

Abstract
The effect of the oxystilbene derivative F3 was tested on nAChRs of whole-cell patch-clamped rat chromaffin cells in vitro and of rat adrenal gland membranes using 125I-epibatidine.
F3 (30 nM) rapidly and reversibly blocked inward currents generated by pulse applications of nicotine, shifting the dose-response curve to the right in a parallel fashion without changing the maximum response. The action of F3 was voltage insensitive and not due to altered current reversal potential.
The R isomer of F3 was more potent (IC50=350±30 nM) than its S-enantiomer (IC50=1.5±0.3 μM). Nicotine-evoked currents were insensitive to 10 μM α-bungarotoxin.
Equi-amplitude currents evoked by nicotine or epibatidine were similarly antagonized by R-F3 in a reversible fashion. Epibatidine-evoked currents readily produced receptor desensitization.
Adrenal membranes specifically bound 125I-epibatidine with a single population of binding sites endowed with high affinity (KD=159 pM) and Bmax of 6.5±1.3 fmol mg−1 of protein.
125I-epibatidine binding was specifically displaced by cytisine (Ki=68 nM) or ACh (Ki=348 nM). F3 specifically displaced 125I-epibatidine binding although with lower affinity (Ki=29.6 μM) than in electrophysiological experiments. 125I-epibatidine binding to rat adrenal tissue was insensitive to α-bungarotoxin which readily antagonized 125I-epibatidine binding to bovine adrenal tissue.
The present results suggest that F3 is a relatively potent and apparently competitive antagonist of nAChRs on rat chromaffin cells. Since previous studies have indicated that F3 targets different subtypes on chick neuronal tissue, it appears that nAChRs display interspecies differences to be considered for drug development studies.
Keywords: Nicotinic receptor, nicotine, epibatidine, chromaffin cell, competitive antagonism, 4-oxystilbene
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are a family of ACh-gated cationic channels consisting of different subtypes with distinct anatomical distribution in the vertebrate central and peripheral nervous systems (for reviews, see Role & Berg, 1996; Gotti et al., 1997; Lindstrom, 1997). The role and function of most of these subtypes are difficult to establish because they may be coexpressed in individual neurons. For this reason tools to improve the pharmacological ‘dissection' of nAChRs would be very useful to elucidate the relative contribution of receptor subtypes to the pathophysiology of neurological disorders like Alzheimer's and Parkinson's diseases, epilepsy, schizophrenia, and Tourette's syndrome (Lena & Changeux, 1997; Lindstrom, 1997).
While searching for new compounds active on nAChRs, we have demonstrated that two oxystilbene derivatives (MG624 and its more recently synthesized derivative F3) are selective ligands for chick nAChRs sensitive to αBgtx (Gotti et al., 1998). MG624 has been found to interact both with the wild and mutated chick α7 receptor subtype (Maggi et al., 1999). Nevertheless, since MG624 was originally developed as a ganglioplegic drug and produces potent ganglion blocking activity causing significant hypotension (Cavallini et al., 1953; Mantegazza & Tommasini, 1955), we have investigated whether, in mammals, oxystilbene derivatives could also affect the α3(α5)β4 receptor subtype which is the principal nAChR class on autonomic ganglia as demonstrated by immunocytochemical and molecular biology techniques (Role & Berg, 1996; Campos Caro et al., 1997) . To this end we studied the action of F3 (see Figure 1 for its chemical formula) as this compound has been reported to be more active than MG624 itself in binding to the β4 receptors (Gotti et al., 1998).
Figure 1.

Chemical formula of N,N,N-trimethyl-1-(4-trans-stilbenoxy)-2-propylammonium iodide (F3).
In addition to mammalian sympathetic, parasympathetic and sensory ganglia, the chromaffin cells of the adrenal medulla (which have the same embryological origin as postganglionic sympathetic neurons; Anderson, 1993) are a major source of α3(α5)β4 nAChRs which are physiologically activated by ACh released from the splanchnic nerve to mediate catecholamine liberation into the blood-stream (for reviews, see Gotti et al., 1997; Lindstrom, 1997). In the present study, we characterized the action of the F3 compound on native nAChRs present on rat chromaffin cells by means of electrophysiological and binding studies.
Methods
Electrophysiology
Cell preparation
Rat chromaffin cells were cultured according to the method of Brandt (Brandt et al., 1976; Giniatullin et al., 1999; Khiroug et al., 1997; 1998). Twenty-five to 35 day old rats were killed with slowly rising levels of CO2 and their adrenal glands were removed, dissected from the cortex, and rinsed in a medium (pH 7.2) containing (mM): NaCl 137, KCl 3, Na2HPO4 0.7, HEPES 25, glucose 10 and 350 units ml−1 of penicillin and streptomycin. Cells were dissociated by treating adrenal tissue fragments at 37°C for about 60 min with collagenase A and DNAse I (0.5 units ml−1 and 10 μg ml−1, respectively; both from Sigma) and drawing them gently up and down inside a Pasteur pipette every 15 min. The cell-containing suspension was centrifuged at 750×g for 5 min, and rinsed twice with the HEPES-buffered medium. Finally, cells were suspended in Dulbecco's modified Eagle's medium supplemented with 10% v v−1 foetal calf serum, plated on poly-Lysine (5 mg ml−1; Sigma)-coated Petri dishes, and cultured at 37°C for 1–2 days under a 5% CO2 containing atmosphere.
