

Abstract
The neurotoxic actions of kainic acid can be partly suppressed by antagonists acting at N-methyl-D-aspartate (NMDA) receptors. The present study examined the possible role of endogenous components of the kynurenine pathway to this phenomenon.
Administration of kainate (2 nmols) into the hippocampus of anaesthetized rats produced damage in the CA1 and CA3 regions. The involvement of NMDA receptors was confirmed by the ability of dizocilpine (1 mg kg−1) to reduce cell loss in the CA1 region from 92 to 42%.
The co-administration of m-nitrobenzoylalanine (20 nmols into the hippocampus), an inhibitor of kynurenine hydroxylase and kynureninase, together with a systemic injection of the compound (100 mg kg−1, i.p.), afforded some protection against kainate, reducing cell loss from 91 to 48%. Protection was not exerted against damage by quinolinic acid or NMDA, excluding a direct interaction between m-nitrobenzoylalanine and NMDA receptors.
The protective effect of m-nitrobenzoylalanine was not prevented by glycine, which would be expected to reverse protection caused by an elevation in the levels of endogenous kynurenic acid, arguing against a major role for increased levels of kynurenic acid.
The results indicate that inhibition of the kynurenine pathway offers protection against kainate-induced damage. One possible mechanism for the protection is that an increased production of quinolinic acid in the brain, possibly from glial cells and macrophages activated by the initial kainate insult, normally contributes to the local activation of NMDA receptors and thus to kainate-induced cerebral insults. This generation of endogenous quinolinic acid would be suppressed by m-nitrobenzoylalanine.
Keywords: Kainic acid, kynurenines, kynurenic acid, quinolinic acid, excitotoxicity, glycine, hippocampus
Introduction
Kainic acid is a glutamate analogue with excitatory (Shinozaki & Konishi, 1970) and neurotoxic activity (Olney et al., 1974) and which is frequently used as an experimental tool in the study of excitotoxic and neurodegenerative processes, partly because of similarities in the pattern of damage with that resulting from ischaemia (Liu et al., 1996; Vecsei et al., 1998). Kainate acts upon several subtypes of glutamate receptor subunits to produce pronounced depolarization, calcium entry into neurones, and cell damage or death after excessive stimulation (Schwob et al., 1980; Michaelis, 1998). It has the advantage over more non-selective neuronal poisons and metabolic inhibitors of producing an axon-sparing lesion in which the cell somata are normally damaged preferentially with respect to the nerve axons (Schwarcz & Coyle, 1977).
The pharmacology of kainate receptors is quite distinct from that of other ionotropic glutamate receptors, including those for N-methyl-D-aspartate (NMDA), there being a range of antagonists available to act on the different receptor subunits to achieve a selective block of kainate and NMDA-induced excitatory effects on neurones. However, there are several reports in which the neurotoxic effects of kainate have been reduced by antagonists acting at the NMDA-sensitive population of glutamate receptors, sites at which kainate is believed to have little or no direct action (Fariello et al., 1989; Wolf et al., 1991; Berg et al., 1993; Virgili et al., 1992; Planas et al., 1995). The usual explanation of this phenomenon is that the depolarizing action of kainate will initiate a cycle of events in which glutamatergic neurons will be activated to release glutamate which can then in turn act upon both NMDA as well as non-NMDA receptors. The antagonism cannot be explained as a suppression of seizure activity, since the behavioural and electrographic correlates of kainate-induced seizures are increased, rather than decreased, by dizocilpine (Fariello et al., 1989).
The neurotoxic action of agents such as kainate is accompanied by a reactive gliosis in which astroglia and microglia proliferate and migrate into the region affected by kainate, and macrophages infiltrate the region (Jorgensen et al., 1993). These activated glia are able to secrete a range of inflammatory mediators which may affect the course of neuronal damage. These include kynurenines produced from tryptophan, with a significant increase (Heyes et al., 1992; 1996) in the secretion of the selective NMDA receptor agonist quinolinic acid (Stone & Perkins, 1981; see Stone, 1993; Espey et al., 1997). The amounts of quinolinic acid secreted by such cells can easily exceed those known to be neurotoxic after prolonged exposure (Pemberton et al., 1997). Quinolinic acid of inflammatory cell origin could, therefore, account at least partly for an indirect activation of NMDA receptors by kainate, contributing to the delayed neurodegeneration produced by this compound. The present study was designed to test this hypothesis.
