

Abstract
Simultaneous measurements of intracellular calcium concentration ([Ca2+]i) and tension were performed to clarify whether the mechanisms which cause the neuropeptide Y (NPY)-elicited contraction and potentiation of noradrenaline contractions, and the NPY inhibition of forskolin responses are linked to a single or different NPY receptor(s) in rat mesenteric small arteries.
In resting arteries, NPY moderately elevated [Ca2+]i and tension. These effects were antagonized by the selective Y1 receptor antagonist, (R)-N2-(diphenacetyl)-N-[(4-hydroxyphenyl)methyl]-D-arginineamide (BIBP 3226) (apparent pKB values of 8.54±0.25 and 8.27±0.17, respectively).
NPY (0.1 μM) caused a near 3 fold increase in sensitivity to noradrenaline but did not significantly modify the tension-[Ca2+]i relationship for this agonist. BIBP 3226 competitively antagonized the contractile response to NPY in arteries submaximally preconstricted with noradrenaline (pA2 7.87±0.20).
In arteries activated by vasopressin, the adenylyl cyclase activator forskolin (3 μM) induced a maximum relaxation and a return of [Ca2+]i to resting levels. NPY completely inhibited these effects. The contractile responses to NPY in arteries maximally relaxed with either sodium nitroprusside (SNP) or nifedipine were not significantly higher than those evoked by the peptide at resting tension, in contrast to the contractions to NPY in forskolin-relaxed arteries. BIBP 3226 competitively antagonized the contraction to NPY in forskolin-relaxed arteries with a pA2 of 7.92±0.29.
Electrical field stimulation (EFS) at 8–32 Hz caused large contractions in arteries relaxed with either forskolin or noradrenaline in the presence of phentolamine. These responses to EFS were inhibited by BIBP 3226. Similar EFS in resting, non-activated arteries did not produce any response.
The present results suggest that different intracellular pathways are linked to a single NPY Y1 receptor in intact rat mesenteric small arteries, and provide little support for involvement of other postjunctional NPY receptors in the contractile responses to NPY. Neurally released NPY also seems to act through Y1 receptors, and may serve primarily as an inhibitor of vasodilatation.
Keywords: Neuropeptide Y, Y1-receptor, BIBP 3226, potentiation, forskolin, nerves, mesenteric small artery
Introduction
Neuropeptide Y (NPY), peptide YY (PYY) and pancreatic polypeptide (PP) belong to the NPY family of neuroendocrine peptides involved in multiple physiological functions, such as vasoconstriction, angiogenesis, regulation of gut motility and gastrointestinal and renal secretion, presynaptic inhibition of neurotransmitter release, regulation of hormone release, and stimulation of food intake and inhibition of anxiety and sexual behaviour in the central nervous system (Grundemar & Håkanson, 1994; Michel et al., 1998). Whereas PYY and PP are synthesized in endocrine cells, NPY is primarily synthesized and released by neurons, and it coexists with noradrenaline in postganglionic sympathetic perivascular and myocardial neurons, playing a central role in the regulation of the cardiovascular function (Grundemar & Håkanson, 1994; Malmström, 1997).
The multiple physiological effects of NPY are mediated by different G protein-coupled receptors. Five distinct NPY receptors have been cloned and designated Y1, Y2, Y4, Y5 and Y6 (Michel et al., 1998). Y1 receptors are predominant in the cardiovascular system, although both prejunctional and postjunctional Y2 receptors have been suggested to mediate in part the functional effects of NPY at the vasculature (McAuley & Westfall, 1992; Malmström, 1997; Michel et al., 1998). Postsynaptic NPY receptors act through pertussis toxin-sensitive G proteins and activate signalling pathways some of which, e.g. inhibition of adenylyl cyclase, are common to all kinds of NPY receptors (Michel et al., 1998). Certain NPY receptors have been shown to couple to different second messenger systems (Aakerlund et al., 1990; Herzog et al., 1992; Selbie et al., 1995; Michel et al., 1998), although in some tissues, such as the rat cardiac myocytes, different NPY receptors mediate different postjunctional actions of the peptide (McDermott et al., 1997).
NPY potentiates noradrenergic contractions through a pertussis toxin-sensitive mechanism which involves Ca2+ influx in mesenteric small arteries (Andriantsitohaina et al., 1990; 1993). The Ca2+ influx is secondary to the moderate depolarization induced by NPY (Prieto et al., 1997). This depolarization is not mediated by the potent inhibition of adenosine 3′,5′-cyclic monophosphate (cyclic AMP) accumulation which NPY also evokes in this preparation (Prieto et al., 1997). Thus, NPY activates different intracellular mechanisms in these small arteries.
Both postjunctional Y1 and Y2 receptors have been proposed to mediate the enhancement by NPY of noradrenaline-elicited vasoconstriction in the mesenteric vascular bed (McAuley & Westfall, 1992; Lew et al., 1996). Therefore, the aim of the present study was to determine whether different postsynaptic NPY receptors are coupled to the different signalling pathways in rat isolated mesenteric small arteries (Prieto et al., 1997). For this purpose we used the non-peptide selective Y1 receptor antagonist, (R)-N2-(diphenacetyl)-N-[(4-hydroxyphenyl)methyl]-D-arginineamide (BIBP 3226) (Rudolf et al., 1994). We directly measured intracellular calcium concentration ([Ca2+]i) and contractile force to assess the receptor involved in the calcium-mobilizing and the potentiating actions of NPY. We also used the adenylyl cyclase activator, forskolin, as a tool to evaluate the effects of NPY on cyclic AMP-mediated relaxations. Finally, we used the antagonist to investigate the involvement of neurally released NPY in neurogenic vasoconstrictor responses under various conditions of tone.
Methods
Dissection and mounting
Male Wistar rats (12–14 weeks) were killed by CO2 inhalation and exsanguination. The mesenteric vascular bed was removed and placed in physiological saline solution (PSS) (for composition see below). Second- or third-order branches of the superior mesenteric artery were dissected free from the surrounding tissue and mounted as ring preparations in a myograph for isometric force recording (Danish Myo Technology, Denmark). The vessels were allowed to equilibrate for 30 min at 37°C in PSS aerated with 5% O2–95% CO2 to maintain pH at 7.4. The relationship between resting wall tension and internal circumference was determined, and from this the internal circumference, L100, corresponding to a transmural pressure of 100 mmHg for a relaxed vessel in situ, was calculated (Mulvary & Halpern, 1977). The arteries were set to the normalized internal circumference L1=0.9L100 where they develop maximal active tension. Their effective lumen diameter was determined as 11=L1/π.
Simultaneous measurements of force and [Ca2+]i
[Ca2+]i was determined in whole arteries by Fura-2 fluorescence, and simultaneous measurements of [Ca2+]i and force were performed as described previously (Nilsson et al., 1994). Briefly, the myograph was mounted on a Nikon inverted microscope equipped for dual excitation wavelength fluorescence microfluorometry. The arteries were loaded with the fluorophore by incubation with 2 μM Fura 2-acetoxymethyl ester (AM) in PSS for 1 h at 37°C, changing the solution after 30 min. At 1 s intervals, the vessels were illuminated with alternating 340- and 380 nm light using a monochromator-based system (Deltascan, Photon Technology International) with the 510 nm emission detected using a photomultiplier. At the end of each experiment, Ca2+-insensitive signals were determined after quenching with Mn2+ and the values obtained were subtracted from those obtained during the experiment. [Ca2+]i was calculated according to the equation: [Ca2+]i=Kd.β.(R−Rmin)/(Rmax−R), using an equilibrium dissociation constant for binding of Ca2+ to Fura-2, Kd of 224 nM (Grynkiewicz et al., 1985). Rmin and Rmax, the minima and maxima for the ratio between the emission intensities at the two wavelengths (R=F340/R380) and β, the ratio between maximal and minimal F380, were determined by using ionomycin as previously described (Nilsson et al., 1994).
