

Abstract
Different α subunits of human γ-aminobutyric acid type A (GABAA) receptors were transiently expressed together with β3 and γ2 subunits in Xenopus oocytes to examine the interactions of various GABAA agonists and representative allosteric modulators. Chloride currents elicited by agonists were measured using two electrode voltage clamp electrophysiology.
Where compounds behaved as full agonists, i.e. GABA on all subtypes and 4,5,6,7-tetrahydroisoxazolo [5,4-c]pyridin-3-ol (THIP) on α2β3γ2 GABAA receptors, agonist concentration-response curves were shifted to the left by the benzodiazepine full agonist chlordiazepoxide and the anticonvulsant loreclezole, or to the right by the inverse agonist 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylic acid methyl ester (DMCM), with no effect on the maximal currents (Imax).
In contrast, maximal responses for different partial GABAA agonists on all benzodiazepine-sensitive αxβ3γ2 GABAA receptors were enhanced by chlordiazepoxide. Imax values for piperidine-4-sulphonic acid (P4S) on α1β3γ2, THIP on α3β3γ2, and 5-(4-piperidyl)isothiazol-3-ol (thio-4-PIOL) on α2β3γ2 and α5β3γ2 GABAA receptors were increased by chlordiazepoxide, while that for P4S on α1β3γ2 receptors was decreased by DMCM.
The Imax values for partial agonists were also enhanced by pentobarbitone, the neurosteroid allopregnanolone and loreclezole irrespective of receptor subtype or the nature of the partial agonist.
In the light of models of ligand-gated ion channel receptor activation we suggest two possible mechanisms of action for the effects of allosteric modulators on partial agonist receptor activation: either selective modulation of agonist affinity for the open/closed state, or direct modulation of the gating process itself.
Keywords: Recombinant GABAA receptors, partial GABAA agonists, THIP, P4S, thio-4-PIOL, chlordiazepoxide, DMCM, pentobarbitone, allopregnanolone, loreclezole
Introduction
γ-Aminobutyric acid is the most important inhibitory neurotransmitter in mammalian brain. GABAA receptors belong to the ligand-gated ion channel superfamily, which include glycine, nicotinic acetylcholine and 5-HT3 serotonin receptors (Barnard et al., 1998). GABAA receptors are assembled from multiple subunits to form chloride ionophores in a presumed pentameric arrangement (Tretter et al., 1997). A great number of different mammalian GABAA receptor subunits have been cloned including α1–6, β1–4, γ1–3, δ, ε, π, Θ and ρ1–3 subunits (reviewed by Barnard et al., 1998). It is thought that GABAA agonists bind at the interface of α and β subunits and critical residues in both these subunits have been shown to be required for agonist-elicited opening of the ionophores (Amin & Weiss, 1993; Sigel & Buhr, 1997). Similarly the type of receptor subunits present also influence the efficacies and EC50 values of GABAA agonists (Ebert et al., 1994). The α subunit is a major determinant of the kinetics of ionophore activity and the efficacy of GABA (Lavoie et al., 1997) as well as of other GABAA agonists (Ebert et al., 1994). Several conformationally restricted analogues of GABA have been developed with different efficacies such as 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol (THIP), piperidine-4-sulphonic acid (P4S) and 5-(4-piperidyl)isothiazol-3-ol (thio-4-PIOL) (Krogsgaard-Larsen et al., 1997).
GABAA receptors have several modulatory sites for a great variety of pharmacologically important agents. The binding sites for anxiolytic benzodiazepines and anxiogenic β-carbolines are probably located at α/γ interfaces as the α1 subunit is photolabelled by [3H]-flunitrazepam and the γ subunit is essential for bidirectional modulation of channel activity by benzodiazepine ligands (Sigel & Buhr, 1997). Several reports confirm the view that ligands of the benzodiazepine site exert their effects via modulating the EC50 of the full agonists GABA and muscimol (Sigel & Baur, 1988; Wafford et al., 1992). Barbiturates in increasing concentrations have three actions on GABAA receptors: potentiation of GABA, direct receptor activation, and blockade of the chloride ionophores (Thompson et al., 1996) via sites distinctly different from that of GABA (Amin & Weiss, 1993). Similar to pentobarbitone, neurosteroids such as allopregnanolone (5α-pregnan-3α-ol-20-one) in increasing concentrations potentiate the effect of GABA, directly activate the receptor and increase the rate of receptor desensitization (Turner & Simmonds, 1989; Puia et al., 1990; Woodward et al., 1992). The anticonvulsant loreclezole is selective for receptors containing β2 or β3 subunits (Wafford et al., 1994). Loreclezole decreases the EC50 and the apparent maximal response for GABA (Wafford et al., 1994) and increases the rate of receptor desensitization (Donnelly & Macdonald, 1996).
Most studies dealing with these allosteric agents have been restricted to interactions with full GABAA agonists such as GABA and muscimol. Here we have used recombinant receptors, different α subunits in combination with β3 and γ2 GABAA subunits, for which the efficacies of different partial agonists have been determined (Ebert et al., 1997). We investigated the allosteric modulation of receptors activated by partial GABAA agonists exerting both high and low efficacies depending on receptor subtype.