Methods
Intrahippocampal injections
Male Wistar rats weighing between 200 and 250 g were used in all experiments. All animals were housed singly and provided with free access to food and water. Animals were anaesthetized with chloral hydrate (360 mg kg−1) and placed in a stereotaxic frame. The scalp was incised and a burr hole made through the skull to permit access of the injection needle into the hippocampus at the desired co-ordinates (anteroposterior: 3.0 mm behind the bregma suture, dorsoventral: 2.8 mm below the cortical surface and lateral: 3.0 mm from the midline suture (Paxinos & Watson, 1986). The needle was then inserted and left in place for 2 min before the injection of test agents. The compounds used in this study were all introduced through a 29 gauge needle, injections being made in a volume of 1 μl at a constant rate of 0.3 μl min−1 using a Sage infusion pump (Jones et al., 1998). The injection needle was allowed to remain in place for 2 min after ending the injection so as to prevent leakage of drug along the needle track. The scalp was then sutured and the animals left to recover for seven days. In order to suppress seizure activity and limit the neuronal damage produced by kainate to direct rather than indirect actions, all animals were treated with clonazepam, 0.1 mg kg−1 at the time of removal from the stereotaxic frame. The ability of this low dose to prevent seizures but not hippocampal toxicity has been described previously (MacGregor et al., 1993).
Quinolinic acid was dissolved in 0.1 N NaOH and then diluted with 165 mM NaCl solution. The pH of the solution was then adjusted using 1 N HCl to between 7 and 7.6 before making up to volume by the addition of further saline. Kainic acid was dissolved in 165 mM NaCl solution. Kainic acid was injected at a dose of 2 nmols, and quinolinic acid was injected at a dose of 120 nmols, doses which have been used frequently in previous studies of excitotoxicity and which we had found in preliminary work to produce a sub-total destruction of the CA1 and CA3 regions of the hippocampus. Meta-nitrobenzoylalanine (mNBA) was co-administered at two dose levels by mixing solutions of the individual agents in the appropriate proportions. A single intraperitoneal injection of mNBA at a dose of 100 mg kg−1 was also administered to all animals receiving intrahippocampal mNBA, 24 h after removal of the animal from the stereotaxic frame, in order to extend the duration of the inhibition of kynurenine synthesis.
Tissue fixing and slicing
Rats were killed by an overdose of sodium pentobarbitone 7 days after recovery from the intrahippocampal injections. The chest was opened to expose the heart and 20 ml of 0.9% NaCl solution was infused over approximately 1 min via a 26 gauge needle inserted into the left cardiac ventricle. This was followed immediately by 20 ml of a solution of 10% formalin buffered to pH 7.2. The brain was then removed and stored in fixative for up to 1 week. At removal, all brains appeared completely white, confirming the flushing of blood from the cerebral vessels and rapid access of the fixative. A slice of brain, 2 mm thick, was prepared to include the location of the injection track, which was normally apparent from the residual dimpling of the cortical surface produced by the needle penetration. The 2 mm block of brain was dehydrated and impregnated with paraffin wax throughout before embedding in wax. Sections were cut 6 μm thick, mounted on slides and stained with cresyl fast violet.
Sections were subsequently examined for damage under a light microscope by an observer blinded to the drug treatment. Normal, intact pyramidal neurones were identified as those with a clearly rounded appearance with a clear nucleus and nucleolus (Figure 1). The damage was quantified in the CA1 region by selecting every third section at a distance of 200–250 μm from the site of the needle track (three sections per hippocampus) and counting the number of intact, surviving neurones in the field of view at a magnification of 100×. The mean number of cells from these three sections was then calculated to give the number of surviving neurons in that area.
Figure 1.

Photomicrographs of the CA1 region of hippocampus in (A) a control animal, (B) 7 days following an intrahippocampal injection of kainic acid, 2 nmols, and (C) 7 days following a combined administration of kainic acid 2 nmols and meta-nitrobenzoylalanine (mNBA) 20 nmols. The control hippocampus shows normal healthy neurons with a full, rounded outline containing a clear rounded nucleus and central necleolus. The damaged section in contrast has no healthy pyramidal neurons, but the stratum pyramidal is now occupied by the small poorly-staining nuclei of degenerating neurons and there is an increase in the number of darkly-staining nuclei of glial cells. In section (C), mNBA has largely protected against kainic acid, the majority of pyramidal cells in this animal being normal and healthy in appearance. Scale bar 100 μm.
Analysis of variance (ANOVA) was followed by the Student-Newman-Keuls post-test for multiple comparisons to determine any statistical significance. Significance refers to results where P<0.05 was obtained.
Results
Effects of dizocilpine
Kainic acid produced substantial damage to the CA3 and CA1 regions of the hippocampus (Figure 1). The dose used produced a mean depletion of cell numbers in CA1 by between 87 and 92% in the different series of animals reported here.