Experimental protocol
In order to investigate the mechanisms of action of NPY at the level of smooth muscle, deendothelialized and chemically denervated mesenteric small arteries were used in the present study. At the beginning of each experiment, the vascular endothelium was mechanically removed by guiding a human hair through the vessel lumen and pushing it gently back and forth several times. The absence of functional endothelium was confirmed by the lack of relaxation to 3 μM acetylcholine in arteries precontracted with noradrenaline. Moreover, except for the experiments with intramural nerve stimulation, chemically sympathectomy of the arteries was performed in vitro by treatment with 10 μM guanethidine for 1 h, changing the solution every 15 min. The viability of the arteries was tested before and after denervation with guanethidine by activation with a high-potassium solution (125 mM K+, KPSS). Only one concentration-response curve to NPY was obtained in each vessel (Gustafsson & Nilsson, 1990; Prieto et al., 1997).
The potentiation by NPY of noradrenaline-evoked contractions was investigated by two different procedures. In the first, the arteries were exposed to a single concentration of NPY (0.1 μM) before a second concentration-response curve to noradrenaline was repeated in the same vessel. In the second, the vessels were given a threshold concentration of noradrenaline (30–50 nM) giving a concentration about 10% of the maximal noradrenaline response, and thereafter a concentration-response curve for NPY was constructed (Lew et al., 1996). The effects of NPY on the cyclic AMP-mediated relaxations were tested in arteries contracted with either vasopressin (0.5–0.8 Ul−1) or KPSS and relaxed by a maximal dose of forskolin (3 μM). Increasing concentrations of NPY (0.1 nM–0.1 μM) were added after the relaxation to forskolin was stable. The contractile responses to NPY in forskolin-relaxed arteries were compared to those in arteries relaxed by a maximal doses of either sodium nitroprusside (SNP) (10 μM) or nifedipine (0.3 μM).
To test the effect of the selective Y1-receptor antagonist, BIBP 3226, the arteries were incubated for 30 min with a single concentration of BIBP 3226 before they were exposed to NPY. Three different concentrations of BIBP 3226 (0.03, 0.1 and 0.3 μM) were used in the experiments designed to determine the role of Y1 receptors in the inhibition by NPY of forskolin-elicited relaxations and in the potentiation of noradrenaline contractions. For that, four arterial segments of the same animal were used. One of them served as a control and the others were incubated with different concentrations of BIBP 3226.
Electrical field stimulation (EFS)
For electrical field stimulation (EFS), arterial rings not subjected to chemical sympathetic denervation were mounted in a double myograph between a pair of platinum electrodes. The preparations were stimulated with long trains (120 s) of stimuli (0.3 ms square pulses, 35 mA) at high frequencies (8, 16 and 32 Hz) every 10 min. EFS was performed on arteries precontracted with vasopressin and relaxed with 3 μM forskolin, which had been previously treated with phentolamine (3 μM), propranolol (3 μM) and suramin (30 μM) in order to block α- and β-adrenoceptors and P2-purinoceptors, respectively. A second series of experiments was performed on baseline tension under the same conditions of receptor blockade. In another set of experiments, in which the arteries were not treated with propranolol, EFS was performed on arteries precontracted with vasopressin and relaxed with 1–3 μM noradrenaline. To investigate the role of Y1 receptors in the EFS-elicited responses, two consecutive arterial segments from the same animal were used. One served as a control and the other was incubated for 30 min with 1 μM BIBP 3226.
Solutions and drugs
PSS had the following composition (mM): NaCl, 119; NaHCO3 25; KCl, 4.7; KH2PO4, 1.18; MgSO4 1.17; CaCl2 2.5; EDTA, 0.026; and glucose, 5.5. In high-potassium solution (KPSS), NaCl was replaced by KCl on an equimolar basis, giving a final concentration of 125 mM K+. Acetylcholine HCl, forskolin, guanethidine, porcine neuropeptide Y, norepinephrine HCl and vasopressin were purchased from Sigma and (Sp)-5,6- dichloro-1-β-D-ribofuranosylbenzimidazole-3′,5′-cyclic-monophosphorothiotate (Sp-5,6-DCl-cBIMPS) from Biolog-Life Science Institute (Bremen, Germany). BIBP 3226 was provided by Dr Klaus Rudolf (Dr Karl Thomae GmbH, Biberach, Germany). Stock solutions of forskolin, Sp-5,6-DCl-BIMPS and BIBP 3226 were made in dimethylsulphoxide and further diluted in distilled H2O.
Analysis of data and statistics
Mechanical responses of the vessels were measured as force and expressed as active wall tension (ΔT) which is the increase in force above baseline (ΔF), divided by twice the segment length. Sensitivity to the agonists is given as pD2 values, where pD2=−log10EC50[M]. The results are expressed as mean±s.e.mean and n represents the number of arteries (1–2 per animal). Statistical differences between groups were tested by the use of Student's paired or unpaired t-test when appropriate. When multiple comparisons were made, values were analysed by one-way analysis of variance (ANOVA), using the Bonferroni method as a post hoc test. Probability levels less than 5% were considered significant.
In the experiments where the potentiating effect of NPY on noradrenaline-induced elevations of [Ca2+]i and force was evaluated, the initial increments of [Ca2+]i and tension elicited by NPY were subtracted from those of noradrenaline in the second noradrenaline concentration-response curve.
To assess the antagonistic effect of BIBP 3226 on the NPY-induced potentiation of noradrenaline contractions and inhibition of forskolin relaxations, at least three different concentrations of antagonist were examined. The pA2 value for this antagonist was determined following the procedure of Arunlakshana & Schild (1959). When the effect of BIBP 3226 was assessed on the NPY-induced increases in [Ca2+]i and tension, only one concentration of antagonist was tested. In this case apparent pKB values were estimated from a single antagonist concentration by use of the Gaddum equation, pKB=log (CR−1)−log [antagonist].
Results
Effect of NPY on basal [Ca2+]i and tension: evidence for functional Y1 receptors
In deendothelialized guanethidine-treated mesenteric small arteries, basal [Ca2+]i was 97±7 nM (n=30). There was no significant change in either resting [Ca2+]i or tension after removal of the endothelium or denervation with guanethidine compared with control conditions. Figure 1a shows the effects of NPY on [Ca2+]i and tension. NPY caused potent, long-lasting moderate elevations of [Ca2+]i accompanied by contractions, maximal increases in [Ca2+]i and tension being 94±15 nM (n=4) and 0.73±0.09 Nm−1 (n=4), respectively, with half-maximal responses at 3 and 5 nM NPY, respectively (Figure 1c). At the highest concentrations applied, NPY evoked oscillations in both [Ca2+]i and tension (Figure 1a,b).
Figure 1.
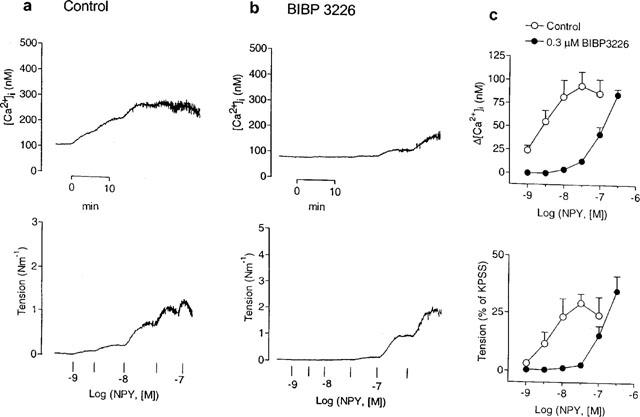
Effects of NPY on deendothelialized, guanethidine-treated rat mesenteric small arteries under resting conditions. (a,b) Simultaneous measurements of [Ca2+]i (upper traces) and tension (lower traces) showing responses to increasing concentrations of NPY of two small arteries (a) under control conditions and (b) after 30 min incubation with the Y1 receptor-selective antagonist, BIBP 3226 (0.3 μM). (c) Average increases in [Ca2+]i (upper) and tension (lower) evoked by NPY in the absence and the presence of 0.3 μM BIBP 3226. Each symbol represents mean±s.e.mean of four arteries.