Methods
Oocyte expression
Oocytes were removed from anaesthetized Xenopus laevis and defolliculated with forceps. After treatment with collagenase (0.5 mg ml−1) for 5 min, a 20 nl aliquot of mixtures of human α1, α2, α3 α5 or α6 with β3 and γ2 GABAA receptor subunit cDNAs (20 ng μl−1) were injected into the nuclei of the oocytes. The cDNAs were engineered into the expression vector pCDM8 or pcDNAAmp. The injection buffers contained (mM): NaCl 88, KCl 1, HEPES 15 at pH 7.0. The oocytes were incubated for 1–3 days at 20°C in modified Barth's solution consisting of (mM): NaCl 88, KCl 1, HEPES 10, MgSO4 0.82, Ca(NO3)2 0.33, CaCl2 0.91 and NaHCO3 2.4 at pH 7.
Electrophysiology
Oocytes were placed in a 50 μl chamber and continuously perfused with modified Barth's solution at a rate of 4–6 ml min−1. Cells were impaled with two 1–4 MΩ capillary electrodes containing 2 M KCl and voltage clamped at −70 mV. GABAA agonists were added to the perfusion medium. All allosteric agents were preapplied for 30 s prior to the coapplication of GABAA agonists, except for chlordiazepoxide for which preapplication was not necessary. Following the observation of the peak current, cells were washed for a minimum of 3 min after returning to baseline, and for at least 5 min between saturating agonist concentrations. Increasing concentrations of the agonists were followed by the addition of 3 mM GABA. The peak agonist responses were normalized to the maximal response of 3 mM GABA which was reached within 10 s.
Data were fitted via the computer program GraphPad Prism 2.0 (San Diego, CA, U.S.A.). Curves were fitted using a non-linear square-fitting program to the equation f(x)=Imax/[1+(EC50/x)n] where x is the agonist concentration, Imax is the maximal current, EC50 is the concentration eliciting a half-maximal response and n is the Hill coefficient. Statistical analysis was performed via Student's t-test for 1g EC50 and the Mann–Whitney nonparametric test for Imax values and considered significant if P⩽0.05.
Drugs
Chlordiazepoxide, collagenase, pentobarbitone and allopregnanolone were purchased from Sigma (Poole, U.K.), 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylic acid methyl ester (DMCM) from Research Biochemicals (Natick, MA, U.S.A.), and P4S from Tocris-Cookson (U.K.). Loreclezole was a gift from Janssen, THIP and thio-4-PIOL were gifts from Prof P. Krogsgaard-Larsen (Copenhagen, Denmark).
Results
Modulation via the benzodiazepine site
GABAA agonists elicited inward chloride currents on oocytes expressing αxβ3γ2 GABAA receptors at a holding potential of −70 mV. GABA concentrations that elicited a response equal to 20% of the maximal response for α1β3γ2 GABAA receptors were combined with different concentrations of chlordiazepoxide, a representative anxiolytic 1,4-benzodiazepine. Figure 1 shows that chlordiazepoxide resulted in concentration dependent potentiation of the current for GABA with an EC50 value of 3.1 (2.3, 4.2) μM (mean±s.e.mean of three experiments). The smaller responses for the partial agonist P4S were similarly potentiated by chlordiazepoxide (Figure 1) with an EC50 of 2.0 (1.6,2.4) μM (mean±s.e.mean of three experiments). However, Figure 1 did not reveal any differences for the allosteric interactions of chlordiazepoxide with the full agonist GABA versus the partial agonist P4S.
Figure 1.
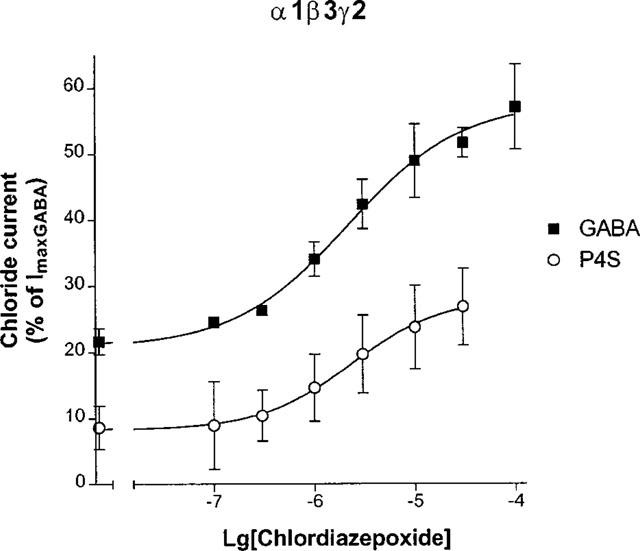
Concentration-response curves of chlordiazepoxide on the chloride currents elicited by GABA and P4S for α1β3γ2 GABAA receptors. The peaks of the chloride currents were expressed as per cent of the maximal currents by 3 mM GABA (ImaxGABA). Agonist concentrations were titrated for each oocyte and equivalent to EC20 values (0.8 μM GABA and 15 μM P4S). The points are mean±s.e.mean of three experiments.