The injection of dizocilpine at a dose of 0.2 mg kg−1 or 1 mg kg−1 tended to reduce the amount of damage produced with the protection being significant at the higher dose level (Figure 2). The amount of cell loss was reduced from 92–41.7% at this dose (P<0.01, n=3).
Figure 2.

Histogram summarizing the number of neurons surviving 7 days after an intrahippocampal injection of kainic acid 2 mols (Kain) either alone or with an intraperitoneal injection of dizocilpine (MK) at a dose of 0.l2 or 1 mg kg−1. Results are shown as mean±s.e. mean (n=3). Analysis was performed by ANOVA followed by the Student-Newman-Keuls test for multiple comparisons. **P<0.01 compared with kainic acid alone. ++P<0.01; +++P<0.001 compared with control.
Effect of mNBA
The administration of mNBA 20 nmols into the hippocampus and 100 mg kg−1 systemically had no effect on neuronal number when administered in the absence of other agents (n=3). The lower of the two doses tested of 5 nmols of mNBA produced no significant changes in the kainate effect. When combined with kainate, however, as an intrahippocampal injection of 20 nmols together with the subsequent intraperitoneal injection, it proved able to suppress the kainate-induced damage, reducing neuronal loss from 90.6% in control animals to 48% after kainate treatment in one set of animals (P<0.05, n=3; Figure 3) and from 87–49% (P<0.01, n=4; Figure 4) in a second group used in testing for the effects of glycine.
Figure 3.

Histograms summarizing the number of neurons surviving after treatment with kainic acid 2 nmols (Kain) administered either alone or together with an intrahippocampal (5 or 20 nmols) and systemic injection of mNBA. Results are shown as mean±s.e.mean (n=3). Analysis was performed by ANOVA followed by the Student-Newman-Keuls test for multiple comparisons. *P<0.05; compared with kainic acid alone. +++P<0.001 compared with control.
Figure 4.

Histograms summarizing the number of neurons surviving after treatment with kainic acid 2 nmols (Kain) administered either alone or together with combined treatments with kainic acid, mNBA and glycine (25 nmols). Results are shown as mean±s.e.mean (n=4). Analysis was performed by ANOVA followed by the Student-Newman-Keuls test for multiple comparisons. **P<0.01 compared with kainic acid alone. ++P<0.01; +++P<0.001 compared with control.
Combination of mNBA and glycine
In order to assess the role of the strychnine-resistant site of the NMDA receptor in the protection afforded by mNBA, a series of experiments was performed in which glycine was administered at a high dose (250 nmols μl−1) along with the kainate or the kainate plus mNBA injections. The dose of 20 nmols mNBA showed a protective effect against kainate (n=4; Figure 4) as in the preceding set of experiments, but this was not changed by the presence of glycine. Indeed, there was a slightly greater mean preservation of neurones with the combined kainate/mNBA/glycine cocktail compared with that produced by mNBA itself though this difference was not statistically significant. (Figure 4).
Combinations of mNBA and quinolinic acid
In order to control for the possibility that mNBA was able to have a direct action on the NMDA receptor, one set of animals was prepared in which damage was induced by quinolinic acid at a dose of 120 nmols. In these animals, the administration of mNBA into the hippocampus and systemically did not change the amount of damage induced by quinolinic acid (n=4; Figure 5). Similarly mNBA did not change the damage produced by the injection of NMDA (2 nmols) into the hippocampus.
Figure 5.
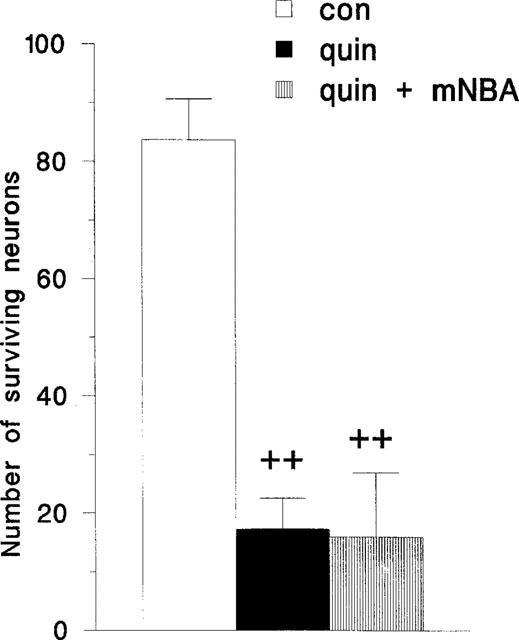
Histogram summarizing the number of neurons surviving 7 days after an intrahippocampal injection of quinolinic acid 120 nmols (quin) either alone or in combination with mNBA at a dose of 20 nmols. Results are shown as mean±s.e.mean (n=4). Analysis was performed by ANOVA followed by the Student-Newman-Keuls test for multiple comparisons. ++P<0.01 compared with control.