The non-peptide selective NPY Y1-receptor antagonist, BIBP 3226 (0.3 μM), inhibited the NPY-evoked rise in [Ca2+]i and tension (Figure 1b), causing parallel rightward shifts of the NPY control curves (Figure 1c). The apparent calculated pKB values for the antagonism of BIBP 3226 against the NPY-elicited increases in [Ca2+]i and tension were 8.54±0.25 and 8.27±0.17 (n=4), respectively.
Effect of NPY on the increases in [Ca2+]i and tension elicited by noradrenaline: evidence for functional Y1 receptors
Figure 2a,b shows the average responses to noradrenaline in the absence and in the presence of NPY. Under control conditions, the maximal rise in [Ca+]i evoked by noradrenaline was achieved at 0.3 μM and averaged 325±21 nM (n=4), whereas the maximal increase in tension (3.7±0.2 Nm−1, n=4) occurred with 3 μM noradrenaline, at slightly lower [Ca2+]i levels (275±30 nM, P<0.05, paired t-test). In the presence of 0.1 μM NPY, the sensitivity of the arteries to noradrenaline was significantly enhanced, pD2 values for the noradrenaline-elicited elevations in [Ca2+]i and tension being 7.20±0.06 and 7.08±0.04 (n=4), respectively, in the absence, and 7.63±0.13 (P<0.05, n=4, paired t-test) and 7.56±0.11 (P<0.01, n=4, paired t-test), respectively, in the presence of NPY (Figure 2a,b). However, NPY did not significantly modify the force-[Ca2+]i ratio for noradrenaline calculated at the EC50 level: 0.35±0.04 and 0.39±0.04 (n=4) in the absence and the presence of NPY, respectively.
Figure 2.
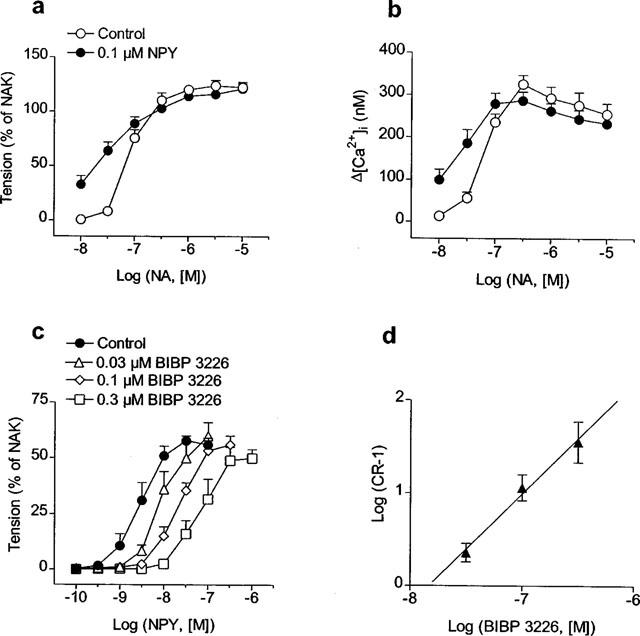
(a,b) Effect of NPY (0.1 μM) on the average increases in (a) tension and (b) [Ca2+]i evoked by noradrenaline (NA) in rat mesenteric small arteries. After constructing an initial concentration-response curve for NA, arteries were washed in PSS for 30 min and exposed to 0.1 μM NPY for 8 min before a second curve for NA was obtained. Remaining increases in [Ca2+]i and tension for NPY after 8 min were subtracted from those of NA in the second NA concentration-response curve. (c) Effect of increasing concentrations of BIBP 3226 on contractile responses to NPY in arteries activated by NA (30–50 nM) to give a contraction about 10% of the maximal NA response. (d) Schild analysis for BIBP 3226 antagonism in mesenteric small arteries. Values represent mean±s.e.mean of 4–7 arteries. In (a) and (c) results are expressed relative to the maximal contraction induced by KPSS plus 10−5 M NA (NAK) in each artery.
A second series of experiments was carried out to evaluate whether Y1 receptors play a role in the potentiation by NPY of NA responses (Figure 2c). The arteries were precontracted with a concentration of noradrenaline (30–50 nM) giving a contraction less than 10% of the maximal response. Cumulative concentration-response curves to NPY were constructed in the absence and the presence of increasing concentrations of BIBP 3226. The Y1-receptor antagonist shifted the dose-response curve for NPY to the right without any significant decrease in the maximal contractile effect (Figure 2c). The pA2 value for BIBP 3226 estimated at the intercept of the abscissa scale was 7.87±0.20 (n=15) (Figure 2d) and the slope of the Schild plot (1.10±0.23) was not significantly different from unity.
Inhibition by NPY of forskolin-induced relaxations: evidence for functional Y1 receptors
Stimulation with vasopressin (0.5–0.8 Ul−1) produced rapid, maximal elevations of both [Ca2+]i and force, which stabilized within the first 10–15 min at slightly lower levels (265±15 nM and 2.98±0.29 Nm−1, n=8, respectively) (Figure 3a). Addition of the adenylyl cyclase activator, forskolin (3 μM), induced a complete relaxation (98±1% of the vasopressin-elicited contraction) that was associated with a decrease in [Ca2+]i to resting or even lower values (74±9 nM, n=8) (Figure 3a). NPY inhibited the forskolin responses on vasopressin-precontracted vessels (Figure 3a,b). Thus, increasing concentrations of the peptide dose-dependently reversed the forskolin response, both in terms of [Ca2+]i reduction (Figure 3a,b, upper panels) and relaxation (Figure 3a,b, lower panels), with half-maximal reversals at 0.7 and 2 nM NPY, respectively (Figure 3b). Moreover, addition of the peptide to forskolin-relaxed arteries evoked marked oscillations in both [Ca2+]i and force, especially at the lower NPY concentrations.
Figure 3.
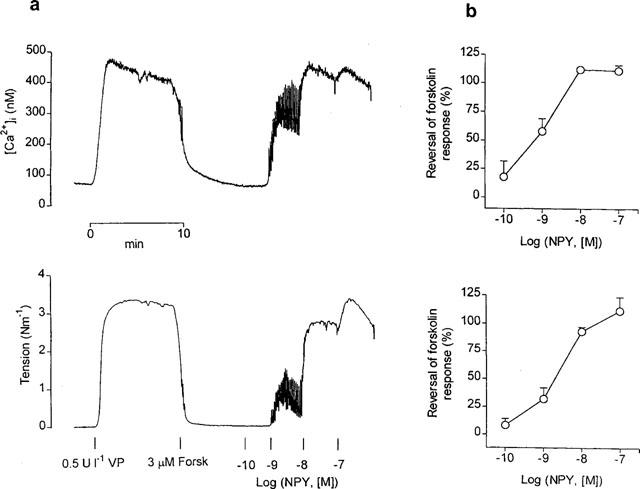
(a) Simultaneous measurements of [Ca2+]i (upper trace) and tension (lower trace) showing effects of increasing concentrations of NPY on a mesenteric small artery precontracted with vasopressin (VP) (0.5 Ul−1) and maximally relaxed with 3 μM forskolin (Forsk). Note large oscillations in [Ca2+]i accompanied by oscillations in tension at 10−9 M NPY. (b) Average inhibition by NPY of forskolin-induced decreases in [Ca2+]i and tension in mesenteric small arteries. Results are expressed as percentage of the rise in [Ca2+]i and tension elicited by vasopressin. Symbols represent mean±s.e.mean of four arteries.