Patch-clamp recording
Cell-containing culture dishes (used at 0–3 days after plating) were mounted on the stage of an inverted Nikon Diaphot microscope and superfused (5–10 ml min−1) with control saline solution containing (mM): NaCl 135, KCl 5, MgCl2 1, CaCl2 2, glucose 15, HEPES 10 (pH adjusted to 7.4 with NaOH, osmolarity 285 mOsm). Patch pipettes pulled from thin glass (1.5 mm o.d.) had 5–6 MΩ resistance when filled with (mM) CsCl 120; HEPES 20; MgCl2 1; Mg2ATP3 3, BAPTA 10 (240 mOsm). The pH of the pipette solution was always adjusted to 7.2 with CsOH. Unless otherwise indicated, cells were voltage clamped at −70 mV. After obtaining the whole-cell configuration a 10 min period of stabilization normally elapsed before membrane currents were recorded, filtered at 1 kHz and acquired on the hard disk of a PC by means of pCLAMP 6.04 software (Axon Instruments Inc., Foster City, CA, U.S.A.).
Drug application
Agonists were usually delivered by pressure application (10–20 p.s.i.) from glass micropipettes positioned about 15–25 μm away from the recorded cell. Previous tests have indicated that with this method there is an approximately 3 fold dilution of the pipette drug concentration (Giniatullin et al., 1996). When this method was also used to apply antagonists, it had the advantage of an almost instantaneous, focal delivery of the drug. However, the strong stream of drug solution caused by the ejection pressure may displace the agonist solution from the extracellular microenvironment immediately around the cell membrane. This phenomenon can magnify the apparent potency of antagonist. In view of this situation, whenever it was necessary to express quantitative data concerning antagonist pharmacology, we employed a PC-controlled rapid solution exchanger (Bio-logic, France) consisting of a multibarrelled array of glass tubes (1 mm o.d.) rapidly rotating to apply the antagonist solution to the recorded cell. In this case the delay between the on command and drug arrival at the cell membrane was 50 ms. Whenever drug concentrations are stated in the results, they refer to experiments based on fast superfusion applications.
Data analysis
Data are presented as mean±s.e.mean (n=number of cells) with statistical significance assessed with Wilcoxon test (for non parametric data) or paired t-test (for normally distributed data). A value of P⩽0.05 was accepted as indicative of a statistically significant difference.
Binding assays
Preparation of homogenates of rat adrenal gland or superior cervical ganglia (SCG)
Adrenal glands were removed from rats killed by terminal anaesthesia with CO2, homogenized in phosphate buffer saline (PBS) containing 2 mM phenylmethysulphonyl fluoride (PMFS) using an Ultraturrax homogenizer and then diluted to 50 ml using the same buffer. The homogenates were centrifuged at 30,000×g for 30 min at 4°C. The dilution and centrifugation steps were then repeated and the homogenates resuspended in buffer containing (mM) Tris-HCl 50 (pH 7), NaCl 150, KCl 5, MgCl2 1, CaCl2 2.5 (buffer A). A mixture of the protease inhibitors leupeptin, bestatin, pepstatin A, aprotinin to a final concentration of 5 μg ml−1 and 2 mM PMFS was added to the homogenate in order to block proteolysis during the assay incubation time. SCG tissue was processed in the same way as adrenal glands.
125I-epibatidine binding
125I-epibatidine (NEN, Boston, U.S.A., specific activity of 2200 Ci mmol1) was used as radioligand for saturation experiments. In preliminary experiments we found that the time required to reach apparent equilibrium conditions for labelled epibatidine binding was 3 h; the standard incubation time used was over four times longer than the one experimentally observed. Aliquots of adrenal gland homogenates were incubated with 125I-epibatidine (0.005–1.5 nM range) diluted in buffer A plus 2 mg ml−1 BSA for 3 h at 25°C or overnight at 4°C.
Non-specific binding was determined in parallel by incubation of tissue samples in the presence of 100–250 nM unlabelled epibatidine. At the end of the incubation period, samples were filtered on GFC filters pre-soaked in polyethylenimine through a Brandell-apparatus, or centrifuged at 10,000×g for 10 min. Radioactivity left on filters was measured with a gamma counter. 125I-epibatidine and 3H-epibatidine (NEN, Boston, U.S.A., specific activity 33.8 Ci mmol−1) binding to SCG homogenates (preincubated in 2 μM αBgtx for 3 h) was performed as described for adrenal gland membranes.
In order to test the ability of nicotinic drugs (ACh, cytisine or F3) to inhibit 125I-epibatidine binding, these agents (dissolved in buffer A just before use) were serially diluted and incubated with rat adrenal homogenates for 30 min at room temperature. After subsequent addition of 125I-epibatidine (final concentration=0.15–0.2 nM), samples were incubated overnight at 4°C. ACh was incubated in the presence of 20 μM physostigmine in order to inhibit cholinesterases. This concentration of physostigmine did not interfere with 125I-epibatidine binding (inhibition constant>100 μM).
125I-α bungarotoxin binding
125I-α Bgtx was obtained from Amersham (specific activity of 200 Ci mmol−1) and used for saturation binding experiments on rat adrenal gland homogenates. Parallel experiments were run using bovine adrenal whole glands and adrenal medulla (fresh bovine glands were obtained from a local abattoir). The 125I-αBgtx concentrations ranged from 0.1 to 20 nM. Non-specific binding was determined using 1 μM unlabelled αBgtx. Binding was performed overnight at 4°C. At least three separate experiments were performed on each tissue.