Using GABA as an agonist, benzodiazepines have been shown not to enhance the maximum current of GABAA receptors (Wafford et al., 1992; Yakushiji et al., 1993), and from the previous experiment the EC50 of chlordiazepoxide was independent of the GABAA agonist. We then studied the effects of modulators on the concentration-response relationship of the partial agonists, particularly the maximal response.
Figure 2a shows the concentration-response curves of P4S at α1β3γ2 GABAA receptors. A saturating concentration (30 μM) of chlordiazepoxide shifted the curve for P4S to the left, and enhanced the maximal current amplitude (Figure 2a). Table 1 summarizes the Imax and EC50 values derived from computer fits, demonstrating significant effects on these parameters. Figure 2a also shows that the concentration-response curve for P4S was depressed by DMCM, a representative inverse agonist β-carboline. DMCM decreased the Imax values for P4S without significantly affecting its EC50 value (Table 1). As a comparison with a full agonist, the allosteric effects on the concentration-response curve of GABA were also examined for the same α1β3γ2 subunit combination. Figure 2b demonstrates that chlordiazepoxide shifted the response curve of GABA to the left, without affecting the maximum. DMCM resulted in an opposite shift (Figure 2b). Table 1 shows that the bidirectional changes in the EC50 value for GABA were statistically significant.
Figure 2.
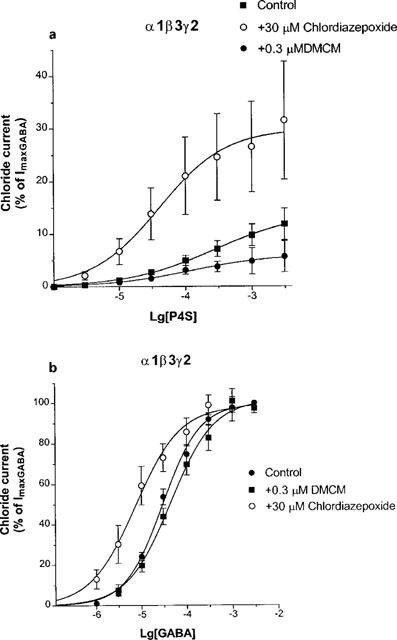
The effects of 30 μM chlordiazepoxide (CDZ) and 0.3 μM DMCM on the concentration-response curves of P4S (a) and GABA (b) for α1β3γ2 GABAA receptors. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of the number of experiments indicated in Table 1. Fitted curves with variable slopes resulted in the Imax and EC50 values in Table 1.
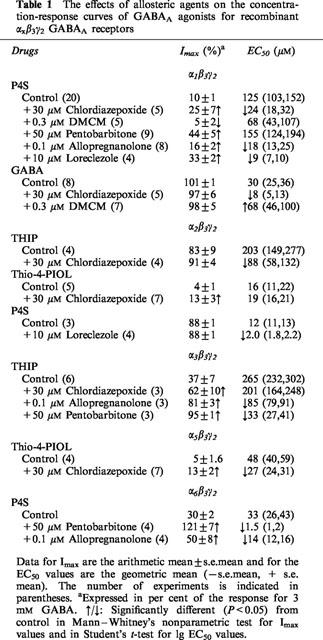
Table 1.
The effects of allosteric agents on the concentration-response curves of GABAA agonists for recombinant αxβ3γ2 GABAA receptors
The α2 subunit enabled us to investigate further partial agonists having either high or low efficacies. THIP has high efficacy at α2β3γ2 GABAA receptors (Figure 3). Chlordiazepoxide shifted the concentration response curve for THIP to the left (Figure 3). Table 1 shows that chlordiazepoxide did not significantly affect the high maximal response for THIP but decreased its EC50 value. In contrast, THIP has low efficacy for α3β3γ2 GABAA receptors (Figure 4b). Chlordiazepoxide enhanced the maximal current elicited by THIP (Figure 4b) but did not significantly decrease the EC50 of THIP for α3β3γ2 receptors (Table 1). The concentration-dependence of chlordiazepoxide was examined against a nearly saturating concentration of THIP (3.3 mM), a concentration which elicited 25% of the maximally activated GABA current on α3β3γ2 receptors (Figure 4c). Chlordiazepoxide enhanced this response with EC50=6 (3,11) μM with a Hill slope value of 0.64 (0.57,0.71) (mean±s.e.mean of five experiments) (Figure 4c). The low slope value and the responses beyond 50% in Figure 4c might indicate an additional response to chlordiazepoxide with potency above 10 μM.
Figure 3.
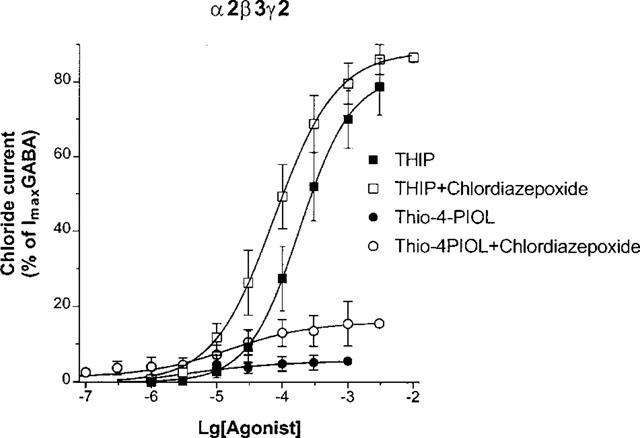
The effects of 30 μM chlordiazepoxide on the concentration-response curves of THIP and thio-4-PIOL for α2β3γ2 GABAA receptors. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of four experiments.