Maximal concentrations of SNP (10 μM) and nifedipine (0.3 μM) relaxed mesenteric small arteries by 80±1% (n=5) and 88±1% (n=4) of the vasopressin-induced contraction, respectively. However, in contrast to the forskolin-relaxed arteries, increasing concentrations of NPY only partially reversed the relaxations to these agents (Figure 4). Thus, the maximal contractile responses to NPY in SNP- and nifedipine-relaxed arteries (39±2% and 5±2% of KPSS, respectively) were not significantly higher than those evoked by the peptide at resting tension (33±6% of KPSS). In arteries relaxed by forskolin, the NPY-induced contraction was 107±9% of KPSS (P<0.01 vs the contraction at resting tension, ANOVA followed by Bonferroni as a posterio test).
Figure 4.
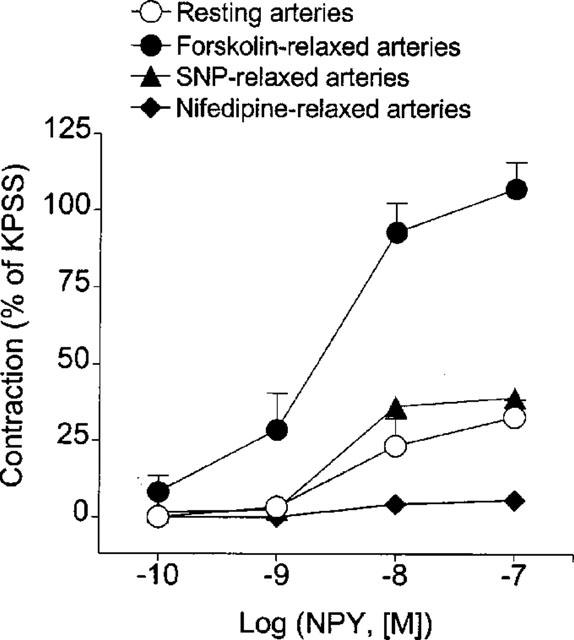
Average contractile responses elicited by increasing concentrations of NPY in mesenteric small arteries at resting tension and in arteries precontracted with vasopressin (0.5–0.8 Ul−1) and relaxed by maximal concentrations of either forskolin (3 μM), SNP (10 μM) or nifedipine (0.3 μM). Responses are expressed as percentage of the response to KPSS in each artery. Values represent mean±s.e.mean of 4–6 arteries.
In order to investigate whether the forskolin-induced relaxations in small mesenteric arteries could be accounted for by mechanisms in addition to decreasing [Ca2+]i, the effects of forskolin (3 μM) on arteries maximally precontracted with vasopressin were compared with those of forskolin on the same arteries depolarized with a high-potassium (KPSS) (Figure 5a). Both vasopressin and KPSS elevated [Ca2+]i to a similar level (258±8 and 266±16 nM, n=4, for vasopressin and KPSS, respectively), although vasopressin caused greater force development (2.7±0.3 Nm−1 vs 1.7±0.1 Nm−1, P<0.05, paired t-test). In contrast to the complete relaxation (97±1%) and reduction of [Ca2+]i (75±3 nM) evoked by forskolin in arteries precontracted with vasopressin, forskolin did not alter [Ca2+]i (330±14 nM) but relaxed by 45±7% the arteries depolarized with KPSS. Forskolin thus not only lowers [Ca2+]i, but causes relaxation also via other mechanisms.
Figure 5.
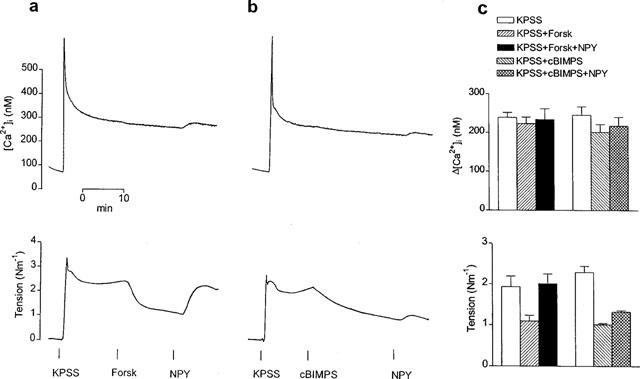
Simultaneous measurements of [Ca2+]i (upper traces) and tension (lower traces) showing effects of NPY (0.1 μM) on the relaxations elicited by (a) forskolin (Forsk) (3 μM) and (b) the protein kinase A activator, Sp-5,6-DCl-cBIMPS (cBIMPS) (0.1 mM) on two mesenteric small arteries depolarized with KPSS. Note that relaxations to cBIMPS, like those to forskolin, were not accompanied by changes in [Ca2+]i. In either case NPY elicited slight further elevations in [Ca2+]i of KPSS-activated arteries, but significant reversal of the relaxations was only achieved in the artery relaxed with forskolin. (c) Average effects of forskolin (3 μM) and cBIMPS (0.1 mM) before and after addition of NPY (0.1 μM) on the increases in [Ca2+]i (upper) and tension (lower) elicited by KPSS in mesenteric small arteries. Bars represent mean±s.e.mean of four arteries.
Figure 5 illustrates also the effects of forskolin (3 μM) (Figure 5a) compared with those of the specific activator of cyclic AMP-dependent protein kinase (protein kinase A, PKA), Sp-5,6-DCl-cBIMPS (0.1 mM) (Figure 5b), on the top of KPSS. Like forskolin, which relaxed the arteries by 43±5% (n=4) without affecting [Ca2+]i (Figure 5a,c), the PKA activator did not significantly alter [Ca2+]i but elicited a slow relaxation that stabilized after 20 min and averaged 55±3% (n=4) of the KPSS response (Figure 5b,c). Thus, both forskolin and 5,6-DCl-cBIMPS decreased the force-[Ca2+]i relationship for KPSS to a similar level, from 0.42±0.02 to 0.26±0.02 (n=4, P<0.01, ANOVA) and from 0.42±0.04 to 0.23±0.01 (n=4, P<0.01, ANOVA), respectively. However, whereas NPY (0.1 μM) increased this ratio back to control levels in the case of forskolin (0.45±0.02, n=4, ns. vs KPSS alone, ANOVA) (Figure 5a,c), the peptide did not significantly alter the force-[Ca2+]i relationship for KPSS in the presence of the PKA activator (0.28±0.01, n=4, P<0.05 vs KPSS alone, ANOVA) (Figure 5b,c). In KPSS-precontracted arteries exposed to both forskolin and Sp-5,6-DCl-cBIMPS, NPY elicited slight increases in [Ca2+]i (Figure 6a,b) averaging 11±3 nM (n=4) and 15±3 nM (n=4) and not significantly different from the elevations in [Ca2+]i elicited by NPY alone on the top of KPSS (11±2 nM, n=6).
Figure 6.