Discussion
The pattern of damage produced by kainic acid is partly dependent on the route of administration. Although any route is able to produce some loss of cells in most regions of the hippocampus, the neurones of the CA3 area are more sensitive to damage after local, intracerebral injections (Nadler et al., 1978; Tauck & Nadler, 1985; Kohler et al., 1979), whereas the CA1 area shows at least as much damage as CA3 after systemic administration (Sperk et al., 1983; Lassman et al., 1984; Clifford et al., 1990; Berg et al., 1993). The relative damage may depend on the concentration of kainate achieved after injection at the various sites (Schwob et al., 1980). In the present study the CA1 region was selected for quantification of cell damage, since preliminary data indicated a greater consistency of damage (less variability) in this region.
In this study, normal intact pyramidal neurones were identified as those with a clearly rounded appearance with a clear nucleus and nucleolus (Figure 1). Kainic acid produced substantial damage to both the CA1 and CA3 areas of the hippocampus. The induction of damage also resulted in the infiltration of the pyramidal cell layers and surrounding tissue by glial cells (Figure 1). Of the damaged areas, the CA1 region was selected for the quantification of the damage and protection.
Dizocilpine
The partial protection produced by dizocilpine supports the view that the neuronal damage produced by intrahippocampally administered kainate is partly attributable to the activation of NMDA-sensitive glutamate receptors (Wolf et al., 1991; Guarnieri et al., 1993; Fariello et al., 1989; Planas et al., 1995; Foster et al., 1988; Lason et al., 1988; Clifford et al., 1990; Lerner-Natoli et al., 1991). While the mechanism of this involvement has in the past been attributed to the stimulated release of glutamate which can then act upon NMDA as well as non-NMDA receptors, the present results suggest an alternative mechanism since the damage can also be reduced by the co-administration of mNBA.
Meta-nitrobenzoylalanine (mNBA)
Meta-nitrobenzoylalanine (mNBA) is one of a series of alanine derivatives which have been developed following the demonstration that nicotinylalanine is able to increase the formation of the amino acid antagonist kynurenic acid in the brain (Connick et al., 1992; Moroni et al., 1991; Russi et al., 1992). MNBA is a potent inhibitor of kynurenine hydroxylase (IC50 value of 0.9 μM) and has weaker activity (IC50 of 100 μM) against kynureninase (Pellicciari et al., 1994; Natalini et al., 1995) two of the key enzymes in the kynurenine metabolic pathway from tryptophan to quinolinic acid and ultimately nicotinic acid (Figure 6; see Stone, 1993; 2000).
Figure 6.

A summary of the major components of the kynurenine pathway relevant to this study, indicating the enzymes, kynurenine hydroxylase and kynureninase which are targets for m-nitrobenzoylalanine.
Since mNBA proved able to reduce the neuronal damage produced by kainate, the present results suggest that the kynurenine pathway may contribute to the neurotoxic activity of kainate. The inhibition of kynurenine hydroxylase activity will decrease the levels of quinolinic acid in the brain but, in addition, inhibition of kynurenine hydroxylase by this compound will also tend to elevate the endogenous levels of kynurenic acid. The administration of mNBA to rats and gerbils, for example, has been shown to raise the levels of kynurenic acid in the blood and brain (Carpenedo et al., 1994; Chiarugi et al., 1995; 1996) as have related kynurenine hydroxylase inhibitors such as 3,4-dichlorobenzoylalanine (Speciale et al., 1996). Kynurenic acid is a component of the kynurenine pathway which was originally described as an antagonist at several types of glutamate receptor (Perkins & Stone, 1982) although it is frequently used for its preferential activity as antagonist at the strychnine-resistant glycine allosteric site on the NMDA receptor (Birch et al., 1988). A rise in kynurenic acid levels could antagonize the effects of kainic acid and NMDA receptor stimulation and account for the protection seen with mNBA. The results show, however, that the administration of a high dose of glycine to reverse the kynurenic acid blockade of the allosteric glycine site, does not modify the protective effect of mNBA, whereas it would be expected to reverse at least that portion of damage attributable to NMDA receptors (and reflected in the protective effect of dizocilpine). This would suggest that an increase of kynurenic acid is not likely to contribute to the protection by mNBA, although the present data cannot exclude this possibility. A definitive comparison between the relative roles of quinolinic acid and kynurenic acid would require direct measurements of their respective concentrations.
