

Abstract
The effect on cholinergic analgesia of inactivation of the M1 gene by an antisense oligodeoxyribonucleotide (aODN) was investigated in the mouse hot plate test. Mice received a single intracerebroventricular (i.c.v.) injection of anti-M1 aODN (0.3, 1.0 or 2.0 nmol per injection), degenerate ODN (dODN) or vehicle on days 1, 4 and 7.
A dose-dependent inhibition of the antinociception induced by the muscarinic agonists oxotremorine (0.1 mg kg−1 s.c.) and McN-A-343 (30 μg per mouse i.c.v.) and the cholinesterase inhibitor physostigmine (0.2 mg kg−1 s.c.) was observed 24 h after the last i.c.v. injection of aODN. Time-course experiments revealed that, after the end of the aODN treatment, sensitivity to analgesic drugs progressively appeared reaching the normal range at 96 h.
The anti-M1 aODN was selective against muscarinic antinociception since the enhancement of pain threshold produced by morphine and baclofen were not affected by the above-mentioned treatment. dODN, used as control, did not affect muscarinic antinociception.
Binding studies evidenced a selective reduction of M1 receptor levels in the hippocampus of aODN-treated mice.
Neither aODN, dODN nor vehicle produced any behavioural impairment of mice as revealed by the rota-rod and Animex experiments.
These results indicate that activation of M1 muscarinic receptor subtype is fundamental to induce central cholinergic analgesia in mice.
Keywords: Analgesia, M1 receptors, antisense oligonucleotides, central cholinergic system, physostigmine, McN-A-343, oxotremorine
Introduction
It is widely accepted that organisms possess endogenous system within the central nervous system that inhibit nociceptive transmission. Several reports have provided evidence for the critical involvement of the cholinergic system in such pain inhibitory pathways. The first observation that the cholinesterase inhibitor physostigmine increased the pain threshold in man was made more than 60 years ago (Pellandra, 1933). Since then, a vast literature has appeared describing the antinociceptive action of both cholinesterase inhibitors and cholinomimetic drugs (Hartvig et al., 1989).
The five types of mammalian muscarinic receptors, m1 through m5, differ in primary structure as determined by molecular cloning (Caufield, 1993). The molecularly defined m1, m2 and M3 receptors correlate with the pharmacologically defined binding M1, M2 and M3 sites in mammalian tissues (Caufield, 1993). Antinociception induced by muscarinic drugs has been reported to be antagonized by pretreatment with either M1, M2 or M3 muscarinic receptor subtype antagonists, even if a major role seems to be played by M1 receptors (Yaksh et al., 1985; Gillbert et al., 1989; Smith et al., 1989; Zhuo & Gebhart, 1991; Bartolini et al., 1992; Ghelardini et al., 1996; Naguib & Yaksh, 1997). It has also been suggested the involvement of activation of α2-adrenoceptors, 5-HT1C/2 and 5-HT3 serotoninergic receptor subtypes (Pavone & Fagioli, 1992; Iwamoto & Marion, 1993), but these results are controversial (Naguib & Yaksh, 1994).
Since the available pharmacological muscarinic receptor antagonists are not completely selective among the muscarinic receptor subtypes (Caufield, 1993), the aim of the present study was to investigate further the role of M1 receptors in the modulation of the pain threshold by using a more selective experimental tool. To this purpose, an antisense oligodeoxyribonucleotide (aODN) able to inactivate M1 gene expression was used. An aODN is a short segment of synthetic DNA having a sequence complementary to a portion of targeted mRNAs which prevents translation and/or mediates mRNA cleavage by the enzyme RNase H. aODNs specifically bind to targeted mRNAs H and, therefore, down-regulate the synthesis of the encoded protein representing useful pharmacological tools for exploring neurobiological processes at the molecular level (Wahlestedt, 1994; Crooke & Bennett, 1996; Galeotti et al., 1997; Ghelardini et al., 1997; Meiri et al., 1997). aODN can transiently inactivate a single gene and, therefore, can inactivate receptor functions in a more specific and selective manner than receptor antagonists. The effects on antinociceptive processes of anti-M1 aODN were evaluated in mice by means of the hot-plate test, and a degenerate ODN (an oligonucleotide accounting for the eventual aODN aspecific effects) was used as control.
Methods
Animals
Male Swiss albino mice (24–26 g) from Morini (San Polo d'Enza, Italy) were used. Fifteen mice were housed per cage. The cages were placed in the experimental room 24 h before the test for acclimatization. The animals were fed a standard laboratory diet and tap water ad libitum and kept at 23±1°C with a 12 h light/dark cycle, light at 07.00 h.