Data analysis
Data are presented as mean±s.e.mean. The experimental data obtained from the saturation binding experiments were analysed with a non-linear least square method using the LIGAND program as previously described (Gotti et al., 1998). The Ki values of test drugs were also obtained with the LIGAND software, using data from three (for F3) and two (for cytisine and ACh) independent competition experiments.
Materials
Lyophilized αBgtx, PMFS, cytisine, ACh, physostigmine, and the anti-protease inhibitors leupeptin, bestatin, pepstatin A, aprotinin were purchased from Sigma, U.S.A.; non-radioactive epibatidine from RBI; 125I-αBgtx from Amersham, U.K.; 125I-epibatidine and 3H-epibatidine were from NEN (Boston, MA, U.S.A.).
Results
Effect of racemic F3 on nAChR evoked currents
Figure 2 illustrates the principal features of the action of racemic F3 on nicotine-induced currents of rat chromaffin cells. Nicotine was applied with brief pulses (5–100 ms) which elicited reproducible responses without apparent desensitization (Khiroug et al., 1997). As shown in Figure 2A, the fast inward current induced by a pressure application of nicotine (50 μM pipette concentration) was reduced by 30% after a 15 s application of F3 (30 nM pipette concentration). This block had thus a relatively rapid onset with puffer-applied F3 which represented a very focal delivery of the antagonist. Full recovery was observed after 45 s washout. F3 per se did not elicit changes in baseline current or input resistance of the cell. Figure 2B shows the average time course of the F3 depressant action for six cells. The extent of the depression did not progressively change during F3 application and recovery was achieved 90 s later.
Figure 2.

Rapid block of nicotine-induced responses by racemic F3. (A) Current records obtained with 20 ms nicotine (50 μM pipette concentration; left), 15 s after starting pressure application of F3 (30 nM pipette concentration; middle) and 45 s after washout of F3. Note reversible reduction in nicotine current amplitude. (B) Time course of depression of nicotine currents (50 μM pipette concentration, 20 ms application) after pressure application of F3 (30 nM pipette concentration) to six cells. (C) Plot of nicotine current amplitude vs increasing duration of nicotine pressure pulses in control solution and in the presence of F3. Ordinate, current amplitude normalized with respect to the response evoked by 20 ms in control solution for each cell. Abscissa, pulse duration of nicotine (50 μM) applications. F3 (30 nM pipette concentration) was applied for ∼15 s before each nicotine response (8–30 cells). Note that the data points for nicotine in F3 solution (filled circles) differed significantly from the corresponding controls (filled squares) with P<0.03 for 5 ms, P<0.01 for 10 ms, P<0.02 for 15 ms, P<0.001 for 20 ms, P<0.006 for 30 ms. The data for 50 and 100 ms responses did not differ from their controls (P>0.05).
Further tests were performed to characterize the mechanism underlying the depression of currents mediated by nAChRs. Figure 2C shows that increasing the duration (5–100 ms) of nicotine pulses (50 μM pipette concentration) caused a progressive increment in current amplitude with apparent saturation at 50 ms pulse. When the same protocol was repeated in the presence of 30 nM F3 (15 s puffer pipette preapplication), evoked currents induced by 5–30 ms application of nicotine were significantly depressed while those induced by 50–100 ms pulses were unaffected (8–30 cells). Thus, the plot was shifted to the right without change in the maximal response. This pattern of action is suggestive of an apparently competitive type of nAChR antagonism. At the midpoint of this curve (20 ms pulse) the average depression was 44±3% (n=30). By applying different concentrations of F3 and using the same test pulses of nicotine (20 ms from 50 μM pipette solution) the current depression was 61±1% for 250 nM F3 (n=5) and 81±3% for 500 nM F3 (n=4). All nicotine-induced currents generated with 50 μM agonist pipette concentration were completely abolished by 10 μM F3.
In view of the report of αBgtx binding sites in bovine adrenal medullary tissue (Garcia-Guzman et al., 1995), we tested if this toxin could interfere with nicotine-evoked currents. Application of αBgtx at concentrations up to 5 μM for 30 min failed to change nicotine-induced currents. Furthermore, in preliminary experiments using an imaging technique to assess changes in intracellular Ca2+ levels (Khiroug et al., 1997; Giniatullin et al., 1999) we did not detect any variations in nicotine-evoked rises in internal Ca2+ in the continuous presence of 1–5 μM αBgtx (data not shown).
The blocking action of F3 did not depend on membrane potential
We next explored if the action of F3 on nAChRs was voltage dependent as this property might provide an indication of its mechanism of action.
The example in Figure 3 shows that, on the same cell held at −70, −100 or −40 mV, F3 (30 nM puffer concentration) elicited a similar reduction in nicotine induced currents. On average the depression at −100 mV was 49±6% (n=4) and 42±5% (n=4) at −40 mV. The reversal potential (about 0 mV) of nicotine currents was unchanged. These data thus indicate that the block exerted by F3 was voltage independent and not caused by a negative shift in current reversal.
Figure 3.

F3-induced depression of nicotine currents is voltage independent. Comparison of currents induced by 20 ms nicotine (50 μM pipette concentration) in control solution and in the presence of F3 (30 nM pipette concentration) at −70 mV (top), −100 mV (middle) and −40 mV (bottom) holding potential (VH). Traces are from the same cell.