An alternative explanation of the results could be that mNBA was acting directly as an antagonist at the NMDA receptors, preventing the activation of these sites either by glutamate released from neurones by kainate (Ferkany et al., 1982), or by quinolinic acid secreted from glial cells. This has been excluded by the finding that mNBA did not modify the neurotoxic effects of quinolinic acid or NMDA themselves.
It is known that any injurious stimulus to the brain parenchyma will result in the proliferation and infiltration of the compromised region both by activated glial cells within the CNS, and by activated peripheral macrophages which are able to penetrate into the CNS across a weakened blood-brain barrier as occurs in response to local injury and will be produced by the physical damage by the intrahippocampal injection needle (Jorgensen et al., 1993). These activated cells exhibit induction of the kynurenine synthetic enzymes (Alberatigiani et al., 1996; 1998; Pemberton et al., 1997) and a greatly enhanced secretion of quinolinic acid synthesized from tryptophan (Heyes et al., 1992; 1996; Espey et al., 1997). Since quinolinic acid is a selective agonist at NMDA-sensitive receptors (Stone & Perkins, 1981), causing excitation (Stone & Perkins, 1981) and neuronal damage (Schwarcz et al., 1983), it is possible that quinolinic acid originating from activated glia and macrophages is at least partly responsible for the apparent involvement of NMDA receptors in the neurotoxic effects of kainic acid. This possibility is strongly supported by the report of Speciale & Schwarcz (1988) that kainic acid administration increased the activity of the quinolinic acid synthesizing enzyme 3-hydroxyanthranilic acid oxygenase in the rat hippocampus, localized to those areas damaged by the kainate injection.
One difficulty with this explanation, however, is that kainic acid-induced damage of neurons in culture also involves NMDA receptors (Jensen et al., 1999), even though kainate has little affinity for NMDA receptors, and in these systems there is clearly no possibility that peripheral inflammatory cells could contribute to the toxicity by releasing quinolinic acid. It may be that the depolarization produced by kainate is sufficient to remove the voltage-dependent blockade of NMDA receptor channels by magnesium, allowing the activation of these receptors by glutamate present in the culture medium. This glutamate could in turn arise as release or leakage from the neurons themselves, or from reversal of the glutamate transporter which is inhibited by kainate. The possibility of a non-specific mechanism of this type in cultured cells is supported by the fact that NMDA receptors also contribute to the excitotoxic effects of quisqualate (Pai & Ravindrath, 1992) and the kainate analogue domoate, for which an increase in the extracellular level of glutamate has been demonstrated directly in neuronal cultures (Berman & Murray, 1997).
A further argument for the involvement of different mechanisms operating in vivo and in vitro is that the concentrations of kainate required for excitotoxicity are different in these situations. In vivo, neuronal death is produced by local injections of around 2 mM kainate (usually in a volume of 1 μl), whereas NMDA and quinolinic acid require to be administered at concentrations of around 100 mM or more. In vitro, the toxic potency of kainate is similar to (Carroll et al., 1998) or less than (Deupree et al., 1996) that of NMDA. Such differences suggest that additional toxic processes, such as the recruitment of inflammatory cells postulated here, which raise the toxic potency of kainate, may be operating in vivo. This enhancement of toxicity may, in turn, reflect the ability of quinolinic acid to cause damage partly, by the generation of oxidative stress (Behan et al., 1999).