Figure 4.
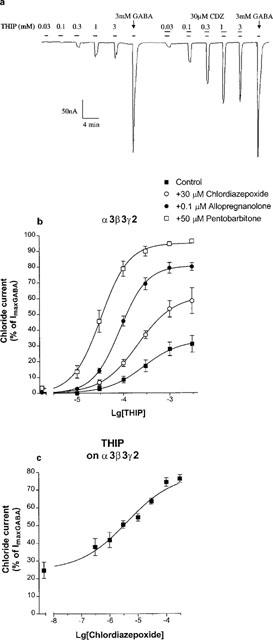
The effects of chlordiazepoxide, allopregnanolone and pentobarbitone on the concentration-response curves of THIP for α3β3γ2 GABAA receptors. (a) Example recording of currents in response to THIP in the absence and presence of 30 μM chlordiazepoxide, showing control responses to a saturating concentration of GABA (3 mM) after each concentration-response curve. Drugs were applied as indicated by the bars. (b) Mean concentration-response curves to THIP in the absence and presence of 30 μM chlordiazepoxide, 100 nM allopregnanolone and 50 μM pentobarbitone. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of 3–6 experiments. (c) The effects of chlordiazepoxide on the chloride currents elicited by a maximally effective concentration (3.3 mM) of THIP for α3β3γ2 GABAA receptors. Data are mean±s.e.mean of five experiments. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. 3.3 mM THIP alone elicited 25±5% of the current peak for 3 mM GABA. It was enhanced by chlordiazepoxide with an EC50 value of 6 (3,11) μM and a Hill slope value of 0.64 (0.57, 0.71).
Thio-4-PIOL has been characterized as a low potency GABAA antagonist with no efficacy of its own for most recombinant GABAA receptors (Ebert et al., 1997). Thio-4-PIOL was able to elicit extremely small currents on α2β3γ2 GABAA receptors but its efficacy was very low (Figure 3), reaching a maximum of 4±1% of a 3 mM GABA current (Table 1). However, chlordiazepoxide strongly enhanced the currents for thio-4-PIOL (Figure 3) by increasing its efficacy without significantly affecting its potency (Table 1).
Thio-4-PIOL elicited very small currents for α5β3γ2 GABAA receptors, too (Figure 5). Chlordiazepoxide strongly enhanced the currents for thio-4-PIOL by increasing not only its efficacy but in this case also its potency (Table 1).
Figure 5.
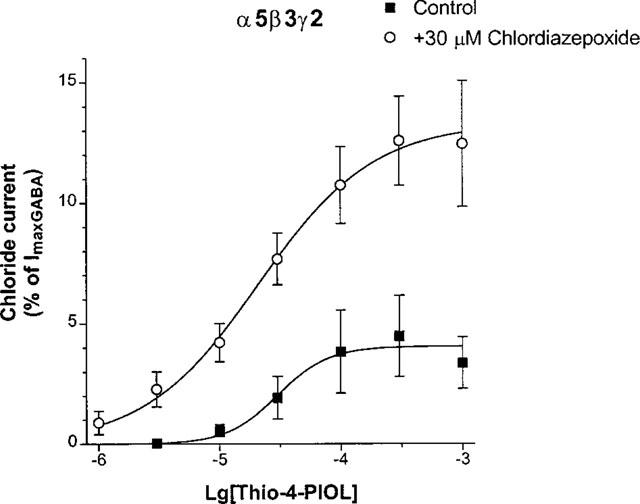
The effects of 30 μM chlordiazepoxide on the concentration-response curves of thio-4-PIOL for α5β3γ2 GABAA receptors. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of the number of experiments indicated in Table 1.
Allosteric potentiating effects of a barbiturate, a neurosteroid and loreclezole
Pentobarbitone, allopregnanolone and loreclezole were applied in concentrations that did not elicit significant direct currents for α1β3γ2 GABAA receptors but strongly potentiated the currents for P4S (see the effects of allopregnanolone in Figure 6a). Pentobarbitone at 50 μM resulted in a great enhancement of the maximal response to P4S (Figure 6b). Table 1 reveals that pentobarbitone did not affect the EC50 of P4S. The neurosteroid allopregnanolone (0.1 μM) produced a large decrease in the EC50 of P4S, as well as significantly increased its efficacy at α1β3γ2 GABAA receptors (Figure 6b and Table 1).
Figure 6.