Blocking potency of the optical stereoisomers of F3
To investigate any stereo-selectivity in the action of F3, we tested its two optically resolved isomers. The R-isomer was found to be more potent than the corresponding S-form enantiomer. For example, 100 nM R-F3 (applied via rapid solution exchanger) depressed by 45±3% responses induced by 20 ms nicotine pulses (100 μM pipette concentration; n=15) whereas 100 nM S-F3 reduced responses by 5±4% only (n=5). Figure 4A shows an example of the action of R-F3 on nicotine induced currents: as with the racemic compound the block displayed a fast onset and a full recovery after a few minutes washout.
Figure 4.

Stereo-selectivity of the two isomers of F3. (A) Inward currents evoked by 20 ms nicotine (0.1 mM pipette concentration) in control solution (left), in the presence of R-F3 (100 nM applied via rapid solution exchanger, middle) and 45 s after washout of R-F3 (right). Note reduction in nicotine current amplitude, fast onset and full recovery. (B) Plot of the fractional reduction in current amplitude against different log concentrations of R-F3 (ranging from 100 nM to 3 μM). The test pulse (20 ms, 100 μM) of nicotine was the same for all concentration of R-F3 (n=4–15 cells). The calculated IC50 value for R-F3 was 350±30 nM. (C) Plot of nicotine current vs increasing duration of nicotine pressure pulses in control solution and in the presence of R-F3. Ordinate, current amplitude normalized with respect to the response evoked by 20 ms in control solution for each cell. Abscissa, pulse duration of nicotine (0.1 mM) applications. R-F3 (100 nM via the rapid solution exchanger) was applied for ∼15 s before each nicotine response (5–15 cells).
Figure 4B shows a plot of the fractional reduction in current amplitude against different log concentrations of R-F3. R-F3 concentrations (ranging from 100 nM to 3 μM) were tested on responses evoked by the same pulse duration of nicotine (20 ms, 100 μM; n=4–15). From these data the calculated IC50 value for R-F3 was 350±30 nM and for S-F3 (n=5) was 1.5±0.3 μM.
By analogy with the observation using racemic F3 (c.f. data in Figure 2C), application of R-F3 on responses evoked by different pulse durations of nicotine also elicited a rightward shift of the curve compared to that obtained with nicotine alone (plot in Figure 4C), indicative of competitive antagonism. Taking the average value at the midpoint of the curve (corresponding to a 20 ms pulse of nicotine), the depression was 45±3% (n=15). No voltage dependence of the R-F3 block was observed.
Action of F3 on nicotine and epibatidine
Amongst nAChR agonists epibatidine is considered to be one of the most potent and is the most commonly used radio-labelled agonist for binding assays (Gotti et al., 1997). To investigate if there was any differential block by F3 of responses induced by nicotine and epibatidine, these agonists were applied by two separate puffer pipettes to the same cell while R-F3 (100 nM) was continuously superfused via the bathing solution.
To elicit inward currents of the same amplitude, i.e. to activate approximately the same number of nAChRs, nicotine was used at 100 μM and epibatidine at 100 nM concentrations (test pulses were 20 ms in all cases). As shown in Figure 5 (left panels) the epibatidine-induced current decayed much slower than that induced by nicotine (Gerzanich et al., 1995; Zhang et al., 1999). The slow time course of epibatidine currents and the strong tendency to desensitize made it difficult to obtain reliable responses over a sustained period of recording. Nevertheless, the block exerted by R-F3 was similar (right panels). Analysis of results from four cells showed 52±1% depression for nicotine and 55±4% for epibatidine, respectively. Note that the extent of nicotine depression was virtually identical to that found with fast perfusion of F3. This finding allowed us to compare patch clamp data routinely obtained with nicotine with binding experiments in which epibatidine was the ligand.
Figure 5.

Action of F3 on different nicotinic agonists. Similar amplitude currents induced by 20 ms nicotine (0.1 mM pipette concentration, top left) or 20 ms epibatidine (0.1 μM pipette concentration, bottom left) before and during bath application of R-F3.
Binding assays
Initial experiments using 3H-epibatidine to test for nAChR binding gave a very low signal from adrenal gland homogenates because of the small number of these receptors (see also Davila-Garcia et al., 1997). We therefore decided to use 125I-epibatidine and compared its binding activity with that of 3H-epibatine in preliminary experiments on rat SCG homogenates (in the presence of αBgtx to saturate possible αBgtx binding sites). 125I-epibatidine binding exibited a KD value of 140 pM (coefficient of variation, CV, =20%) and a Bmax value of 210±20 fmol mg−1 protein. Corresponding data for 3H-epibatine were 127 pM (CV=20%) and 230±25 fmol mg−1 protein. Hence, the two ligands had similar KD and Bmax values. Since the radioactivity signal was much greater with 125I-epibatidine, we decided to use this substance for binding studies of adrenal gland homogenates.
Figure 6A shows a representative experiment in which 125I-epibatidine bound adrenal gland membranes in a specific and saturable manner. These data were fitted with a linear Scatchard plot, thus demonstrating the presence of a single class of high affinity sites (see Figure 6B). The apparent KD value of 125I-epibatidine calculated from five separate experiments performed in duplicate was 159 pM (CV=37%) and Bmax was 6.5±1.3 fmol mg−1 protein. Non-specific binding was determined in parallel by incubating samples in the presence of 100 nM unlabelled epibatidine, and averaged 5–40% of total binding. Pilot tests with membranes isolated from rat adrenal medulla showed that, in these samples, the KD value for 125I-epibatidine binding was similar to that measured using whole gland tissue, although the actual number of receptors was almost double. Nevertheless, in view of the very small amount of tissue provided by isolated adrenal medulla, we decided to perform all subsequent binding experiments in whole gland tissue.