Abbreviations
- mNBA
meta-nitrobenzoylalanine
- NMDA
N-methyl-D-aspartate
References
- ALBERATIGIANI D., CESURA A.M. Expression of the kynurenine enzymes in macrophages and microglial cells: regulation by immune modulators. Amino Acids. 1998;14:251–255. doi: 10.1007/BF01345271. [DOI] [PubMed] [Google Scholar]
- ALBERATIGIANI D., RICCIARDICASTAGNOLI P., KOHLER C., CESURA A.M. Regulation of the kynurenine metabolic pathway by interferon-gamma in murine cloned macrophages and microglial cells. J. Neurochem. 1996;66:996–1004. doi: 10.1046/j.1471-4159.1996.66030996.x. [DOI] [PubMed] [Google Scholar]
- BEHAN W.M.H., MCDONALD M., DARLINGTON L.G., STONE T.W. Oxidative stress as a mechanism for quinolinic acid induced hippocampal damage: protection by melatonin and deprenyl. Brit. J. Pharmacol. 1999;128:1754–1760. doi: 10.1038/sj.bjp.0702940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERG M., BRUHN T., JOHANSEN F.F., DIEMER N.H. Kainic acid-induced seizures and brain damage in the rat: different effects of NMDA- and AMPA receptor antagonist. Pharmacol. Toxicol. 1993;73:262–268. doi: 10.1111/j.1600-0773.1993.tb00582.x. [DOI] [PubMed] [Google Scholar]
- BERMAN F.W., MURRAY T.F. Domoic acid neurotoxicity in cultured cerebellar granule neurons is mediated predominantly by NMDA receptors that are activated as a consequence of excitatory amino acid release. J. Neurochem. 1997;69:693–703. doi: 10.1046/j.1471-4159.1997.69020693.x. [DOI] [PubMed] [Google Scholar]
- BIRCH P.J., GROSSMAN C.J., HAYES A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Europ. J. Pharmacol. 1988;154:85–87. doi: 10.1016/0014-2999(88)90367-6. [DOI] [PubMed] [Google Scholar]
- CARPENEDO R., CHIARUGI A., RUSSI P., LOMBARDI G., CARLA V., PELLICCIARI R., MORONI F., MATTOLI L. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anti-convulsant activities. Neuroscience. 1994;61:237–244. doi: 10.1016/0306-4522(94)90227-5. [DOI] [PubMed] [Google Scholar]
- CAROLL F.Y., CHEUNG N.S., BEART P.M. Investigations of non-NMDA receptor-induced toxicity in serum-free antioxidant-rich primary cultures of murine cerebellar granule cells. Neurochem. Intern. 1998;33:23–28. doi: 10.1016/s0197-0186(05)80004-x. [DOI] [PubMed] [Google Scholar]
- CHIARUGI A., CARPENEDO R., MOLINA M.T., MATTOLI L., PELLICCIARI R., MORONI F. Comparison of the neurochemical and behavioural effects resulting from the inhibition of kynurenine hydroxylase and/or kynureninase. J. Neurochem. 1995;65:1176–1183. doi: 10.1046/j.1471-4159.1995.65031176.x. [DOI] [PubMed] [Google Scholar]
- CHIARUGI A., CARPENEDO R., MORONI F. Kynurenine disposition in blood and brain of mice: effects of selective inhibitors of kynurenine hydroxylase and kynurenase. J. Neurochem. 1996;67:692–698. doi: 10.1046/j.1471-4159.1996.67020692.x. [DOI] [PubMed] [Google Scholar]
- CLIFFORD D.B., OLNEY J.W., BENZ A.M., FULLER T.A., ZORUMSKI C.F. Ketamine, phencyclidine and MK-801 protect against kainic acid-induced serizure-related brain damage. Epilepsia. 1990;31:382–390. doi: 10.1111/j.1528-1157.1990.tb05492.x. [DOI] [PubMed] [Google Scholar]
- CONNICK J.H., HEYWOOD G.C., SILLS G.J., THOMPSON G.G., BRODIE M.J., STONE T.W. Nicotinylalanine increases cerebral kynurenic acid content and has anticonvulsant activity. Gen. Pharmacol. 1992;23:235–239. doi: 10.1016/0306-3623(92)90017-e. [DOI] [PubMed] [Google Scholar]
- DEUPREE D.L., TANG X.W., YAROM M., DICKMAN E., KIRCH R.D., SCHLOSS J.V., WU J-Y. Studies of NMDA and non-NMDA-mediated neurotoxicity in cultured neurons. Neurochem. Intern. 1996;29:255–261. doi: 10.1016/0197-0186(96)00003-4. [DOI] [PubMed] [Google Scholar]
- ESPEY M.G., CHERNYSHEV O.N., REINHARD J.F., Jr, NAMBOODIRI M.A.A., COLTON C.A. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8:431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- FARIELLO R.G., GOLDEN G.T., SMITH G.G., REYES P.F. Potentiation of kainic acid epileptogenicity and sparing from neuronal damage by an NMDA receptor antagonist. Epilepsy Res. 1989;3:206–213. doi: 10.1016/0920-1211(89)90025-9. [DOI] [PubMed] [Google Scholar]
- FERKANY J.W., ZACZEK R., COYLE J.T. Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature. 1982;298:757–759. doi: 10.1038/298757a0. [DOI] [PubMed] [Google Scholar]