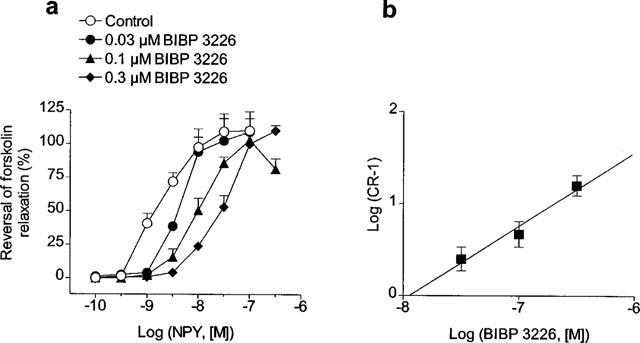
(a) Effect of increasing concentrations of the Y1 receptor antagonist, BIBP 3226, on the reversal by NPY of forskolin-induced relaxations in rat mesenteric small arteries. Arteries were precontracted with vasopressin and maximally relaxed with 3 μM forskolin before addition of NPY. Since concentration-response curves for NPY could not be repeated in the same artery, four arterial segments from the same animal were used, one serving as control and the others being incubated for 30 min with increasing concentrations of BIBP 226 (0.03–0.3 μM). (b) Schild plot for the antagonism of BIBP 3226 against the NPY responses in forskolin-relaxed mesenteric small arteries. Points represent mean±s.e.mean of 4–8 arteries.
In order to evaluate whether Y1-receptors are involved in the inhibition by NPY of forskolin-elicited relaxations, the effects of increasing concentrations of BIBP 3226 (0.03–0.3 μM) were examined in the reversal by NPY of the maximal relaxations elicited by 3 μM forskolin on vasopressin-precontracted arteries (Figure 6). BIBP 3226 competitively antagonized the effect of NPY without suppressing the maximal response (Figure 6a). The pA2 value for BIBP 3226 was 7.92±0.29 (n=16) and the slope of the Schild plot was not significantly different from unity (0.81±0.23) (Figure 6b).
Inhibition by EFS of forskolin-elicited relaxations: evidence for functional Y1 receptors
In view of the potent inhibitory action of applied NPY on forskolin relaxation, it was of interest to examine the influence of endogenous NPY under conditions where it elicits contraction indirectly (by inhibiting relaxation), and comparing this to its direct contractile effect. For this purpose, stimulation of periarterial nerves was carried out in arteries precontracted with vasopressin and maximally relaxed with forskolin (Figure 7), under conditions of α- and β-adrenoceptor and P2-purinoceptor blockade with phentolamine, propranolol and suramin, respectively. Electrical field stimulation (EFS) at high frequency (8, 16 and 32 Hz) was performed for 2 min at 10 min intervals. EFS reversed forskolin-induced relaxations, with maximal reversal (78±6%, n=5) being produced at a frequency of 16 Hz (Figures 7a and 8a). The peak of the EFS responses occurred within 2–3 min, and force returned to prestimulation levels within 8–10 min after stimulation. These responses were inhibited by BIBP 3226 (1 μM) (Figures 7b and 8a). Moreover, EFS performed at resting tension under conditions of adrenoceptor and purinoceptor blockade, did not elicit significant contractions (Figure 7c). In a last set of experiments where mesenteric small arteries were precontracted with vasopressin and treated with phentolamine and suramin to block α-adrenoceptors and P2-purinoceptors, noradrenaline relaxed the arteries by 91±2% (n=4) of the vasopressin-induced contraction. EFS reversed noradrenaline relaxant responses (maximal effect at 16 Hz: 101±1%), this effect being markedly inhibited by BIBP 3226 (Figure 8).
Figure 7.
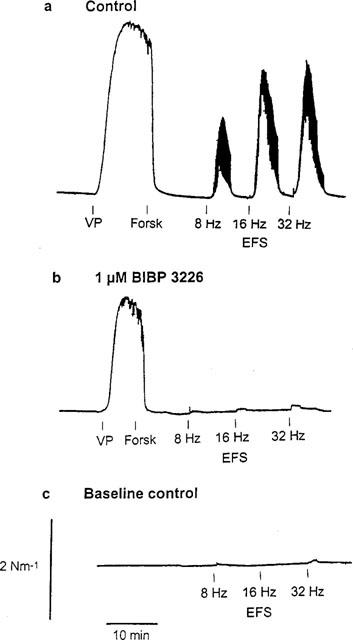
Effect of electrical field stimulation (EFS) (0.3 ms pulses, 120 s trains, 35 mA output current) on mesenteric small arteries either (a,b) contracted with vasopressin (VP) and maximally relaxed with 3 μM forskolin or (c) at resting tension. In (a), (b) and (c) arteries were previously incubated with 3 μM phentolamine, 3 μM propanolol and 30 μM suramin for 30 min to block α- and β-adrenoceptors and P2-purinoceptors, respectively. In (b) the artery was also incubated with 1 μM of the Y1 receptor antagonist, BIBP 3226.
Figure 8.
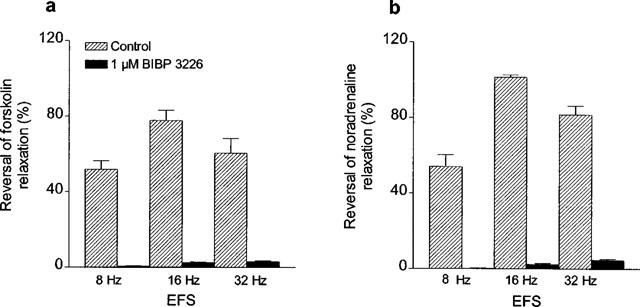
Average reversal by EFS of relaxations induced by (a) forskolin and (b) noradrenaline in mesenteric small arteries precontracted with vasopressin, and inhibitory effects of BIBP 3226 (1 μM). In (b), arteries were preincubated with phentolamine (3 μM) and suramin (30 μM), precontracted with vasopressin and relaxed with noradrenaline (1–3 μM), in the absence and presence of 1 μM BIBP 3226. In both (a) and (b) two consecutive arterial segments were used, one which served as a control and the other which was treated with the Y1 receptor antagonist. Bars represent mean±s.e.mean of 3–5 experiments.
Discussion
In the present study, we have shown that NPY elevates [Ca2+]i and thereby causes contraction and potentiation of noradrenaline contractions, and inhibits forskolin-induced vasodilation, thus reversing the decreases in [Ca2+]i and contraction elicited by activation of adenylyl cyclase. Moreover, NPY also reversed the part of the forskolin-induced relaxations which did not depend on changes in [Ca2+]i, which represents an indirect mechanism of Ca2+ sensitization. The use of the Y1 receptor-selective antagonist, BIBP 3226, has allowed us to demonstrate that the latter signalling mechanisms are linked to a single Y1 postsynaptic receptor in intact mesenteric small arteries, thus confirming the observation that this receptor subtype is coupled to several second messengers systems upon heterologous expression (Aakerlund et al., 1990; Herzog et al., 1992; Selbie et al., 1995).
Involvement of Y1 receptors in the contractile and potentiating effects of NPY
The ability of NPY to promote rises in [Ca2+]i is well documented in vascular smooth muscle (Mihara et al., 1989; Erdbrüger et al., 1993; Shigeri et al., 1995; Tanaka et al., 1995). In the mesenteric small arteries studied here, NPY elicits moderate increases in basal [Ca2+]i and tension which probably are consequent to depolarization (Prieto et al., 1997), in particular since these responses are markedly inhibited by the L-type calcium channel blocker, nifedipine (Prieto et al., 1997). Thus, the major [Ca2+]i mobilizing action of NPY in mesenteric small arteries is due to a transmembrane influx of Ca2+. This is in agreement with results reported for canine basilar (Tanaka et al., 1995) and bovine retinal (Prieto et al., 1995) arteries, where the NPY-induced contraction also is produced mainly by Ca2+ influx through L-type channels. However, data obtained in porcine aortic smooth muscle cells (Mihara et al., 1989; Erdbrüger et al., 1993; Shigeri et al., 1995) and other cell systems (Herzog et al., 1992; Selbie et al., 1995), indicate that the NPY-elicited rises in [Ca2+]i are fully or partially due to mobilization of Ca2+ from intracellular stores. These differences in the Ca2+ handling by NPY may be ascribed to the different cell types and vascular beds, but might also reflect differences in the interactions between NPY and other vasoconstrictor agents.