Antisense oligonucleotides
Low cell permeability and the high degradation of natural phosphodiester oligomers are considerable drawbacks in the application of aODNs both in vitro and in vivo. To overcome these drawbacks, phosphorothioate-capped phosphorodiester oligonucleotides were used. The above-mentioned compounds are a class of ODN derivatives shown to maintain more stable and effective concentrations in the brain when compared with their unmodified counterpart (Whitesell et al., 1993). Phosphodiester oligonucleotides (ODNs) protected by terminal phosphorothioate double substitution (capped ODNs) against possible exonuclease-mediated degradation were purchased from Genosys (Cambridge, England) and purified by high-performance liquid chromatography (HPLC). The 18-mer antisense ODN (aODN) 5′-CAC TGA GGT GTT CAT TGC-3′ (phosphorothioate residues are underlined) complementary to the residues 112-129 of the published mouse M1 cDNA sequence (Shapiro et al., 1988) and the 18-mer fully degenerated ODN (dODN) 5′-NNN NNN NNN NNN NNN NNN-3′ (where N is G, or C, or A, or T and phosphorothioate residues are underlined) were vehiculated intracellularly by an artificial cationic lipid (DOTAP, Boehringer-Mannheim, Germany) to enhance both uptake and stability, as described previously (Capaccioli et al., 1993; Quattrone et al., 1994). aODN or dODN (100–400 μM) were preincubated at 37°C for 30 min with 13 μM DOTAP, sterilized through a 0.2 μm filter and supplied to mice by i.c.v. injection of a 5 μl solution as described in the next section.
GenBank accession numbers
The accession number of the cDNA sequence for the mouse muscarinic receptor subtype reported in this paper (M1) is J04192.
I.c.v. injection of oligonucleotides
Mice were randomly assigned to anti-M1 aODN, dODN, vehicle, saline or naive group. The antisense and dODNs were dissolved in a vehicle constituted by DOTAP. Each group received a single i.c.v. injection on days 1, 4 and 7 whereas naive animals did not receive any treatment. I.c.v. administration was performed under ether anaesthesia with isotonic saline as solvent, according to the method described by Haley & McCormick (1957). During anaesthesia, mice were grasped firmly by the loose skin behind the head. A hypodermic needle (0.4 mm external diameter) attached to a 10 μl syringe was inserted perpendicularly through the skull and no more than 2 mm into the brain of the mouse, where 5 μl ODNs were then administered. The injection site was 1 mm to the right or left from the midpoint on a line drawn through to the anterior base of the ears. Injections were performed randomly into the right or left ventricle. To ascertain that ODNs were administered exactly into the cerebral ventricle, some mice were injected with 5 μl of diluted 1 : 10 India ink and their brains were examined macroscopically after sectioning. The accuracy of the injection technique was evaluated with 95% of injections being correct.
Hot-plate test
The method adopted was described by O'Callaghan & Holtzman (1975). Mice were placed inside a stainless steel container, which was set thermostatically at 52.5±0.1°C in a precision water-bath from KW Mechanical Workshop, Siena, Italy. Reaction times (s), were measured with a stopwatch before and 15, 30 and 45 min after administration of the analgesic drugs. The endpoint used was the licking of the fore or hind paws. Those mice scoring less than 12 and more than 18 s in the pretest were rejected (30%). An arbitrary cut-off time of 45 s was adopted.
Rota-rod test
The test was performed according to the method described by Kuribara et al. (1977). Briefly, the apparatus consisted of a base platform and a rotating rod of 3 cm diameter with a non-slippery surface. The rod was placed at a height of 15 cm from the base. The rod, 30 cm in length, was divided into five equal sections by six disks. Thus, up to five mice were tested simultaneously on the apparatus, with a rod-rotating speed of 16 r.p.m. The integrity of motor coordination was assessed on the basis of endurance time of the animals on the rotating rod. One day before the test, the animals were trained twice. On the day of the test only the mice that were able to stay balanced on the rotating rod between 90 and 120 s (cut-off time) were selected for testing. The performance time was measured before and 15, 30 and 45 min after treatment.
Spontaneous activity meter (Animex)
Locomotor activity in mice was quantified using an Animex activity meter Type S (LKB, Farad, Sweden) set to maximum sensitivity. Every movement of mice, which were placed on the top of the Animex activity meter, produced a signal due to variation in inductance and capacity of the apparatus resonance circuit. Then signals were automatically converted to numbers. On the day of the experiment the mice were treated and then the cage, containing five mice, was put on the measuring platform. Activity counts were made for 10 min starting 30 min after the beginning of the test. Because of the arbitrary scale adopted to quantify movements, drug-treated mice were always compared with saline-treated ones.
Binding assay – membrane preparation
Male mice used for the [3H]-telenzepine binding assays were killed by decapitation 24 h after the last i.c.v. injection of aODN, dODN or vehicle. The brain was removed, rinsed with ice-cold 0.9% saline solution and dissected into two regions: cortex and hippocampus. Tissues were homogenized in 40 vol HEPES buffer containing (mM): HEPES 20, NaCl 100, MgCl2 10 and phenyl-methyl-sulphonil-fluoride (PMSF) 0.01, pH 7.4 for 15 s using an Ultraturrax homogenizer at half of the maximum speed. Homogenates were centrifugated at 1000×g for 5 min; supernatants were again centrifuged at 48,000×g for 30 min and the resultant sediments were immediately frozen at −80°C until binding experiments were performed.