Figure 6.
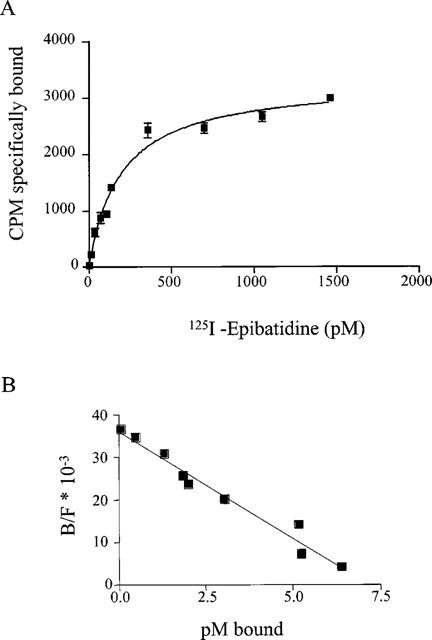
Saturation of specific 125I-epibatidine binding to adrenal gland homogenates. (A) plot of bound radioactivity against concentration of 125I-epibatidine. The homogenates were incubated overnight with 125I-epibatidine (5–1500 nM) at 4°C. Non specific binding was determined in the presence of 100–250 nM cold epibatidine. The data shown are the mean values±s.e.mean of one representative experiment performed in triplicate. Similar data were obtained in five experiments performed in duplicate. (B) Scatchard plot of data from experiments shown in (A).
Some of the basic pharmacological characteristics of rat adrenal gland nAChRs were examined in binding competition experiments using 150–200 pM 125I-epibatidine (Figure 7). Cytisine was more potent than ACh in inhibiting binding as the Ki value for cytisine was 68 nM (CV 13%) vs 348 nM (CV 39%) Ki for ACh. F3 was much less potent with 29.6 μM Ki. Almost no difference in potency was found between the two stereoisomers of F3, S-F3 and R-F3, which had Ki values of respectively 17.2 (CV 29%) and 25.7 (CV 30%) μM.
Figure 7.
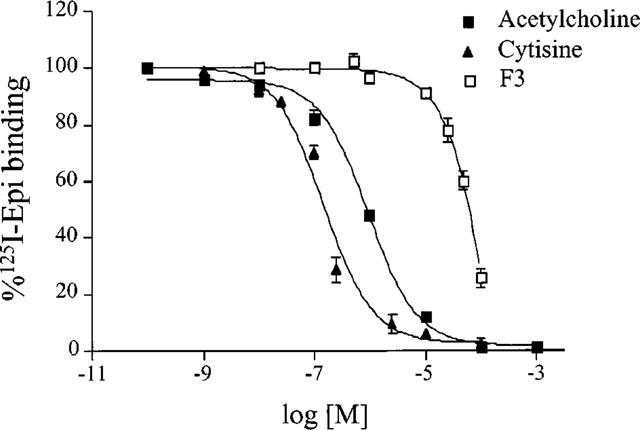
Displacement of bound 125I-epibatidine by ACh, cytisine or F3. Homogenates were preincubated for 30 min with the indicated drug concentrations and then incubated with 0.2 nM 125I-epibatidine overnight at 48°C. The data are the mean±s.e.mean of three experiments for F3 and of two experiments each for ACh and cytisine. All experiments were performed in duplicate or triplicate.
In order to establish whether rat adrenal glands possess α7 receptors that bind αBgtx, we performed saturation binding experiments using rat adrenal homogenates and 125I-αBgtx. In these experiments no specific 125I-αBgtx binding could be detected. Lack of these binding sites prompted further tests to find out if our experimental conditions allowed detection of αBgtx binding. Bovine chromaffin cells are known to express high affinity αBgtx binding sites containing the α7 subunit (Wilson & Kirshner, 1977; Garcia-Guzman et al., 1995) and were thus used for comparison. In three separate experiments we found that on bovine whole adrenal gland or adrenal medulla the affinity of 125I-αBgtx was 1.22 nM (CV 37%) and 1.4 nM (CV 30%), respectively. In the same samples Bmax was 16±5 and 57±4 fmol mg−1 protein, respectively. These data indicate that absence of αBgtx binding in rat tissue samples was not due to inappropriate experimental conditions.
Discussion
The principal finding of the present study is that the oxystilbene derivative, F3, acts as an apparently competitive antagonist on native nAChRs of rat chromaffin cells. This action was manifested as a rapid onset and agonist-surmountable block of inward currents evoked by pulse application of nicotine.
Characteristics of the fast action of F3 on nicotine-induced currents
When F3 was rapidly superfused onto a single chromaffin cell for up to 15 s before nicotine application, it evoked no change in baseline current (indicating lack of partial agonist activity or of non-selective effects on membrane permeability) but did strongly depress the inward currents induced by nicotine. The extent of this block did not intensify during continuous application of F3 and was readily reversible on washout.