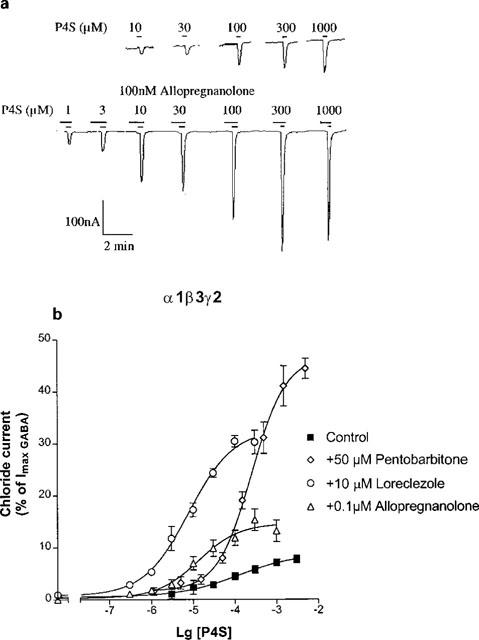
The effects of pentobarbitone, allopregnanolone and loreclezole on the concentration-response curves of P4S for α1β3γ2 GABAA receptors. (a) Example traces of a concentration-response curve to P4S on a cell expressing α1β3γ2 GABAA receptors in the absence and presence of 100 nM allopregnanolone. Drugs were applied as indicated by the bars. The current to 1 mM P4S was equivalent to 10% of that to 3 mM GABA (not indicated). (b) The effects of 50 μM pentobarbitone, 0.1 μM allopregnanolone and 10 μM loreclezole on the concentration-response curves of P4S for α1β3γ2 GABAA receptors. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of the number of experiments indicated in Table 1.
Loreclezole (10 μM) produced a much greater increase in maximal response for P4S on α1β3γ2 GABAA receptors than the neurosteroid (Figure 6b). Table 1 shows that loreclezole enhanced both the efficacy and potency of P4S. For comparison, the effects of loreclezole were also examined on α2β3γ2 GABAA receptors for which P4S has higher potency and almost full efficacy (Table 1). In this case loreclezole did not change the efficacy of P4S but further enhanced its high potency (Table 1).
On α6β3γ2 GABAA receptors pentobarbitone at 50 μM exerted significant currents in the absence of GABA agonists (Figure 7). However, pentobarbitone resulted in a huge enhancement of the maximal current and potency of P4S (Figure 7), the maximal response surpassing that for 3 mM GABA alone (Figure 7). Allopregnanolone at 100 nM did not elicit chloride currents but also enhanced strongly the efficacy and potency of P4S on α6β3γ2 GABAA receptors (Figure 7).
Figure 7.
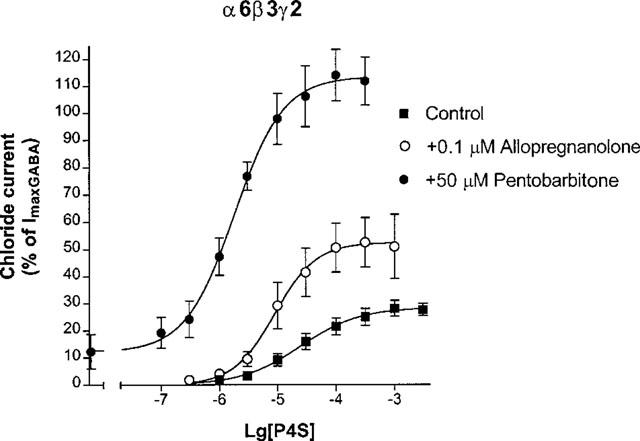
The effects of 100 nM allopregnanolone and 50 μM pentobarbitone on the concentration-response curves of P4S for α6β3γ2 GABAA receptors. Chloride currents are expressed as per cent of the peak current elicited by 3 mM GABA. Data are mean±s.e.mean of 3–4 experiments.
Discussion
Benzodiazepine site ligands affect the efficacy and potency of partial and full GABAA agonists, respectively
Analysis of the allosteric modulation of GABAA receptor-ionophore function has primarily been carried out using GABA (Lavoie et al., 1997) and another full agonist muscimol (Macdonald & Twyman, 1992). The allosteric effects of the ligands of the benzodiazepine site or other allosteric binding sites, and ionophore function of GABAA receptors have been very thoroughly studied because of their pharmacological importance. Several studies support the view that benzodiazepine agonists facilitate, while inverse agonists inhibit GABAergic neurotransmission, via mutual allosteric interactions to modulate the affinity of GABA binding rather than subsequent gating of the channel (Macdonald & Twyman, 1992; Serfozo & Cash, 1992). Recent evidence however suggests that benzodiazepines may be more closely linked to the ion channel itself, and can modulate spontaneously open channels in the absence of GABA (Thompson et al., 1999).