Ligand binding assay
Frozen pellets were dispersed in HEPES buffer and then spun at 48,000×g for 30 min. The resulting pellets were rehomogenized in the above-mentioned buffer and used for the binding assays; an aliquot was taken for protein determinations. Aliquots of membranes (0.4–0.6 mg ml−1) were incubated in a final volume of 0.5 ml ligands in HEPES buffer at 37°C for 2 h; [3H]-telenzepine was present at 0.4 nM in tubes containing increasing concentrations (0.3–10,000 nM) of unlabelled telenzepine and at 0.05–0.4 nM in tubes without unlabelled ligand. All measurements were obtained in duplicate. After incubation, bound radioactivity was separated by filtration through Whatman GF/C filters presoaked in 0.1% polyethyleneimine (PEI) using the Brandel M-48R cell harvester (Gaithersburg, MD, U.S.A.). Filters were washed twice with 5 ml of 50 mM Tris-HCl buffer, pH 7.4 containing 10 mM MgCl2 and counted in 5 ml of FilterCount (Packard) in a liquid scintillation counter TRI-CARB 1900TR (Packard). Protein content was measured using the Pierce protein assay reagent (Pierce Chemical Co., Rockford, IL, U.S.A.), according to the standard assay procedure recommended by the manufacturer and based on the method of Bradford (1976) using bovine serum albumin as a standard.
Drugs
The following drugs were used: baclofen (β-p-chlorophenyl GABA), physostigmine hemisulphate (Sigma, Italy); morphine hydrochloride (U.S.L. 10/D, Firenze); McN-A-343 (4-(N-[3-chlorophenyl]-carbamoyloxy)-2-butynyl-trimethylammonium chloride), oxotremorine methiodide (R.B.I.); [3H]-telenzepine (DuPont NEN, Italy). Other chemicals were of the highest quality commercially available. Drugs were dissolved in isotonic (NaCl 0.9%) saline solution immediately before use. Drug concentrations were prepared so that the necessary dose could be administered in a volume of 5 μl per mouse by intracerebroventricular (i.c.v.) injection and 10 ml kg−1 by subcutaneous (s.c.) injection.
Data analysis
All experimental results are given as the mean±s.e.mean. An analysis of variance ANOVA, followed by Fisher's Protected Least Significant Difference procedure for post hoc comparison, were used to verify significance between two means of behavioural results. Data were analysed with the StatView software for the Macintosh (1992). The binding data were evaluated quantitatively using the Ligand (Munson & Rodbard, 1980) computer program; this type of analysis provides optimal estimates of binding parameters (affinity constants, binding capacities, non specific binding). P values of less than 0.05 were considered significant.
Results
Effect of aODN on muscarinic antinociception
Mice were pretreated with a single i.c.v. injection of antisense ODN (aODN) to M1 gene, degenerate ODN (dODN) or vehicle on days 1, 4 and 7. The effect of aODN pretreatment on oxotremorine (0.1 mg kg−1 s.c.), physostigmine (0.2 mg kg−1 s.c.) and McN-A-343 (30 μg per mouse i.c.v.) induced antinociception was then evaluated in the mouse hot-plate test.
aODN, at the dose of 0.3 nmol per i.c.v. injection, did not significantly affect oxotremorine (Figure 1A), physostigmine (Figure 2A) and McN-A-343 (Figure 3A) analgesia. At the dose of 1.0 and 2.0 nmol per i.c.v. injection, aODN dose-dependently prevented oxotremorine (Figure 1B,C), physostigmine (Figure 2B,C) and McN-A-343 (Figure 3B,C) antinociception. This antagonistic effect was detected 24 h after the last i.c.v. injection.
Figure 1.
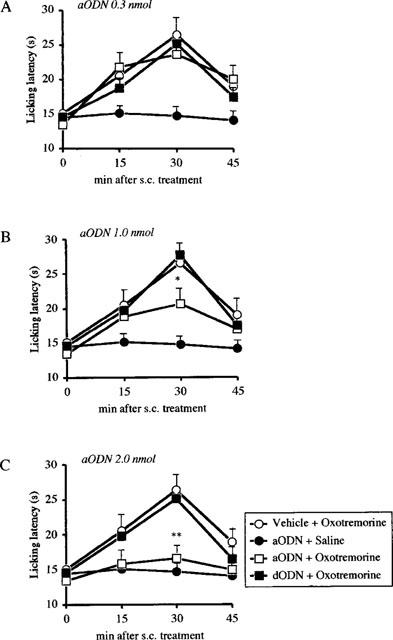
Prevention of oxotremorine (0.1 mg kg−1 s.c)-induced antinociception by pre-treatment with an antisense ODN (aODN) to M1 gene in the mouse hot-plate test. Mice were i.c.v. injected with vehicle, aODN or degenerated ODN(dODN) at the dose of 0.3 (A), 1.0 (B) and 2.0 nmol (C) per single i.c.v. injection on days 1, 4 and 7. The hot plate test was performed 24 h after the last i.c.v. injection. Vertical lines give s.e.mean. Each point represents the mean of 12–18 mice. *P<0.05; **P<0.01 in comparison with dODN+oxotremorine-treated mice.
Figure 2.