- FOSTER A.C., GILL R., WOODRUFF G.N. Neuroprotective effects of MK-801 in vivo: selectivity and evidence for delayed degeneration mediated by NMDA receptor activation. J. Neurosci. 1988;8:4745–4754. doi: 10.1523/JNEUROSCI.08-12-04745.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUARNIERI T., VIRGILI M., VILLANI L., FACCHINETTI F., CONTESTABILE A., MIGNANI P. Pharmacological manipulation of the NMDA receptor differentially protects from kainic acid neuropathology: evaluation through ornithine decarboxylase induction, morphology and GFAP immunohistochemistry. Resto. Neurol. Neurosci. 1993;5:327–335. doi: 10.3233/RNN-1993-55603. [DOI] [PubMed] [Google Scholar]
- HEYES M.P., ACHIM C.L., WILEY C.A., MAJOR E.O., SAITO K., MARKEY S.P. Human microglia convert L-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEYES M.P., SAITO K., MARKEY S.P. Human macrophages convert L-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1992;283:633–635. doi: 10.1042/bj2830633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JENSEN J.B., SCHOUSBOE A., PICKERING D.S. Role of desensitization and subunit expression for kainate receptor-mediated neurotoxicity in murine neocortical cultures. J. Neurosci. Res. 1999;55:208–217. doi: 10.1002/(SICI)1097-4547(19990115)55:2<208::AID-JNR8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- JONES P.A., SMITH R.A., STONE T.W. Protection against hippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res. 1998;800:328–335. doi: 10.1016/s0006-8993(98)00540-x. [DOI] [PubMed] [Google Scholar]
- JORGENSEN M.B., FINSEN B.R., JENSEN M.B., CASTELLANO B., DIEMER N.H., ZIMMER J. Microglial and astroglial reactions to ischemic and kainic acid induced lesions of the adult rat hippocampus. Exp. Neurol. 1993;120:70–88. doi: 10.1006/exnr.1993.1041. [DOI] [PubMed] [Google Scholar]
- KOHLER C., SCHWARCZ R., FUXE K. Hippocampal lesions indicate differences between the excitotoxic properties of acidic amino acids. Brain Res. 1979;175:366–371. doi: 10.1016/0006-8993(79)91018-7. [DOI] [PubMed] [Google Scholar]
- LASON W., SIMPSON J.N., MCGINTY J.F. Effects of D-(−)-2-aminophosphonovalerate on behavioural and histological changes induced by systemic kainic acid. Neurosci. Lett. 1988;87:23–28. doi: 10.1016/0304-3940(88)90139-5. [DOI] [PubMed] [Google Scholar]
- LASSMANN H., PETSCHE U., KITZ K., BARAN H., SPERK G., SEITELBERGER F., HORNYKIEWICZ O. The role of brain edema in epileptic brain damage induced by systemic kainic acid injection. Neuroscience. 1984;13:691–704. doi: 10.1016/0306-4522(84)90089-7. [DOI] [PubMed] [Google Scholar]
- LERNER-NATOLI M., RONDOUIN G., BELAIDI M., BALDY-MOULINIER M., KAMENKA J.M. TCP does not block kainic acid induced status epilepticus but reduces secondary hippocampal damage. Neurosci. Lett. 1991;122:174–178. doi: 10.1016/0304-3940(91)90851-j. [DOI] [PubMed] [Google Scholar]
- LIU H.M., DE LIANG L., DONG L.Y. Kainate-induced brain lesion: similar local and remote histopathological and molecular changes as in ischemic brain infarct. J. Neuropathol. Exp. Neurol. 1996;55:787–794. [PubMed] [Google Scholar]
- MACGREGOR D.G., MILLER W., STONE T.W. Mediation of the neuroprotective action of R-phenylisopropyladenosine by a centrally located adenosine A1 receptor. Brit. J. Pharmacol. 1993;110:470–476. doi: 10.1111/j.1476-5381.1993.tb13834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICHAELIS E.K. Molecular Biology of glutamate receptors in the CNS and their role in excitotoxicity, oxidative stress and aging. Progr. Neurobiol. 1998;54:369–415. doi: 10.1016/s0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- MORONI F., RUSSI P., GALLO-MEZO M.A., MONETI G., PELLICCIARI R. Modulation of quinolinic and kynurenic acid content in the rat brain: effects of endotoxins and nicotinylalanine. J. Neurochem. 1991;57:1630–1635. doi: 10.1111/j.1471-4159.1991.tb06361.x. [DOI] [PubMed] [Google Scholar]
- NADLER J.V., PERRY B.W., COTMAN C.W. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature. 1978;271:676–677. doi: 10.1038/271676a0. [DOI] [PubMed] [Google Scholar]