Several studies have suggested that the interactions between NPY and other vasoactive agents to involve a synergistic activation of second messengers associated with the sensitization of the contractile apparatus to Ca2+. Thus, in porcine aortic smooth muscle cells (Lobaugh & Blackshear, 1990) and in cells from a line expressing the Y1 receptor (Selbie et al., 1995), NPY potentiated the angiotensin II- and phenylephrine-elicited activation of protein kinase C (PKC), respectively. The present results show, however, that in mesenteric small arteries the potentiation of the contractions elicited by low concentrations of noradrenaline primarily is accounted for by a synergistic rise in [Ca2+]i. Any Ca2+ sensitizing action of NPY under these conditions was not large enough to reach statistical significance. Therefore Ca2+ sensitization appears to play only a minor role in the potentiating effect of NPY on adrenergic responses.
The effects of NPY on both Ca2+ and tension, as well as on the noradrenaline-elicited contractions, were competitively inhibited by the Y1 receptor-selective antagonist, BIBP 3226. Thus, both the direct contractile effect and the potentiating action seem to be mediated primarily by Y1 receptors. These have been suggested previously to mediate the enhancement of contractions elicited by NPY in pressurized mesenteric arteries activated with threshold concentrations of noradrenaline (Lew et al., 1996). An uncertainty with the latter studies is the fact that the NPY antagonist used, 1229U91, possesses high affinity not only for the Y1 but also for the Y4 receptor at which it has agonist properties (Michel et al., 1998), in contrast to the Y1-selective BIBP 3226 used in the present study. However, the conclusions by Lew et al. (1996) are supported by our present findings in the mesenteric small arteries, where the pKB and pA2 values obtained for the antagonism of BIBP 3226 against the NPY-elicited increases in [Ca2+]i and tension and the potentiation of noradrenaline contractions were quite similar (8.5, 8.3 and 7.9, respectively), and in the agreement with the affinity values around 8 reported for this compound at vascular Y1 receptors in isolated cerebral (8.5) (Abounader et al., 1995) and small coronary (7.9) (Prieto et al., 1998) arteries, perfused rabbit ear artery (7.5) and rat kidney (7.5) (Doods et al., 1995), and guinea-pig vena cava (8.0) (Malmström, 1997). This indicates that the various NPY responses studied here are mediated by a single type of NPY receptor, the Y1 receptor. Moreover, our data confirm in vitro and in vivo studies which show BIBP 3226 inhibiting the potentiation by NPY of the increases in perfusion pressure and vascular resistance, respectively, evoked by α-adrenergic agonists in the mesenteric vascular bed (Doods et al., 1995; Bischoff et al., 1997).
Involvement of Y1 receptors in the inhibition by NPY of forskolin relaxation
We have previously shown that the direct contractile effect as well as the potentiating action of NPY are not related to an effect on cyclic AMP production (Prieto et al., 1997). However, we have also demonstrated that NPY is very potent in inhibiting cyclic AMP production in these arteries–at a concentration of 0.1 μM it inhibits by 94% the 21 fold increase in cyclic AMP levels elicited by 1 μM forskolin (Prieto et al., 1997). A functional demonstration of this inhibiting action on adenylyl cyclase activity is the complete reversal of forskolin-induced [Ca2+]i reduction and relaxation in vasopressin-precontracted arteries seen in the present study. Since BIBP 3226 competitively antagonized the effect of NPY on forskolin relaxations (pA2 7.9), these findings suggest that also inhibition of adenylyl cyclase by NPY is linked to a Y1 receptor and confirm earlier observations on other cell lines expressing this receptor (Aakerlund et al., 1990; Herzog et al., 1992). In fact, similar correlations between inhibition of adenylyl cyclase and inhibition of relaxation have been found for α2-adrenergic (Roberts et al., 1998) and M2-muscarinic (Hedge et al., 1997) receptors. Activation of these receptors induces little direct contraction, but since they inhibit adenylyl cyclase activity, relaxation will be inhibited and thus contraction produced indirectly. In the present study, it seems unlikely that either direct effects of applied NPY on basal [Ca2+]i and tension or a specific counteraction of the relaxant mechanism of forskolin by NPY account for the peptide-induced complete reversal of forskolin responses. Thus, in arteries where the contractions of vasopressin were maximally inhibited by either blockade of calcium influx with nifedipine or by relaxation with SNP, NPY only partially reversed these relaxations and the contractions elicited by the peptide under these conditions were not significantly higher than those evoked by NPY at resting tension. Moreover, NPY did not significantly reverse the relaxations elicited by activation of PKA. Along with our earlier observations (Prieto et al., 1997), the present results thus suggest that the greater NPY contractile responses seen on forskolin-relaxed arteries are mainly related to its inhibitory influence on intracellular cyclic AMP levels. It cannot be excluded, however, that this effect of NPY might involve a mechanism other than inhibition of cyclic AMP formation.
Increased intracellular cyclic AMP levels can produce relaxation of vascular smooth muscle by decreasing either [Ca2+]i, the sensitivity of the contractile apparatus to Ca2+, or both (Murray, 1990; McDaniel et al., 1991). The present results demonstrate that both mechanisms underlie the forskolin-elicited relaxations in mesenteric small arteries. Thus forskolin caused both [Ca2+]i-dependent and independent responses in arteries precontracted with vasopressin and KPSS. The decreases in [Ca2+]i accompanying forskolin relaxations in vasopressin-precontracted arteries are probably accounted for by a repolarization of the smooth muscle, with a subsequent decrease in the Ca2+ influx through L-type channels. This is based on our previous demonstration that stimulation of adenylyl cyclase with forskolin evokes marked hyperpolarizations of small mesenteric arteries which involve activation of KATP channels (Prieto et al., 1997). On the other hand, the relaxations to forskolin in the absence of significant changes in [Ca2+]i suggest that increased cyclic AMP levels may also induce a desensitization of the contractile proteins to Ca2+, thus supporting earlier studies in α-toxin permeabilized mesenteric arteries (Nishimura & Van Breemen, 1989).
Although most effects of cyclic AMP in vascular smooth muscle cells are assumed to be mediated by activation of PKA, large increases in intracellular cyclic AMP levels can cross-activate cyclic GMP-dependent protein kinase (PKG; Lincoln et al., 1990). However, in the present study, the specific activator of PKA, Sp-5,6-DCl-cBIMPS, mimicked the relaxations elicited by forskolin in KPSS-activated arteries and decreased to a similar extent the ratio force/[Ca2+]i for KPSS. This suggests that the Ca2+ desensitization underlying the relaxations to forskolin primarily involves activation of PKA rather than PKG. The fact that NPY completely reversed the relaxations to forskolin in KPSS-contracted small mesenteric arteries, but not those to the PKA activator, again suggests that the large contractions evoked by NPY in forskolin-relaxed vessels are mainly due to its influence on cyclic AMP levels, and not to changes distal to the cyclic AMP accumulation. As a whole, these findings suggest that although NPY may have a small direct sensitizing effect to Ca2+, a more important action is inhibition of adenylyl cyclase which causes indirect Ca2+ sensitization and contraction through reversal of cyclic AMP-mediated desensitizing effects in mesenteric small arteries.