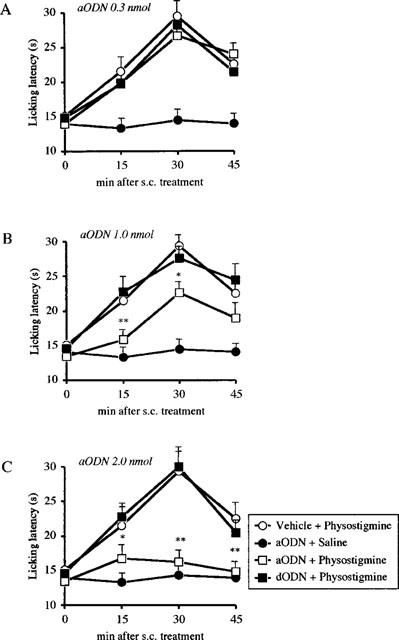
Prevention of physostigmine (0.2 mg kg−1 s.c)-induced antinociception by pre-treatment with an antisense ODN(aODN) to M1 gene in the mouse hot-plate test. Mice were i.c.v. injected with vehicle, aODN or degenerated ODN (dODN) at the dose of 0.3 (A), 1.0 (B) and 2.0 nmol (C) per single i.c.v. injection on days 1, 4 and 7. The hot plate test was performed 24 h after the last i.c.v. injection. Vertical lines give s.e.mean. Each point represents the mean of 10–14 mice. *P<0.05; **P<0.01 in comparison with dODN+physostigmine-treated mice.
Figure 3.
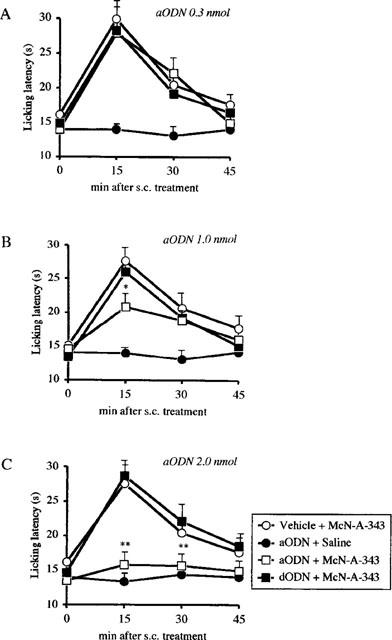
Prevention of McN-A-343 (0.03 mg per mouse i.c.v.)-induced antinociception by pretreatment with an antisense ODN (aODN) to M1 gene in the mouse hot-plate test. Mice were i.c.v. injected with vehicle, aODN or degenerated ODN (dODN) at the dose of 0.3 (A), 1.0 (B) and 2.0 nmol (C) per single i.c.v. injection on days 1, 4 and 7. The hot plate test was performed 24 h after the last i.c.v. injection. Vertical lines give s.e.mean. Each point represents the mean of 12–15 mice. *P<0.05; **P<0.01 in comparison with dODN+McN-A343-treated mice.
The regression lines which show the dose-dependent reduction of oxotremorine, physostigmine and McN-A-343 antinociception produced by increasing concentrations of aODN are shown in Figure 4A–C respectively. The percentage of the maximum analgesic effect was evaluated in correspondence with the maximum effect of analgesic compounds that occurred 15 min after McN-A-343 and 30 min after oxotremorine and physostigmine administration. The hot-plate test was performed 24 h after the end of the ODNs treatments.
Figure 4.
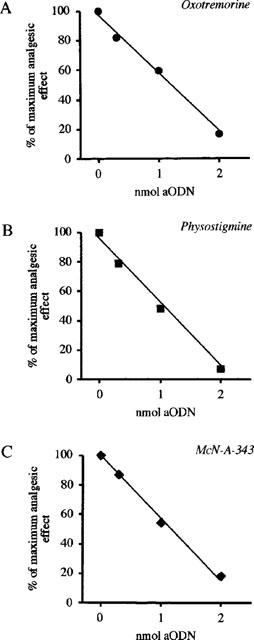
Effect of increasing concentrations of aODN to M1 gene on oxotremorine (0.1 mg kg−1 s.c.; A), physostigmine (0.2 mg kg−1 s.c.; B) and McN-A-343 (0.3 mg per mouse i.c.v.; C)-induced antinociception in the mouse hot-plate test. Mice received a single i.c.v. injected of aODN (0.3, 1.0 or 2.0 nmol per injection) on days 1, 4 and 7. The hot plate test was performed 24 h after the last i.c.v. injection. The evaluation of the analgesic effect was carried out 15 min after McN-A-343 and 30 min after oxotremorine and physostigmine administration. Each point represents the mean of at least 15 mice.
A time-course study for aODN antagonistic effect showed that 48 h after the last i.c.v. injection of ODNs the antinociception produced by oxotremorine, physostigmine and McN-A-343 was completely prevented (Figure 5). At 72 h the aODN effect was still detectable against physostigmine and McN-A-343 antinociception (Figure 5B,C), but not against oxotremorine antinociception (Figure 5A). At 96 h, the three analgesic compounds were able to enhance the pain threshold at the same intensity in aODN-, dODN- and vehicle-treated mice indicating the loss of antagonistic activity by the anti-M1 aODN (Figure 5).
Figure 5.