The effects of chlordiazepoxide and DMCM on GABA for α1β3γ3 and on THIP for α2β3γ2 GABAA receptors are in agreement with previous reports which suggest a shift in apparent agonist potency but no effect on maximum response when full agonists are employed to activate the receptor complex (Sigel & Baur, 1988; Wafford et al., 1992). Similar studies for partial GABAA agonists have not been reported previously. Chlordiazepoxide exerted 2–3 fold enhancements of the maximal responses of three partial agonists for αxβ3γ2 GABAA receptors containing all types of α subunits apart from the benzodiazepine-insensitive α4 and α6 subtypes. This included thio-4-PIOL, which had a maximum efficacy of only 4–5% on α2β3γ2 and α5β3γ2 receptors, and has previously been described as an antagonist (Ebert et al., 1997). This allosteric modulation of the efficacies of partial agonists therefore seems to be a general phenomenon, valid for different structures of both agonists and GABAA receptor subtypes. The concentration-response curves of chlordiazepoxide resulted in similar EC50 values (2–6 μM), using both different agonist concentrations (EC20 or maximally effective concentration) and different chemical structures of GABAA agonists (GABA, P4S and THIP). Its effects are mediated by common benzodiazepine binding sites on the GABAA receptor complex, whose behaviour appears not to depend on the choice of the GABA agonist applied. The effects on efficacy of partial agonists seems also to be the case of inverse agonists, as the low efficacy of P4S on α1β3γ2 was further decreased by DMCM, a β-carboline inverse agonist.
The effects of the ligands of the benzodiazepine sites on the EC50 values of partial agonists were not so consistent. Chlordiazepoxide increased the potencies of P4S for α1β3γ2 and of thio-4-PIOL for α5β3γ2 receptors but its effects on thio-4-PIOL for α2β3γ2 as well as on THIP for α3β3γ2 GABAA receptors were not significant. As the effects of benzodiazepines on GABA EC50 are 2–3 fold at maximum, it is likely that the small shifts in EC50 produced by the benzodiazepine and β-carboline, some of which do not reach significance, are overwhelmed by the larger effects observed on the efficacy of partial agonists.
The effects of a barbiturate, a neurosteroid and loreclezole
Low concentrations of pentobarbitone up to 50 μM facilitate the effect of GABA and enhance the potencies rather than the efficacies of GABAA agonists (Akaike et al., 1985; Zhang & Simmonds, 1997). In contrast, 50 μM pentobarbitone did not affect the potency of the partial agonist P4S but strongly enhanced its efficacy for α1β3γ2 GABAA receptors.
Pentobarbitone increased the maximal responses for the low efficacy agonists THIP on α3β3γ2 and P4S on α6β3γ2 receptors close to the maximal level of GABA. The enhancement of the maximal response of P4S on α6β3γ2 receptors beyond 100% was unusual. Maximal responses beyond 100% for THIP (Wafford et al., 1996) and pentobarbitone on α6β3γ2 receptors have been reported previously (Thompson et al., 1996). It is possible that the rapid desensitization of α6β3γ2 receptors when activated by GABA results in a significant reduction in the observed peak response, which may be different when activated by P4S which elicits less desensitization. It is unlikely however that this could account for the large changes in efficacy observed here as shown by compounds such as THIP which can behave as a full or partial agonist. Pentobarbitone also enhanced the potencies of THIP and P4S by about an order of magnitude. It is interesting however, that the low EC50 value of P4S on α6β3γ2 receptors was strongly decreased by pentobarbitone while the high EC50 value on α1β3γ2 receptors was not affected by it.
The effects of the neurosteroid allopregnanolone up to 100 nM are also restricted to potentiation of GABA (Turner & Simmonds, 1989; Puia et al., 1990), i.e. to decrease the EC50 value of the full agonists (Woodward et al., 1992). In contrast, 100 nM allopregnanolone enhanced not only the potencies of P4S on α1β3γ2 and α6β3γ2 receptors and THIP on α3β3γ2 receptors but also their maximal responses.
The anticonvulsant loreclezole (10 μM) enhanced the affinity of GABA (Wafford et al., 1994), muscimol (Zhang & Simmonds, 1997) and P4S (this study). Further, it resulted in an apparent decrease in the maximal response for GABA on recombinant α1β2γ2 GABAA receptors (Wafford et al., 1994) which might be attributed to the enhancement of desensitization (Donnelly & Macdonald, 1996). In contrast, the high efficacy of P4S (88%) for α2β3γ2 GABAA receptors was not affected by 10 μM loreclezole. Consequently, loreclezole does not necessarily decrease the maximal response for full agonists. This may be related to receptor subtypes and distinct agonists differing in their rates of desensitization. In contrast, for α1β3γ2 receptors at which P4S is an agonist with low efficacy, 10 μM loreclezole greatly enhanced the maximal response to P4S. In conclusion, the effects of the representative barbiturate, neurosteroid and loreclezole in GABA-facilitating concentrations on partial GABAA agonists are different from their effects on full agonists.