Involvement of Y1 receptors in the neurogenic vasoconstriction of relaxed arteries
The demonstration of a functional role of endogenously released NPY in sympathetic vasoconstriction has been hampered for a long time by the lack of selective NPY receptor antagonists. The recent availability of BIBP 3226 has allowed clarification of the relative contribution of NPY to the contractile responses to stimulation of periarterial sympathetic nerves in the mesenteric arterial bed (Donoso et al., 1997; Han et al., 1998). Such experiments have confirmed earlier suggestions that non-adrenergic non-purinergic neurogenic contractions seen upon intense field stimulation of precontracted small mesenteric arteries are due to NPY release (Sjöblom-Widfeldt et al., 1990). It is known that high-frequency electrical stimulation potentiates the responses to contractile agents and mimics the effects of exogenous NPY (Sjöblom-Widfeldt et al., 1990; McAuley & Westfall, 1992; Donoso et al., 1997), and also induces release of NPY-like immunoreactivity (Donoso et al., 1997; Han et al., 1998). Blocking Y1 receptors with BIBP 3226 not only abolishes this potentiation (Donoso et al., 1997), but also attenuates EFS-induced contractions (Han et al., 1998). This indicates that NPY is released upon stimulation of periarterial sympathetic nerves and contributes to vasoconstriction by potentiating noradrenaline contractions. In agreement with these observations, we found that contractions to EFS after adrenergic and purinergic blockade in forskolin-relaxed arteries to be inhibited by BIBP 3226, indicating that they were due to neurally released NPY. These results furthermore show that endogenous NPY primarily interacts with postjunctional Y1 receptors, in a similar manner as exogenous NPY.
In earlier studies, the magnitude of the neurogenic contractions that could be attributed to NPY was, however, limited (Han et al., 1998). In contrast, a dramatic increase in the NPY-related response to neural stimulation was seen during vasodilatation induced by cyclic AMP-related mechanisms (forskolin administration or β-adrenergic stimulation). This could be ascribed to a facilitatory effect of cyclic AMP on NPY release from the sympathetic nerve terminals, as earlier reported for noradrenaline in several vascular preparations (Bucher et al., 1990; Nozaki & Speralakis, 1991). However, the action of neurally-release NPY on forskolin-induced or β-receptor-mediated relaxations is similar to that of exogenous NPY and selectively blocked by BIBP 3226. This suggests that the contractile responses evoked by EFS in forskolin- and noradrenaline-relaxed arteries are likely due to a postjunctional inhibition of adenylyl cyclase by NPY released from periarterial nerves. Therefore, the vasoconstrictor action of the peptide under physiological conditions may be highly dependent on the prevailing activity of the cyclic AMP system.
In summary, the present results demonstrate that NPY binding to postsynaptic Y1 receptors activates different signalling pathways that regulate [Ca2+]i and contraction in mesenteric small arteries. There was no evidence to suggest that other smooth muscle receptors were involved in the contractile responses to NPY. NPY promotes a direct Ca2+ influx which leads to moderate contraction and potentiates noradrenaline-contractions, with only a minor influence on Ca2+ sensitizing mechanisms. On the other hand, NPY strongly interferes with forskolin vasodilatation by inhibiting both the decreases in [Ca2+]i and the Ca2+ desensitization. Neurally released NPY mimics the inhibitory effects of exogenous NPY on both forskolin and β-adrenergic relaxations, which may represent an important mechanism for vasoconstriction under physiological conditions.
Acknowledgments
The present study was supported by Danish Medical Research Council and Danish Heart Foundation. We are grateful to Dr K. Rudolf of Karl Thomae, GmbH, Biberach, Germany, for the generous supply of BIBP 3226. We also thank Dr U. Simonsen for helpful advice in the experiments with electrical field stimulation.
Abbreviations
- BIBP 3226
(R)-N2-(diphenacetyl)-N-[(4-hydroxyphenyl)methyl]-D-arginineamide
- EFS
electrical field stimulation
- NPY
neuropeptide Y
- PKA
protein kinase A
- SNP
sodium nitroprusside
- Sp-5,6-DC1-cBIMPS
(Sp)-5,6-dichloro-1-β-D-ribofuranosylbenzimidazole-3′,5′-cyclic-monophosphorothiotate
References
- AAKERLUND L., GETHER U., FUHLENDORFF J., SCHWARTZ T.W., THASTRUP O. Y1 receptors are coupled to mobilization of intracellular calcium and inhibition of adenylate cyclase. FEBS Lett. 1990;260:73–78. doi: 10.1016/0014-5793(90)80069-u. [DOI] [PubMed] [Google Scholar]
- ABOUNADER R., VILLEMURE J.-G., HAMEL E,. Characterization of neuropeptide (NPY) receptors in human cerebral arteries with selective agonists and the new Y1 antagonists BIBP 3226. Br. J. Pharmacol. 1995;116:2245–2250. doi: 10.1111/j.1476-5381.1995.tb15060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDRIANTSITOHAINA R., ANDRE P., STOCLET J.-C. Pertussis toxin abolishes the effect of neuropeptide Y on rat resistance arteriole contraction. Am. J. Physiol. 1990;259:H1427–H1432. doi: 10.1152/ajpheart.1990.259.5.H1427. [DOI] [PubMed] [Google Scholar]
- ANDRIANTSITOHAINA R., BIAN K., STOCLET J.-C., BUKOSKI R. Neuropeptide Y increases force development through a mechanism that involves calcium entry in resistance arteries. J. Vasc. Res. 1993;30:309–314. doi: 10.1159/000159011. [DOI] [PubMed] [Google Scholar]
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Ther. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISCHOFF A., FREUND A., MICHEL M.C. The Y1 antagonist BIBP 3226 inhibits potentiation of the methoxamine-induced vasoconstriction by neuropeptide Y. Naunyn Schmiedeberg's Arch Pharmacol. 1997;356:653–640. doi: 10.1007/pl00005100. [DOI] [PubMed] [Google Scholar]
- BUCHER B., PAIN L., STOCLET J.C., ILLES P. Role of cyclic AMP in the prejunctional alpha2-adrenoceptor modulation of noradrenaline release from the rat tail artery. Naunyn Schmiedebergs Arch. Pharmacol. 1990;342:640–649. doi: 10.1007/BF00175706. [DOI] [PubMed] [Google Scholar]
- DONOSO M.V., BROWN N., CARRASCO C., CORTES V., FOURNIER A., RUIDOBRO J.P. Stimulation of sympathetic perimesenteric arterial nerves releases neuropeptide Y potentiating the vasomotor activity of noradrenaline: involvement of neuropeptide Y-Y1 receptors. J. Neurochem. 1997;69:1048–1059. doi: 10.1046/j.1471-4159.1997.69031048.x. [DOI] [PubMed] [Google Scholar]
- DOODS H.N., WIENEN W., ENTZROTH M., RUDOLF K., EBERLEIN W., ENGEL W., WIELAND H.A. Pharmacological characterization of the selective nonpeptide neuropeptide Y Y1 receptor antagonist. J. Pharmacol. Exp. Ther. 1995;275:136–142. [PubMed] [Google Scholar]
- ERDBRÜGER W., VISCHER P., BAUCH H.-J., MICHEL M.C. Norepinephrine and neuropeptide Y increase intracellular Ca2+ in cultured porcine aortic smooth muscle cells. J. Cardiovasc. Pharmacol. 1993;22:97–102. doi: 10.1097/00005344-199307000-00016. [DOI] [PubMed] [Google Scholar]