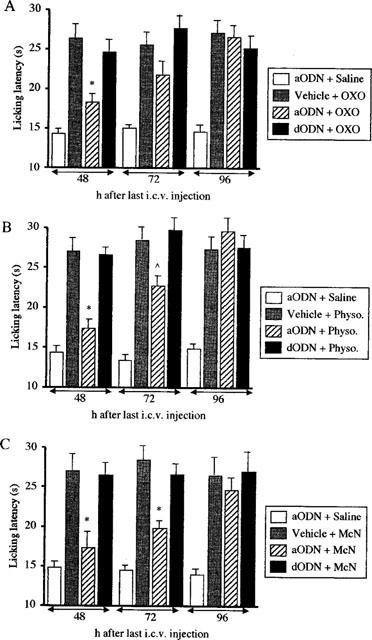
Effect of antisense ODN (aODN) to M1 gene on oxotremrine (0.1 mg kg−1 s.c.; A), physostigmine (0.2 mg kg−1 s.c.; B) and McN-A-343 (0.3 mg per mouse i.c.v.; C)-induced antinociception 48, 72 and 96 h after the end of the aODN treatment. Mice were i.c.v. injected with vehicle, a aODN or degenerated ODN (dODN) at the dose of 2.0 nmol per single i.c.v. injection on days 1, 4 and 7. Modification of pain threshold was evaluated by using the mouse hot-plate test. The licking latency was detected 15 min after McN-A-343 and 30 min after oxotremorine and physostigmine administration. Vertical lines give s.e.mean. Each column represents the mean of 10–14 mice. ⁁P<0.05; *P<0.01 in comparison with vehicle-oxotremorine/physostigmine/MCN-A-343-treated mice.
The aODN pretreatment (2.0 nmol per i.c.v. injection) did not prevent the analgesia induced by morphine (8 mg kg−1 i.p.; Figure 6A) and baclofen (4 mg kg−1 s.c., Figure 6B) 24 h after the end of ODNs administration.
Figure 6.
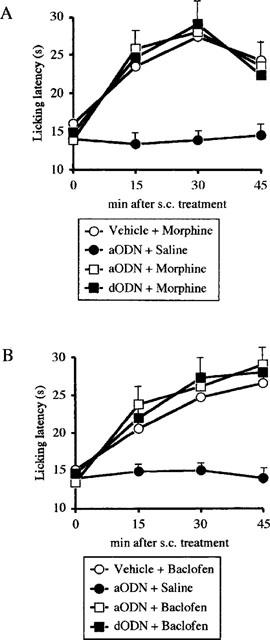
Lack of prevention of morphine (8 mg kg−1 s.c.; A) and baclofen (4 mg kg−1 s.c.; B)-induced antinociception by pretreatment with an antisense ODN (aODN) to M1 gene in the mouse hot-plate test. Mice were i.c.v. injected with vehicle, aODN or degenerated ODN (dODN) at the dose of 2.0 nmol per single i.c.v. injection on days 1, 4 and 7. The hot plate test was performed 24 h after the last i.c.v. injection. Vertical lines give s.e.mean. Each point represents the mean of 12–15 mice.
The aODN pretreatment (2.0 nmol per i.c.v. injection) did not reduce the pain threshold in mice showing a lack of any hyperalgesic effect (Figures 1, 2, 3, 4 and 5).
The pretreatment with the dODN never modified oxotremorine-, physostigmine and McN-A-343-induced antinociception in comparison with mice injected with vehicle as shown in Figures 1, 2, 3 and 4.
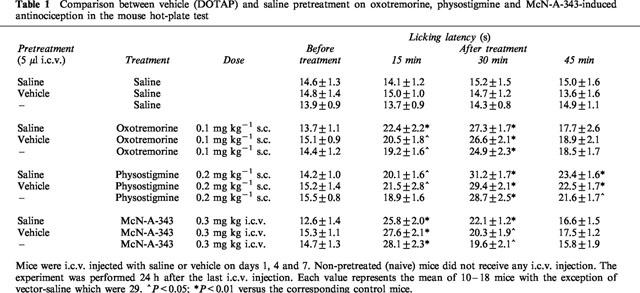
The i.c.v. injection of a solution of DOTAP, used as vehicle, did not modify the licking latency of mice in comparison with naive and saline-treated animals (Table 1). Furthermore, DOTAP did not influence the sensitivity of mice to the analgesic treatments (Table 1).
Table 1.
Comparison between vehicle (DOTAP) and saline pretreatment on oxotremorine, physostigmine and McN-A-343-induced antinociception in the mouse hot-plate test
Determination of M1 receptor protein brain levels
Saturation analysis demonstrates that [3H]-telenzepine binds with high affinity and saturable manner to a homogeneous class of sites in membranes from brain, hippocampus and cortex of dODN- and aODN-treated mice. A decrease in [3H]-telenzepine binding was observed in both cortex and hippocampus even if statistical significance was reached only in the hippocampus. [3H]-telenzepine binding in the whole brain was not appreciably affected by the treatment and, in any case, KD values remained unaffected (data not shown).
Effect of aODN on mouse behaviour
Mice pretreated with aODN (2.0 nmol per injection) or dODN (2.0 nmol per injection) were evaluated for motor coordination and spontaneous motility respectively by use of the rota-rod test (Table 2) and Animex Apparatus. Both tests were performed 24 h after the last i.c.v. injection.
Table 2.
Lack of effect of repeated i.c.v. injections of an antisense ODN (aODN) to M1 gene in the mouse rota-rod test.
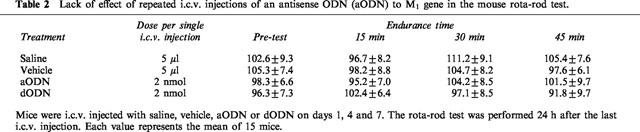
The endurance time, evaluated before and 15, 30 and 45 min after the beginning of the rota-rod test, showed the lack of any impairment in the motor coordination of animals pretreated with aODN and dODN in comparison with saline- and vehicle-treated mice (Table 2).