Allosteric model of receptor activation
Electrophysiological and 36Cl− flux data on GABAA receptor-ionophores have been analysed in terms of a sequential model (Macdonald & Twyman, 1992; Serfozo & Cash, 1992; Lavoie et al., 1997) or a ‘bifurcation' model of channel activation (Johnes et al., 1998). Spontaneous openings of nicotinic acetylcholine (Galzi et al., 1996) and GABAA receptor channels (Neelands et al., 1999) and changes in their pharmacological profiles cannot be reconciled with these sequential models. Recent findings for the phenotypes of neurotransmitter receptor-ionophores have been better explained within the framework of the Monod–Wyman–Changeux allosteric receptor model (Galzi et al., 1996). The simplest two-state model is characterized by an isomerization equilibrium between an inactive close state B and an active open state A of the ionophore. Ligands can be differentiated by the ratio of distinct dissociation constants to these states: C=KA/KB. This model can also be applied for GABAA receptors. Full agonists are supposed to bind preferentially to the open state. Partial GABAA agonists such as P4S and THIP might then result in lower selectivities of binding to the open state. This seems to be in agreement with shorter channel opening duration for THIP and P4S in comparison to GABA and muscimol as concluded from the fluctuation analysis of cultured mouse spinal neurons (Barker & Mathers, 1981). The antagonism of thio-4-PIOL for all recombinant αxβ3γ2 GABAA receptors (Ebert et al., 1997) can be reconciled with preferential binding to the closed state. Benzodiazepine agonists such as chlordiazepoxide and other allosteric modulators are likely to increase, while the inverse agonist DMCM decreases the receptor's ability to isomerize to the open state (the E value or efficacy in del-Castillo & Katz's two-state model) (Colquhoun, 1998). This is supported by evidence showing modulation of constitutive GABAA receptor activity by benzodiazepines in the absence of a GABAA agonist (Thompson et al., 1999).
If allosteric agents affect the affinities of the agonist for both states A and B and the ratio c=KA/KB remains constant, the concentration-response curves of the agonists are shifted horizontally in a parallel manner (Galzi et al., 1996). This seems to be valid for the bidirectional effects of benzodiazepine site ligands on full GABAA agonists. However, chlordiazepoxide resulted in nonparallel shifts of the concentration-response curves for partial agonists, suggesting that benzodiazepine potentiation is either being mediated via a change in gating, or the KA/KB ratio is not constant. Modulation of spontaneous activity by allosteric modulators suggests the mechanism to be due to direct changes in channel gating (Thompson et al., 1999; Neelands et al., 1999).
As to other neurotransmitter receptor-ionophores, a similar effect has been reported for α7 neuronal nicotinic acetylcholine receptors (Krause et al., 1998). Ivermectin enhanced the potency and efficacy of the partial agonist 1,1-dimethyl-4-phenylpiperazinium. It has been attributed to an allosteric effect on the isomerization from a closed to open state of the ionophore (Krause et al., 1998).
In conclusion, several allosteric agents affect differently the response curves of partial versus full GABAA agonists suggesting the mechanism to be more than just a shift in agonist affinity. The differential effects appear to be independent of the type of α7 subunit and are not compound specific. Single channel analysis will hopefully elucidate further the differences in molecular mechanism of the interaction between GABAA agonists and allosteric agents.
Acknowledgments
Discussions with Dr Bjarke Ebert (Royal Danish School of Pharmacy, Copenhagen, Denmark) are acknowledged. This study was supported by grants T 029723 from the Hungarian Science Research Fund OTKA, the Soros Foundation and by the British Council for Gábor Maksay.
Abbreviations
- DMCM
6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylic acid methyl ester
- P4S
piperidine-4-sulphonic acid
- Thio-4-PIOL
5-(4-piperidyl)isothiazol-3-ol
- THIP
4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol
References
- AKAIKE N., HATTORU K., INOMATA N., OOMURA Y. γ-Aminobutyric acid- and Pentobarbitone-gated chloride currents in internally perfused frog sensory neurons. J. Physiol. 1985;360:367–386. doi: 10.1113/jphysiol.1985.sp015622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMIN J., WEISS D.S. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by Pentobarbitone. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- BARKER J.L., MATHERS D.A. GABA analogues activate channels of different duration on cultured mouse spinal neurons. Science. 1981;212:358–361. doi: 10.1126/science.6259733. [DOI] [PubMed] [Google Scholar]
- BARNARD E.A., SKOLNICK P., OLSEN R.W., MOHLER H., SIEGHART W., BIGGIO G., BRAESTRUP C., BATESON A.N., LANGER S.Z. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- COLQUHOUN D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and the effects of mutating receptors. Br. J. Pharmacol. 1998;125:923–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONNELLY J.L., MACDONALD R.L. Loreclezole enhances apparent desensitization of recombinant GABAA receptor currents. Neuropharmacology. 1996;35:1233–1241. doi: 10.1016/s0028-3908(96)00053-6. [DOI] [PubMed] [Google Scholar]