- GRUNDEMAR L., HÅKANSON R. Neuropeptide Y effector systems: perspective for drug development. TIPS. 1994;15:153–159. doi: 10.1016/0165-6147(94)90076-0. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENI M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GUSTAFSSON H., NILSSON H. Endothelium-independent potentiation by neuropeptide Y of vasoconstrictor responses in isolated arteries from rat and rabbit. Acta Physiol. Scand. 1990;138:503–507. doi: 10.1111/j.1748-1716.1990.tb08878.x. [DOI] [PubMed] [Google Scholar]
- HAN S., YANG C.-L., CHEN X., NAES L., COX F., WESTFALL T. Direct evidence for the role of neuropeptide Y in sympathetic nerve stimulation-induced vasoconstriction. Am. J. Physiol. 1998;274:H290–H294. doi: 10.1152/ajpheart.1998.274.1.H290. [DOI] [PubMed] [Google Scholar]
- HEDGE S.S., CHOPPIN A., BONHUS D., BRIAUD S., LOEB M., MOY T.M., LOURY D., EGLEN R.M. Functional role of M2 and M3 muscarinic receptors in the urinary bladder of rats in vitro and in vivo. Br. J. Pharmacol. 1997;120:1409–1418. doi: 10.1038/sj.bjp.0701048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERZOG H., HORT Y.J., BALL H.J., HAYES G., SHINE J., SELBIE L.A. Cloned human neuropeptide Y receptor couples to two different second messenger systems. Proc. Natl. Acad. Sci. U.S.A. 1992;89:5794–5798. doi: 10.1073/pnas.89.13.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEW M., MURPHY R., ANGUS J.A. Synthesis and characterization of a selective peptide antagonists of neuropeptide Y vascular postsynaptic receptors. Br. J. Pharmacol. 1996;117:1768–1772. doi: 10.1111/j.1476-5381.1996.tb15352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINCOLN T.M., CORNWELL T.L., TAYLOR A.E. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. Am. J. Physiol. 1990;258:C399–C407. doi: 10.1152/ajpcell.1990.258.3.C399. [DOI] [PubMed] [Google Scholar]
- LOBAUGH L.A., BLACKSHEAR P.J. Neuropeptide Y stimulation of myosin light chain phosphorylation in cultured aortic smooth muscle cells. J. Biol. Chem. 1990;265:18393–18399. [PubMed] [Google Scholar]
- MALMSTRÖM R. Neuropeptide Y1 receptor mechanisms in sympathetic vascular control. Acta Physiol. Scand. 1997;636 Suppl.:1–55. [PubMed] [Google Scholar]
- MCAULEY M.A., WESTFALL T. Possible location and function of neuropeptide Y receptor subtypes in the rat mesenteric arterial bed. J. Pharmacol. Exp. Ther. 1992;261:863–868. [PubMed] [Google Scholar]
- MCDANIEL N.L., REMBOLD C.M., RICHARD H.M., MURPHY R.A. Cyclic AMP relaxes swine arterial smooth muscle predominantly by decreasing cell Ca2+ concentration. J. Physiol. 1991;439:147–160. doi: 10.1113/jphysiol.1991.sp018661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCDERMOTT B.J., MILLAR C., DOLAN F.M., BELL D., BALASUBRAMANIAM A. Evidence for Y1 and Y2 subtypes of neuropeptide Y receptors linked to opposing postjunctional effects observed in rat cardiac myocytes. Eur. J. Pharmacol. 1997;336:257–265. doi: 10.1016/s0014-2999(97)01258-2. [DOI] [PubMed] [Google Scholar]
- MICHEL M.C., BECK-SICKINGER A., COX H., DOODS H.N., HERZOG H., LARHAMAR D., QUIRION R., SCHWARTZ T., WESTFALL T. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY and pancreatic polypeptide receptors. Pharmacol. Rev. 1998;50:143–150. [PubMed] [Google Scholar]
- MIHARA S., SHIGERI Y., FUJIMOTO M. Neuropeptide Y-induced intracellular Ca2+ increases in vascular smooth muscle cells. FEBS Lett. 1989;259:79–82. doi: 10.1016/0014-5793(89)81499-1. [DOI] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- MURRAY K.J. Cyclic AMP and mechanisms of vasodilation. Pharmac. Ther. 1990;47:329–345. doi: 10.1016/0163-7258(90)90060-f. [DOI] [PubMed] [Google Scholar]
- NILSSON H., JENSEN P., MULVANY M.J. Minor role for direct adrenoceptor-mediated calcium entry in rat resistance arteries. J. Vasc. Res. 1994;31:314–321. doi: 10.1159/000159059. [DOI] [PubMed] [Google Scholar]
- NISHIMURA J., VAN BREEMEN C. Direct regulation of smooth muscle contractile elements of second messengers. Biochem. Biophys. Res. Commun. 1989;163:929–935. doi: 10.1016/0006-291x(89)92311-5. [DOI] [PubMed] [Google Scholar]
- NOZAKI M., SPERALAKIS N. Chloera toxin and Gs protein modulation of synaptic transmission in guinea pig mesenteric artery. Eur. J. Pharmacol. 1991;197:57–62. doi: 10.1016/0014-2999(91)90364-v. [DOI] [PubMed] [Google Scholar]
- PRIETO D., BUUS C., MULVANY M.J., NILSSON H. Interactions between neuropeptide Y and adenylate cyclase pathway in rat mesenteric small arteries: role of membrane potential. J. Physiol. 1997;502:281–292. doi: 10.1111/j.1469-7793.1997.281bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIETO D., GARCÍA-SACRISTÁN A., SIMONSEN U. Characterization of NPY receptors mediating contraction in rat intramyocardial arteries. Regul. Pept. 1998;75:155–160. doi: 10.1016/s0167-0115(98)00063-9. [DOI] [PubMed] [Google Scholar]
- PRIETO D., SIMONSEN U., NYBORG N.C.B. Regional involvement of an endothelium-derived contractile factor in the vasoactive actions of neuropeptide Y in bovine isolated retinal arteries. Br. J. Pharmacol. 1995;116:2729–2737. doi: 10.1111/j.1476-5381.1995.tb17234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS R.E., TOMLINSON A.E., KENDALL D.A., WILSON V.G. α2-adrenoceptor-mediated contractions of the porcine isolated ear artery: evidence for a cyclic AMP-dependent and a cyclic AMP-independent mechanism. Br. J. Pharmacol. 1998;124:1107–1114. doi: 10.1038/sj.bjp.0701935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUDOLF K., EBERLEIN W., ENGEL W., WIELAND H.A., WILLIM K.D., ENTZEROTH M., WIENE W., BECK-SICKINGER A.G., DOODS H.N. The first highly potent and selective non-peptide neuropeptide Y Y1 receptor antagonist: BIBP 3226. Eur. J. Pharmacol. 1994;271:R11–R13. doi: 10.1016/0014-2999(94)90822-2. [DOI] [PubMed] [Google Scholar]
- SELBIE L.A., DARBY K., SCHMITZ-PEIFFER C., BROWNE C.L., HERZOG H., SHINE J., BIDEN T.J. Synergistic interaction of the Y1-neuropeptide Y and a1b-adrenergic receptors in the regulation of phospholipase C, protein kinase C and arachidonic acid production. J. Biol. Chem. 1995;270:11789–11796. doi: 10.1074/jbc.270.20.11789. [DOI] [PubMed] [Google Scholar]
- SHIGERI Y., NAKAJIMA S., FUJIMOTO M. Neuropeptide Y Y1 receptors-mediated increase in intracellular Ca2+ concentration via phospholipase C-dependent pathways in porcine aortic smooth muscle cells. J. Biochem. 1995;118:515–520. doi: 10.1093/oxfordjournals.jbchem.a124938. [DOI] [PubMed] [Google Scholar]
- SJÖBLOM-WIDFELDT N., GUSTAFSSON H., NILSSON H. Transmitter characteristics of small mesenteric arteries from rats. Acta Physiol. Scand. 1990;138:203–212. doi: 10.1111/j.1748-1716.1990.tb08834.x. [DOI] [PubMed] [Google Scholar]
- TANAKA Y., NAKAZAWA T., ISHIRO H., SAITO M., UNEYAMA H., IWATA S., ISHII K., NAKAYAMA K. Ca2+ handling mechanisms underlying neuropeptide Y-induced contraction in canine basilar artery. Eur. J. Pharmacol. 1995;289:59–66. doi: 10.1016/0922-4106(95)90168-x. [DOI] [PubMed] [Google Scholar]