The spontaneous motility of mice, expressed as counts in 10 min, was unmodified by pretreatment with aODN 2.0 nmol per injection (604±47) or dODN 2.0 nmol per injection (518±89) as revealed by the Animex Apparatus in comparison with saline- (622±83) and vector-treated (561±54) mice.
Discussion
Our results clearly demonstrate that M1 muscarinic receptor subtype plays an essential role in the modulation of pain perception. Cholinergic antinociception induced both directly, through muscarinic agonists, and indirectly, by enhancing ACh extracellular levels through cholinesterase inhibitors, is prevented, in a dose-related manner, by i.c.v. administration of an antisense to the M1 gene coding for the mouse M1 receptor.
The present data indicate that cholinergic antinociception in mice is mediated by M1 receptor stimulation confirming and extending previous results which, by using pharmacological agonists and antagonists, evidenced the involvement of M1 receptors in muscarinic analgesia (Bartolini et al., 1992; Iwamoto & Marion, 1993; Ghelardini et al., 1996; Naguib & Yaksh, 1997). Furthermore, the employment of an aODN to the M1 receptor allows clear elucidation of the role of the M1 receptor in pain perception without any potential interference due to interaction with the other muscarinic receptor subtypes. The above-mentioned literature data were obtained by using pharmacological M1 receptor antagonists which, even if they are the most selective M1/M2-M3 compounds available, are endowed with a quite weak selectivity for M1 receptors. The selectivity ratio values are three for dicyclomine (Giachetti et al., 1986), 29 for pirenzepine and 176 for S-(−)-ET 126 (Ghelardini et al., 1996). These values do not exclude that, at doses active on antinociceptive processes, these compounds can also interact with other muscarinic receptor subtypes such as M2 and M3, that has been reported to be involved in modulation of pain perception (Iwamoto & Marion, 1993; Naguib & Yaksh, 1997).
The aODN treatment induces a transient prevention of muscarinic antinociception since the inhibition of oxotremorine, McN-A-343 and physostigmine antinociception disappeared 96 h after the last i.c.v. injection of the aODN. This return to normal sensitivity to analgesic treatments implies both the total reversal of aODN-induced specific inhibition of M1 gene expression and a lack of damage or toxicity associated with aODN treatment.
In these experimental conditions, anti-M1 aODN did not modified the licking latency values of mice in comparison with control groups excluding that the prevention of oxotremorine, McN-A-343 and physostigmine antinociception is due to hyperalgesic effect of the treatment used. Furthermore, the antinociception induced by activation of other neurotransmitter systems able to enhance the pain threshold, such as opioid and GABAergic, were not modified by anti-M1 aODN treatment. These data indicate not only the specificity of the aODN treatment, but also lead us to exclude a correlation between these two antinociceptive systems and muscarinic analgesia.
In comparison with naive and saline groups, dODN and vehicle treatments did not modify the enhancement of pain threshold produced by both muscarinic agonists and cholinesterase inhibitors, ruling out the possibility that the antagonism exerted by aODN could be caused by sequence-independent effects on cerebral structures. This claim is supported by results obtained from the quantitative RT–PCR analysis of ODN effects on M1 gene expression. Quantitative results of M1 mRNA brain levels following aODN treatment confirmed that phenotypic effects of anti-M1 aODN on pain modulation were actually due to the specific inhibition of M1 gene expression since dODN did not modify M1 mRNA brain levels (Ghelardini et al., 1999).
Through molecular cloning, five muscarinic receptor subtypes have been identified and renamed m1-m5 based on their order of discovery (Bonner, 1989). Muscarinic receptors can be generally grouped on the basis of the signal transduction mechanism used in two main groups: those which mobilize intracellular calcium (m1, m3 and m5) and those which inhibit adenylate cyclase (m2 and m4) (Felder, 1995). Considering the similarity among the structures of the muscarinic receptors, a specific anti-M1 aODN was employed in order to inhibit selectively the expression of this receptor subtype without interfering with the other subtypes. The sequence encompassing the translation start sites of mRNAs, which is considered particularly prone to aODN action (Goodchild, 1989; Stein & Cheng, 1993), was compared among the murine known muscarinic receptors gene family, to design a specific antimouse M1 aODN with a very low sequence homology even with the nearest other members of the muscarinic receptor family. An homology search in the Genbank database confirmed the absolute specificity of the employed aODN. Furthermore, considering the described sequence-independent, non-antisense effects of ODNs (Storey et al., 1991; Gao et al., 1992; Blagosklonny & Neckers, 1994; Schick et al., 1995), a fully degenerated phosphorothioate capped phosphodiester ODN (dODN) was used as the most suitable control for these potentially confusing effects. The dODN used is a collection of about 3×1014 different molecular species. At concentrations achieved in the nanomolar to micromolar range in in vitro antisense experiments, every species, i.e. every ODN of defined sequence, was present at the site of action at a concentration less than 10−18 M, which is totally insufficient to achieve any antisense, or generally sequence-dependent, cellular effect. Therefore, if ODN i.c.v. administration per se had achieved any biological response, this would have been present in ODN-treated controls.
aODN treatment produced a reduction at both M1 mRNA and protein levels. By performing binding experiments in hippocampus and cortex of anti-M1 treated mice, we documented a reduction in the binding sites of [3H]-telenzepine to M1 receptors in both cerebral structures. We investigated the effects produced by aODN treatment in M1 receptor levels in these two areas since M1 receptors are expressed at the highest levels in particular brain areas, such as the cortex and hippocampus (Cortes & Palacios, 1986; Cortes et al., 1986). Present binding results are in agreement with previous experiments performed in the rat in which a reduction of [3H]-QNB binding in the cortex and hippocampus was observed after treatment with an aODN to the m1 muscarinic receptor mRNA (Zang et al., 1994). Fewer M1 receptors in the hippocampus of anti-M1 aODN-treated mice confirms the importance of a complete integrity and functionality of M1 receptors in the modulation of pain perception.