The electrophysiological protocol which relied on pulse applications of nicotine was used to minimize rapid-onset receptor desensitization (developing with a time constant of about 100 ms; see Khiroug et al., 1997; 1998). Nevertheless, the use of non-equilibrium responses to nicotine and the puffer application protocol precluded obtaining strictly quantitative pharmacological data and accordingly it has not been possible to analyse the nature of the antagonism by F3 in detail. Even with these constraints, it is apparent that F3 preferentially blocked small (and short) responses to nicotine and that increasing the amount of nicotine delivered to the cell counteracted the inhibitory effect of F3. Indeed, the graph in which fractional response amplitude was plotted against the amount of nicotine delivered by pressure pulse showed a rightward shift in the presence of F3. This observation is therefore consistent with an apparently competitive antagonism of nicotinic receptors by F3.
Another possibility is that F3 acted as a channel blocker (Neher & Steinbach, 1978) on nAChRs of chromaffin cells by analogy with results obtained using other cholinoceptor antagonists (e.g. mecamylamine; Nooney et al., 1992). Two observations clearly contradict this interpretation, namely the absence of any use dependence of the block and its voltage independence over a wide range of membrane potentials.
It is worth noting that the present electrophysiological experiments on rat tissue samples could not detect any nAChR activity with pharmacological properties typical of α7 receptors since αBgtx was inactive. Even if bovine α7 receptors display rapid desensitization capable of making them insensitive over a period of nearly one second (Lopez et al., 1998), the present electrophysiological tests based on 20 ms pulse application of nicotine should have been able to demonstrate their functional presence. The most likely interpretation of the present data is that on rat chromaffin cells any homomeric α7 receptors present were not functional and/or their density was extremely low. Evidence for the second possibility was provided using the 125I-αBgtx binding assay. Indeed, we found no 125I-αBgtx specific binding to rat adrenal gland samples whereas the same experimental protocol readily revealed specific 125I-αBgtx binding sites in bovine adrenal gland tissue with affinity very similar to that previously reported (Wilson & Kirshner, 1977). We therefore believe that in the rat adrenal gland the effect of F3 is only exerted on the α3(α5)β4 subtype without contamination by α7 receptors.
Stereo-selectivity of the antagonism
When the two optical isomers were tested on nicotine evoked responses, the R form was the more potent. This observation was obtained by using the fast superfusion system which enabled us to avoid pressure artefacts due to puffer application and to use known concentrations of the antagonist compounds to express their potency in quantitative terms. The antagonism profile by R-F3 was the same as that of the racemic compound. We observed apparently competitive antagonism with no voltage dependence of the block and a similar time course of action. The fact that we observed stereo-selectivity in the action of F3 suggests that ganglion-type nAChRs preferentially recognize one stereo-isomer of the antagonist. Stereoselectivity has also been shown for the antagonist methyl-epibatidine on recombinant α3β4 receptors (Bertrand et al., 1999), which are the subtype predominantly expressed by chromaffin cells (Garcia-Guzman et al., 1995). In the present experiments the difference in potency between the two isomers was approximately 5 fold.
Epibatidine and nicotine are similarly antagonized
When epibatidine was applied to chromaffin cells, it generated inward currents of different shape (slower onset and offset) from those observed with nicotine. The most glaring difference between epibatidine- and nicotine-induced currents was the slow off-rate of the epibatidine response. Epibatidine behaved as an extremely potent agonist on native nAChRs expressed in chromaffin cells. Nevertheless, pipette doses of epibatidine higher than 1 μM elicited inward currents smaller than those evoked by 100 nM (see also Gerzanich et al., 1995; Zhang et al., 1999) presumably because these receptors are prone to desensitization (Khiroug et al., 1997; 1998) and epibatidine is very effective in promoting desensitization (Zhang et al., 1999). At concentrations of epibatidine 1000 fold less than nicotine, the peaks of the current evoked by either agonist were virtually the same, indicating that the number of activated nAChRs was similar. In this case the extent of the block exerted by R-F3 was the same suggesting that the antagonism by F3 was not peculiar to the action of nicotine but was also seen (at the same potency) with another nicotinic agonist.
Binding of F3 to adrenal nicotinic receptors
Binding studies using adrenal gland homogenates demonstrated the presence of a single class of 125I-epibatidine high affinity receptors. The KD value for 125I-epibatidine binding to this receptor subtype is similar to that previously reported for the α3β4 subtype expressed in oocytes (Parker et al., 1998) or in a transfected cell line (Xiao et al., 1998). In agreement with the data already reported by Davila-Garcia et al. (1997), we found that the amount of adrenal nAChRs was quite low compared with rat SCG membranes which expressed approximately 30–40 times more receptor sites per mg of protein.
To characterize the nAChRs expressed by adrenal tissue further, binding studies were also carried out with the agonists ACh and cytisine (Ki=348 and 68 nM, respectively), which displayed receptor affinity close to values (Ki=560 and 56 nM, respectively) previously observed for binding to oocytes expressing the rat α3β4 subtype (Parker et al., 1998). The present experiments indicate that the Ki value for the antagonist F3 was much larger than its IC50 value obtained with patch clamp recording. It is interesting that, when agonist activity was considered, agonist affinity in binding experiments was much higher (pM range) than in electrophysiological tests.