- NATALINI B., MATTOLI L., PELLICCIARI R., CARPENEDO R., CHIARUGI A., MORONI F. Synthesis and activity of enantiopure (S) (m-nitrobenzoyl) alanine, potent kynurenine-3-hydroxylase inhibitor. Biorg. Med. Chem Lett. 1995;5:1451–1454. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- OLNEY J.W., RHEE V., HO O.L. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- PAI K.S., RAVINDRATH V. Quisqualic acid-induced neurotoxicity is protected by NMDA and non-NMDA receptor antagonists. Neurosci. Lett. 1992;143:177–180. doi: 10.1016/0304-3940(92)90260-e. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The rat brain in stereotaxic co-ordinates. Academic Press, London; 1986. [Google Scholar]
- PELLICCIARI R., NATALINI B., COSTANTINO G., MAHMOUD M.R., MATTOLI L., SADEGHPOUR B.M. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J. Med. Chem. 1994;37:647–655. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- PEMBERTON L.A., KERRM S.J., SMYTHE G., BREW B.J. Quinolinic acid production by macrophages stimulated with IFN-gamma, TNF-alpha and IFN-alpha. J. Inter. Cytokine Res. 1997;17:589–595. doi: 10.1089/jir.1997.17.589. [DOI] [PubMed] [Google Scholar]
- PERKINS M.N., STONE T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982;247:184–187. doi: 10.1016/0006-8993(82)91048-4. [DOI] [PubMed] [Google Scholar]
- PLANAS A.M., SORIANO M.A., FERRER I., FARRE E.R. Kainic acid-induced heat shock protein-70, mRNA and protein expression is inhibited by MK-801 in certain rat brain regions. Europ. J. Neurosci. 1995;7:293–304. doi: 10.1111/j.1460-9568.1995.tb01065.x. [DOI] [PubMed] [Google Scholar]
- RUSSI P., ALESIANI M., LOMBARDI G., DAVOLIO P., PELLICCIARI R., MORONI F. Nicotinylalanine increases the formation of kynurenic acid in the brain and antagonizes convulsions. J. Neurochem. 1992;59:2076–2080. doi: 10.1111/j.1471-4159.1992.tb10097.x. [DOI] [PubMed] [Google Scholar]
- SCHWARCZ R., COYLE J.T. Striatal lesions with kainic acid: neurochemical characteristics. Brain Res. 1977;127:235–249. doi: 10.1016/0006-8993(77)90538-8. [DOI] [PubMed] [Google Scholar]
- SCHWARCZ R., WHETSELL W.O., MANGANO R.M. Quinolinic acid: an endogenous metabolite that causes axon-sparing lesions. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- SCHWOB J.E., FULLER T., PRICE J.L., OLNEY J.W. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- SHINOZAKI H., KONISHI S. Actions of several anthelmintics and insecticides on rat cortical neurons. Brain Res. 1970;24:368–371. doi: 10.1016/0006-8993(70)90122-8. [DOI] [PubMed] [Google Scholar]
- SPECIALE C., SCHWARCZ R. Effect of systemic kainate administration on cerebral quinolinic acid metabolism in the rat. Exp. Neurol. 1988;99:213–218. doi: 10.1016/0014-4886(88)90140-9. [DOI] [PubMed] [Google Scholar]
- SPECIALE C., WU H.Q., CINI M., MARCONI M., VARASI M., SCHWARCZ R. (R,S)-3,4-dichlorobenzoylalanine (FCE28833A) causes a large and persistent increase in brain kynurenic acid levels in rats. Europ. J. Pharmacol. 1996;315:263–267. doi: 10.1016/s0014-2999(96)00613-9. [DOI] [PubMed] [Google Scholar]
- SPERK G., LASSMANN H., BARAN H., KISH S.J., SEITELBERGER F., HORNYKIEWICZ O. Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- STONE T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Revs. 1993;45:309–379. [PubMed] [Google Scholar]
- STONE T.W.The development and therapeutic potential of kynurenic acid and kynurenine derivatives for CNS neuroprotection Trends Pharmacol. Sci. 2000. in press [DOI] [PubMed]
- STONE T.W., PERKINS M.N. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Europ. J. Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- TAUCK D.L., NADLER J.V. Evidence of functional mossy fibre sprouting in hippocampal formation of kainic acid-treated rats. J. Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VECSEI L., DIBO G., KISS C. Neurotoxins and neurodegenerative disorders. Neurotoxicology. 1998;19:511–514. [PubMed] [Google Scholar]
- VIRGILI M., MIGANI P., CONTESTABILE A, BARNABEI O. Protection from kainic acid neuropathological syndrome by NMDA receptor antagonists: effects of MK-801 and CGP 39551 on neurotransmitter and glial markers. Neuroscience. 1992;31:469–474. doi: 10.1016/0028-3908(92)90085-4. [DOI] [PubMed] [Google Scholar]
- WOLF G., FISCHER S., HASS P., ABICHT K., KEILHOFF G. Magnesium sulphate subcutaneously injected protects against kainate-induced convulsions and neurodegeneration: in vivo study on the rat hippocampus. Neuroscience. 1991;43:31–34. doi: 10.1016/0306-4522(91)90413-i. [DOI] [PubMed] [Google Scholar]