The anti-M1 aODN does not cause any detectable modification in mouse gross behaviour. At active doses, aODN to M1 receptor did not impair motor coordination nor modify spontaneous motility in comparison with dODN- and vehicle-treated groups. Since animals were trained to balance on the rotating rod before performing the rota rod test, the lack of variation of the endurance time represents an index of the absence of motor incoordination. By contrast, a reduction of the endurance time after treatment indicates impaired mouse motor coordination that could lead to a misinterpretation of the results obtained in the hot-plate test. Mice, even if still sensitive to the thermal stimulus used in the hot plate test, may have no reaction to pain (licking of paws) because of their impaired motility.
Seen as a whole, our data indicate that activation of M1 muscarinic receptor subtype plays a fundamental role in the mechanism responsible for the induction of cholinergic analgesia in mice.
Acknowledgments
This work was supported by MURST. The authors wish to thank Mary Forrest for linguistic revision of the manuscript.
References
- BARTOLINI A., GHELARDINI C., FANTETTI L., MALCANGIO M., MALMBERGAIELLO P., GIOTTI A. Role of muscarinic receptor subtypes in central antinociception. Br. J. Pharmacol. 1992;105:77–82. doi: 10.1111/j.1476-5381.1992.tb14213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLAGOSKLONNY M.V., NECKERS L.M. Oligonucleotides protect cells from the cytotoxicity of several anti-cancer chemotherapeutic drugs. Anti-Cancer Drugs. 1994;5:437–442. [PubMed] [Google Scholar]
- BONNER T.I. The molecular basis of muscarinic receptor diversity. Trends Neurosci. 1989;12:148–151. doi: 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. A rapid and sensitive method for the quantification of microgram of protein in utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CAPACCIOLI S., DI PASQUALE G., MINI E., MAZZEI T., QUATTRONE A. Cationic lipids improve antisense oligonucleotide uptake and prevent degradation in cultured cells and in human serum. Biochem. Biophys. Res. Comm. 1993;107:818–825. doi: 10.1006/bbrc.1993.2552. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P. Muscarinic receptors-Characterization, coupling and function. Pharmacol. Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- CORTES R., PALACIOS J.M. Muscarinic cholinergic receptor subtypes in the rat brain: I Quantitative autoradiographic studies. Brain Res. 1986;362:227–238. doi: 10.1016/0006-8993(86)90448-8. [DOI] [PubMed] [Google Scholar]
- CORTES R., PROBST A., TOBLER H.J., PALACIOS J.M. Muscarinic receptor subtypes in the human brain: II Quantitative autoradiographic studies. Brain Res. 1986;362:239–253. doi: 10.1016/0006-8993(86)90449-x. [DOI] [PubMed] [Google Scholar]
- CROOKE S.T., BENNETT C.F. Progress in antisense oligonucleotide therapeutics. Annu. Rev. Pharmacol. Toxicol. 1996;36:107–129. doi: 10.1146/annurev.pa.36.040196.000543. [DOI] [PubMed] [Google Scholar]
- FELDER C.C. Muscarinic acetylcholine receptors: signal transduction through multiple effector. FASEB J. 1995;9:619–625. [PubMed] [Google Scholar]
- GALEOTTI N., GHELARDINI C., PAPUCCI L., QUATTRONE A., CAPACCIOLI S., BARTOLINI A. An antisense oligonucleotide to the mouse Shaker-like potassium channel Kv1.1 gene prevents the antinociception induced by morphine and baclofen. J. Pharmacol. Exp. Ther. 1997;281:941–949. [PubMed] [Google Scholar]
- GAO W.-Y., HAN F.-S., STORM C., EGAN W., CHEN Y.-C. Phosphorothioate oligonucleotides are inhibitors of human DNA polymerases and RNase H: implications for antisense technology. Mol. Pharmacol. 1992;41:223–229. [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., GUALTIERI F., ROMANELLI M.N., BARTOLINI A. S-(−)-ET126: a potent and selective M1 antagonist in vivo and in vitro. Life Sci. 1996;58:991–1000. doi: 10.1016/0024-3205(96)00047-1. [DOI] [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., PECORI VETTORI A., CAPACCIOLI S., QUATTRONE A., BARTOLINI A. Effect of potassium channels modulation on mouse feeding behaviour. Eur. J. Pharmacol. 1997;329:1–8. doi: 10.1016/s0014-2999(97)10102-9. [DOI] [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., MATUCCI R., BELLUCCI C., GUALTIERI F., CAPACCIOLI S., QUATTRONE A., BARTOLINI A. Antisense ‘knockdowns' of M1 receptors induces transient anterograde amnesia in mice. Neuropharmacology. 1999;38:339–348. doi: 10.1016/s0028-3908(98)00194-4. [DOI] [PubMed] [Google Scholar]