Like muscle AChRs, nAChRs rapidly undergo transitions to various interconvertible conformational states (closed, open or desensitized) characterized by different affinities for nicotinic agonists; in particular, the desensitized state has a much higher affinity for the agonist than the open or closed states (reviewed by Changeux & Edelstein, 1998). Receptor affinities measured with binding experiments using radiolabelled agonists under equilibrium conditions are generally much higher than those determined by functional assays, a finding borne out by the present study. This result suggests that radioligand binding activity reflects agonist interaction with desensitized receptors whereas data obtained with electrophysiological assays presumably estimate agonist occupancy and activation of closed receptors. In the present binding studies the affinity of F3 was much lower than its IC50 value measured electrophysiologically. These data might therefore be interpreted as an indication that binding affinity of F3 for the desensitized receptor was lower than its affinity to the closed receptor. Thus, desensitized receptors might preferentially bind agonists over competitive antagonists. Further studies will be necessary to clarify this issue.
Species-specificity of the F3 action
We have previously demonstrated that, in chick tissues, oxystilbene derivatives are selective antagonists for the α7 receptor subtype and possess a low activity against brain-type nAChRs (Gotti et al., 1998; Maggi et al., 1999). Conversely, in the present study, we show that F3 has antagonist activity (with high affinity) for ganglion-type nAChRs of rat chromaffin cells which lacked α7 receptors. We have also seen that MG624 and F3 display nM affinity and antagonist activity against human and rat α7 receptors expressed in Xenopus oocytes (C. Gotti and R. Zwart, unpublished). Thus, we conclude that, in mammals, oxystilbene derivatives are very active but do not retain subtype selectivity.
Since the precise identification of the agonist/antagonist binding sites on nAChRs is not yet available, any speculation on the identity of the aminoacids responsible for conferring species-specificity is currently unwarranted. Nonetheless, the present results suggest that caution is required in assuming that pharmacological data obtained from receptors in one animal species can be applied to similar receptors in another species, especially when dealing with drugs potentially targeted for future clinical development.
Acknowledgments
We are very grateful to Drs L. Villa and M. Pallavicini (Institute of Pharmaceutical Chemistry, University of Milan) for providing us with the oxystilbene derivatives. We thank Dr Massimo Righi for his help in preparing chromaffin cell cultures. This work was supported by grants from Istituto Nazionale di Fisica della Materia (PRA CADY), Fabriques de Tabac Reunies, Neuchatel, Switzerland, the Italian Ministry for Universities and for Scientific and Technological Research (co-finanziamento MURST) and the European Programme ‘Training and Mobility of Researchers', Contract n. ERB4061PL97-0790.
Abbreviations
- ACh
acetylcholine
- αBgtx
α Bungarotoxin
- CV
coefficient of variation
- F3
N,N,N-trimethyl-1-(4-trans-stilbenoxy)-2-propylammonium iodide
- KD
dissociation constant
- Ki
inhibition constant
- IC50
concentration of inhibitor blocking agonist response by 50%
- nAChR
neuronal nicotinic ACh receptor
References
- ANDERSON D.J. Molecular control of cell fate in the neural crest: the symphatoadrenal lineage. Annu. Rev. Neurosci. 1993;16:129–158. doi: 10.1146/annurev.ne.16.030193.001021. [DOI] [PubMed] [Google Scholar]
- BERTRAND S., PATT J.T., SPANG J.S., WESTERA G., SCHUBIGER P.A., BERTRAND D. Neuronal nAChR stereoselectivity to non-natural epibatidine derivatives. FEBS Lett. 1999;450:273–279. doi: 10.1016/s0014-5793(99)00473-1. [DOI] [PubMed] [Google Scholar]
- BRANDT B.L., HAGIWARA S., KIDOKORO Y., MIYAZAKI S. Action potentials in the rat chromaffin cell and effects of ACh. J. Physiol. 1976;263:417–439. doi: 10.1113/jphysiol.1976.sp011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMPOS-CARO A., SMILLIE F.I., DOMINGUEZ DEL TORO E., ROVIRA J.C., VICENTE-AGULLO F., CHAPULI J., JUIZ J.M., SALA F., BALLESTA J.J., CRIADO M. Neuronal nicotinic ACh receptors on bovine chromaffin cells: cloning, expression and genomic organization of receptor subunits. J. Neurochem. 1997;68:488–497. doi: 10.1046/j.1471-4159.1997.68020488.x. [DOI] [PubMed] [Google Scholar]
- CAVALLINI G., MANTEGAZZA P., MASSARINI E., TOMMASINI R. Sull'attivita' ganglioplegica di alcuni derivati alchilaminici dello stilbene e del difenile. Il Farmaco. 1953;6:317–331. [PubMed] [Google Scholar]
- CHANGEUX J.P., EDELSTEIN S.J. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- DAVILA-GARCIA M., MUSACHIO J., PERRY D., XIAO Y., HORTI A., LONDON E., DANNALS R., KELLAR J. 125I-IPH, an epibatidine analog, binds with high affinity to neuronal nicotinic cholinergic receptors. J. Pharmacol. Exp. Therap. 1997;282:445–451. [PubMed] [Google Scholar]
