of gastro-oesophageal reflux disease. Alimentary Pharmacol. Ther. 1993;7:5–20. doi: 10.1111/j.1365-2036.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- RILEY R.J., ROBERTS P., KITTERINGHAM N.R., PARK B.K. Formation of cytotoxic metabolites from phenytoin, imipramine, desimipramine, amitryptiline and mianserin by mouse and human hepatic microsomes. Biochem. Pharmacol. 1990;39:1951–1958. doi: 10.1016/0006-2952(90)90614-q. [DOI] [PubMed] [Google Scholar]
- ROWBOTHAM D.J., MILLIGAN K., MCHUGH P. Effect of cisapride on morphine absorption after oral administration of sustained-release morphine. Br. J. Anaesth. 1991;67:421–425. doi: 10.1093/bja/67.4.421. [DOI] [PubMed] [Google Scholar]
- SHAW P.M., BARNES T.S., CAMERON D. Purification and characterisation of an anti-convulsant-induced human cytochrome P-450 catalysing cyclosporin metabolism. Biochem. J. 1989;263:653–663. doi: 10.1042/bj2630653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHULMAN R.J. Cisapride and the attack of the P-450s. J. Pediatr. Gastroenterol. Nutr. 1996;23:395–397. doi: 10.1097/00005176-199611000-00003. [DOI] [PubMed] [Google Scholar]
- SUTTON D., BUTLER A.M., NADIN L., MURRAY M. Role of CYP3A4 in human hepatic diltiazem N-demethylation: inhibition of CYP3A4 activity by oxidized diltiazem metabolites. J. Pharmacol. Exp. Ther. 1997;282:294–300. [PubMed] [Google Scholar]
- TASSANEEYAKUL W., BIRKETT D.J., VERONESE M.E., MCMANUS M.E., TUKEY R.H., QUATTROCHI L.C., GELBOIN H.V., MINERS J.O. Specificity of substrate and inhibitor probes for human cytochromes P450 1A1 and 1A2. J. Pharmacol. Exp. Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- THOMAS A.R., CHAN L.-N., BAUMAN J.L., OLOPADE C.O. Prolongation of the QT interval related to cisapride-diltiazem interaction. Pharmacotherapy. 1998;18:381–385. [PubMed] [Google Scholar]
- UGES D.R.A. Referentiewaarden van xenobiotica in humaan materiaal. Pham. Weekbl. 1996;130:180–204. [Google Scholar]
- VAN HAARST A.D., VAN T KLOOSTER G.A.E., VAN GERVEN J.M.A., SCHOENMAKER R.C., VAN OENE J.L., BURGGRAAF J., COENE M.C., COHEN A.F. The influence of cisapride and clarithromycin on QT intervals in healthy volunteers. Clin. Pharmacol. Ther. 1998;64:542–546. doi: 10.1016/S0009-9236(98)90137-0. [DOI] [PubMed] [Google Scholar]
- VON MOLTKE L.L., GREENBLATT D.J., COURT M.H., DUAN S.X., HARMATZ J.S., SHADER R.I. Inhibition of alprazolam and desipramine hydroxylation in vitro by paroxetine and fluvoxamine: comparison with other selective serotonin reuptake inhibitor antidepressants. J. Clin. Psychopharmacol. 1995;15:125–131. doi: 10.1097/00004714-199504000-00008. [DOI] [PubMed] [Google Scholar]
- VERLINDEN M., REYNTJENS A., SCHUERMANS V.Safety profile of cisapride. Progress in the treatment of gastrointestinal motility disorders: the role of cisapride Excerpta Medica 1988Amsterdam; 30–36.Ed. Johnson & Lux. pp [Google Scholar]
- WALKER A.M., SZNEKE P., WEATHERBY L.B., DICKER L.W., LANZA L.L., LOUGHLIN J.E., YEE C.L., DREYER N. The risk of serious cardiac arrhythmias among cisapride users in the United Kingdom and Canada. Amer. J. Med. 1999;107:356–362. doi: 10.1016/s0002-9343(99)00241-7. [DOI] [PubMed] [Google Scholar]
- WARD S., BACK D.J. Metabolism of gestodene in human liver cytosol and microsomes in vitro. J. Steroid. Biochem. Molec. Biol. 1993;46:235–243. doi: 10.1016/0960-0760(93)90299-c. [DOI] [PubMed] [Google Scholar]
- WISEMAN L.R., FAULDS D. Cisapride: An updated review of its pharmacology and therapeutic efficacy as a prokinetic agent in gastrointestinal motility disorders. Drugs. 1994;47:116–152. doi: 10.2165/00003495-199447010-00008. [DOI] [PubMed] [Google Scholar]
- WYSOWSKI D.K., BACSANYI J. Cisapride and fatal arrhythmia. N. Engl. J. Med. 1996;335:290. doi: 10.1056/NEJM199607253350416. [DOI] [PubMed] [Google Scholar]
- YUN C.H., SHIMADA T., GUENGERICH F.P. Purification and characterisation of human liver microsomal cytochrome P-450 2A6. Am. Soc. Pharmacol. Exp. Ther. 1991;40:679–685. [PubMed] [Google Scholar]