- EBERT B., THOMPSON S.A., SAOUNATSOU K., MCKERNAN R., KROGSGAARD-LARSEN P., WAFFORD K.A. Differences in agonist/antagonist binding affinity and receptor transduction using recombinant human γ-aminobutyric acid type A receptors. Mol. Pharmacol. 1997;52:1150–1156. [PubMed] [Google Scholar]
- EBERT B., WAFFORD K.A., WHITING P.L., KROGSGAARD-LARSEN P., KEMP J.A. Molecular pharmacology of γ-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different α, β and γ receptor subunit combinations. Mol. Pharmacol. 1994;46:957–963. [PubMed] [Google Scholar]
- GALZI J.L., EDELSTEIN S.J., CHANGEUX J.P. The multiple phenotypes of allosteric receptor mutants. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1853–1858. doi: 10.1073/pnas.93.5.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNES M.V., SAHARA Y., DZUBAY J.A., WESTBROOK G.L. Defining affinity with the GABAA receptor. J. Neurosci. 1998;18:8590–8604. doi: 10.1523/JNEUROSCI.18-21-08590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRAUSE R.M., BUISSON B., BERTRAND S., CARRINGER P.J., GALZI J.L., CHANGEUX J.P., BERTRAND D. Ivermectin: a positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1998;53:283–294. doi: 10.1124/mol.53.2.283. [DOI] [PubMed] [Google Scholar]
- KROGSGAARD-LARSEN P., FROLUND B., EBERT B.GABAA receptor agonists, partial agonists, and antagonists The GABA Receptors 1997Humana Press: Totowa, NJ; 37–81.Enna, S.J. & Bovery, N.G. (eds) [Google Scholar]
- LAVOIE A.M., TINGEY J.J., HARRISON N.L., PRITCHETT D.B., TWYMAN R.E. Activation and deactivation rates of recombinant GABAA receptor channels are dependent on α− subunit isoform. Biophys. J. 1997;73:2518–2526. doi: 10.1016/S0006-3495(97)78280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDONALD R.L., TWYMAN R.E.Kinetic properties and regulation of GABAA receptor channels Ion Channels 19923Plenum Press: New York; 315–343.Narahashi T. (ed.) [DOI] [PubMed] [Google Scholar]
- NEELANDS T.R., FISCHER J.L., BIANCHI M., MACDONALD R.L. Spontaneous and γ-aminobutyric acid (GABA)-activated GABAA receptor channels formed by ε subunit-containing isoforms. Mol. Pharmacol. 1999;54:168–178. doi: 10.1124/mol.55.1.168. [DOI] [PubMed] [Google Scholar]
- PUIA G., SANTI M., VICINI S., PRITCHETT D.B., PURDY R.H., PAUL S.M., SEEBURG P.H., COSTA E. Neurosteroids act on recombinant human GABAA receptors. Neuron. 1990;4:757–765. doi: 10.1016/0896-6273(90)90202-q. [DOI] [PubMed] [Google Scholar]
- SERFOZO P., CASH D. Effect of a benzodiazepine (chlordiazepoxide) on a GABAA receptor from rat brain: requirement of only one bound GABA molecule to channel opening. FEBS Lett. 1992;310:55–59. doi: 10.1016/0014-5793(92)81145-c. [DOI] [PubMed] [Google Scholar]
- SIGEL E., BAUR R. Allosteric modulation by benzodiazepine receptor ligands of the GABAA receptor expressed in Xenopus oocytes. J. Neurosci. 1988;8:289–295. doi: 10.1523/JNEUROSCI.08-01-00289.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIGEL E., BUHR A. The benzodiazepine binding site of GABAA receptors. TIPS. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- THOMPSON S.A., SMITH M.Z., WINGROVE P.B., WHITING P.J., WAFFORD K.A. Mutation at the putative GABAA ion-channel gate reveals changes in allosteric modulation. Br. J. Pharmacol. 1999;127:1349–1358. doi: 10.1038/sj.bjp.0702687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON S.A., WHITING P.B., WAFFORD K.A. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br. J. Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRETTER V., EHYA N., FUCHS K., SIEGHART W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURNER J.P., SIMMONDS M.H. Modulation of the GABAA receptor complex by steroids in slices of rat cuneate nucleus. Br. J. Pharmacol. 1989;96:409–417. doi: 10.1111/j.1476-5381.1989.tb11832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAFFORD K.A., BAIN C.J., QUIRK K., MCKERNAN R.M., WINGROVE P.B., WHITING P.J., KEMP J.A. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12:775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- WAFFORD K.A., THOMPSON S.A., THOMAS D., SIKELA J., WILCOX A.S., WHITING P.J. Functional characterization of human γ-aminobutyric acidA receptors containing the α4 subunit. Mol. Pharmacol. 1996;50:670–678. [PubMed] [Google Scholar]
- WAFFORD K.A., WHITING P.J., KEMP J.A. Differences in affinity and efficacy of benzodiazepine receptor ligands at recombinant γ-aminobutyric acidA receptor subtypes. Mol. Pharmacol. 1992;43:240–244. [PubMed] [Google Scholar]
- WOODWARD R.M., POLENZANI L., MILEDI R. Effects of steroids on γ-aminobutyric acid receptors expressed in Xenopus oocytes by poly(A)+ RNA from mammalian brain and retina. Mol. Pharmacol. 1992;41:89–103. [PubMed] [Google Scholar]
- YAKUSHIJI T., SHIRASAKI T., MUNAKATA M., HIRATA A., AKAIKE N. Differential properties of type I and type II benzodiazepine receptors in mammalian CNS neurons. Br. J. Pharmacol. 1993;109:819–825. doi: 10.1111/j.1476-5381.1993.tb13648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y., SIMMONDS M. Interactions between loreclezole, chlormethiazole and pentobarbitone at GABAA receptors: functional and binding studies. Br. J. Pharmacol. 1997;121:1392–1399. doi: 10.1038/sj.bjp.0701269. [DOI] [PMC free article] [PubMed] [Google Scholar]