- GARCIA-GUZMAN M., SALA F., SALA S., CAMPOS CARO A., STUHMER W., GUTIERREZ L., CRIADO M. αBungarotoxin-sensitive nicotinic receptors on bovine chromaffin cells: molecular cloning, functional expression and alternative splicing of the α7 subunit. Eur. J. Neurosci. 1995;7:647–655. doi: 10.1111/j.1460-9568.1995.tb00668.x. [DOI] [PubMed] [Google Scholar]
- GERZANICH V., PENG X., WANG F., WELLS G., ANAND R., FLETCHER S., LINDSTROM J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic ACh receptors. Mol. Pharmacol. 1995;48:774–782. [PubMed] [Google Scholar]
- GINIATULLIN R., DI ANGELANTONIO S., MARCHETTI C., SOKOLOVA E., KHIROUG L., NISTRI A. Calcitonin gene-related peptide rapidly downregulates nicotinic receptor function and slowly raises intracellular Ca2+ in rat chromaffin cells in vitro. J. Neurosci. 1999;19:2945–2953. doi: 10.1523/JNEUROSCI.19-08-02945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GINIATULLIN R., KHIROUG L., TALANTOVA M., NISTRI A. Fading and rebound of currents induced by ATP on PC12 cells. Br. J. Pharmacol. 1996;119:1045–1053. doi: 10.1111/j.1476-5381.1996.tb15776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOTTI C., BALESTRA B., MORETTI M., ROVATI G.E., MAGGI L., ROSSONI G., BERTI F., VILLA L., PALLAVICINI M., CLEMENTI F. 4-oxystilbene compounds are selective ligands for neuronal nicotinic αBungarotoxin receptors. Br. J. Pharmacol. 1998;124:1197–1206. doi: 10.1038/sj.bjp.0701957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOTTI C., FORNASARI D., CLEMENTI F. Human neuronal nicotinic ACh receptors. Prog. Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- KHIROUG L., GINIATULLIN R., SOKOLOVA E., TALANTOVA M., NISTRI A. Imaging of intracellular calcium during desensitization of nicotinic acetylcholine receptors of rat chromaffin cells. Br. J. Pharmacol. 1997;122:1323–1332. doi: 10.1038/sj.bjp.0701518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHIROUG L., SOKOLOVA E., GINIATULLIN R., AFZALOV R., NISTRI A. Recovery from desensitization of neuronal nicotinic acetylcholine receptors of rat chromaffin cells is modulated by intracellular calcium through distinct second messengers. J. Neurosci. 1998;18:2458–2466. doi: 10.1523/JNEUROSCI.18-07-02458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENA C., CHANGEUX J-P. Pathological mutations of nicotinic receptors and nicotine-based therapies for brain disorders. Curr. Opin. Neurobiol. 1997;7:674–682. doi: 10.1016/s0959-4388(97)80088-8. [DOI] [PubMed] [Google Scholar]
- LINDSTROM J. Nicotinic acetylcholine receptors in health and disease. Mol. Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- LOPEZ M.G., MONTIEL C., HERRERO C.J., GARCIA-PALOMERO E., MAYORGAS I., HERNANDEZ-GUIJO J.M., VILLAROYA M., OLIVARES R., GANDIA L., MCINTOSH J.M., OLIVERA B.M., GARCIA A.G. Unmasking the functions of the chromaffin cell α7 nicotinic receptor by using short pulses of acetylcholine and selective blockers. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14184–14189. doi: 10.1073/pnas.95.24.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGGI L., PALMA E., EUSEBI F., MORETTI M., BALESTRA B., CLEMENTI F., GOTTI C. Selective effects of a 4-oxystilbene derivative on wild and mutant neuronal chick α7 nicotinic receptor. Br. J. Pharmacol. 1999;126:285–295. doi: 10.1038/sj.bjp.0702299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANTEGAZZA P., TOMMASINI R. Central antinicotinic activity of 4-oxystilbene and 4-oxydiphenylethane derivatives. Arch. Int. Pharmacodyn. 1955;4:371–403. [PubMed] [Google Scholar]
- NEHER E., STEINBACH J.H. Local anaesthetics transiently block currents through single acetylcholine receptor channels. J. Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOONEY J.M., PETERS J.A., LAMBERT J.J. A patch clamp study of the nicotinic acetylcholine receptor of bovine adrenomedullary chromaffin cells in culture. J. Physiol. 1992;455:503–527. doi: 10.1113/jphysiol.1992.sp019314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKER M., BECK A., LUETJE W. Neuronal nicotinic receptor β2 and β4 subunits confer large differences in agonist binding affinity. Mol. Pharmacol. 1998;54:1132–1139. [PubMed] [Google Scholar]
- ROLE L.W., BERG K.D. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- WILSON S.P., KIRSHNER N. The acetylcholine receptor of the adrenal medulla. J. Neurochem. 1977;28:687–693. doi: 10.1111/j.1471-4159.1977.tb10615.x. [DOI] [PubMed] [Google Scholar]
- XIAO Y., MEYER E.L., THOMPSON J.M., SURIN A., WROBLEWSKI J., KELLAR K.J. Rat α3/β4 subtype of neuronal nicotinic acetylcholine receptor stably expressed in a transfected cell line: pharmacology of ligand binding and function. Mol. Pharmacol. 1998;54:322–333. doi: 10.1124/mol.54.2.322. [DOI] [PubMed] [Google Scholar]
- ZHANG J., XIAO Y., ABDRAKHMANOVA G., WANG W., CLEEMANN L., KELLAR K.J., MORAD M. Activation and Ca2+ permeation of stably transfected α3/β4 neuronal nicotinic acetylcholine receptor. Mol. Pharmacol. 1999;55:970–981. doi: 10.1124/mol.55.6.970. [DOI] [PubMed] [Google Scholar]