- GIACHETTI A., GIRALDO E., LADINSKY H., MONTAGNA E. Binding and functional profiles of the selective M1 muscarinic selective antagonists trihexyphenidyl and dicyclomine. Br. J. Pharmacol. 1986;89:83–90. doi: 10.1111/j.1476-5381.1986.tb11123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILLBERT P.G., GORDH T. , JR, HARTVIG P., JANSSON I., PETTERSON J., POST C. Characterization of the antinociception induced by intrathecally administered carbachol. Pharmacol. Toxicol. 1989;64:340–343. doi: 10.1111/j.1600-0773.1989.tb00660.x. [DOI] [PubMed] [Google Scholar]
- GOODCHILD J. Oligodeoxynucleotides. Antisense Inhibitors of Gene Expression 1989Macmillan Press: London; 53–77.Cohen, J. (ed) [Google Scholar]
- HALEY T.J., MCCORMICK W.G. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTVIG P., GILLBERG P.G., GORDH T., POST C. Cholinergic mechanisms in pain and analgesia. Trends Pharmacol. Sci. 1989. pp. 75–79. [PubMed]
- IWAMOTO E.T., MARION L. Characterization of the antinociception produced by intrathecally administered muscarinic agonists in rats. J. Pharmacol. Exp. Ther. 1993;266:329–338. [PubMed] [Google Scholar]
- KURIBARA H. , HIGUCHI Y., TAKADORO S. Effects of central depressants on rota-rod and traction performances in mice. Jpn. J. Pharmacol. 1977;27:117–126. doi: 10.1254/jjp.27.117. [DOI] [PubMed] [Google Scholar]
- MEIRI N., GHELARDINI C., TESCO G., GALEOTTI N., DAHAL D., TOMSIC D., CAVALLARO S., QUATTRONE A., CAPACCIOLI S., BARTOLINI A., ALKON D.L. Antisense inhibition of the rodent Shaker-like potassium channel Kv1.1: disruption of memory without effects on LTP. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4430–4434. doi: 10.1073/pnas.94.9.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. LIGAND: a versatile computerized approach for characterization of ligand-binding system. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- NAGUIB M., YAKSH T.L. Antinociceptive effects of spinal cholinesterase inhibition and isobolographic analysis of the interaction with μ and α2 receptor systems. Anesthesiology. 1994;80:1338–1348. doi: 10.1097/00000542-199406000-00022. [DOI] [PubMed] [Google Scholar]
- NAGUIB M., YAKSH T.L. Characterization of muscarinic receptor subtypes that mediate antinociception in the rat spinal cord. Anesth. Analg. 1997;85:847–853. doi: 10.1097/00000539-199710000-00025. [DOI] [PubMed] [Google Scholar]
- O'CALLAGHAN J.P., HOLTZMAN S.G. Quantification of the analgesic activity of narcotic antagonists by a modified hot-plate procedure. J. Pharmacol. Exp. Ther. 1975;192:497–505. [PubMed] [Google Scholar]
- PAVONE F., FAGIOLI S. Serotoninergic influence on cholinergic-induced analgesia: differences in two inbred strains of mice. Brain Res. 1992;577:347–350. doi: 10.1016/0006-8993(92)90296-l. [DOI] [PubMed] [Google Scholar]
- PELLANDRA C.L. La geneserine-morphine adjuvant de l'anesthesia generale. Lyon. Med. 1933;151:653. [Google Scholar]
- QUATTRONE A., PAPUCCI L., SCHIAVONE N., MINI E., CAPACCIOLI S. Intracellular enhancement of intact antisense oligonucleotide steady-state levels by cationic lipids. Anti-Cancer Drug Design. 1994;9:549–553. [PubMed] [Google Scholar]
- SCHICK B.P., ERAS J.L., MINTZ P.L. Phosphorothioate oligonucleotides cause degradation of secretory but not intracellular serglycin proteoglycan core protein in a sequence-independent manner in human megacaryocytic tumor cells. Antisense Res. Dev. 1995;5:59–65. doi: 10.1089/ard.1995.5.59. [DOI] [PubMed] [Google Scholar]
- SHAPIRO R.A., SCHERER N.M., HABECKER B.A., SUBERS E.M., NATHANSON N.M. Isolation, sequence, and functional expression of the mouse M1 muscarinic acetylcholine receptor gene. J. Biol. Chem. 1988;263:18397–18403. [PubMed] [Google Scholar]
- SMITH M.D., YANG X. , NHA J.-Y., BUCCAFUSCO J.J. Antinociceptive effect of spinal cholinergic stimulation: interaction with substance P. Life Sci. 1989;45:1255–1261. doi: 10.1016/0024-3205(89)90127-6. [DOI] [PubMed] [Google Scholar]
- STEIN C.A., CHENG Y.-C. Antisense oligonucleotides as therapeutic agents – Is the bullet really magic. Science. 1993;261:1004–1011. doi: 10.1126/science.8351515. [DOI] [PubMed] [Google Scholar]
- STOREY A., OATES D., BANKS L., CRAWFORD L., CROOK T. Antisense phosphorothioate oligonucleotides have both specific and non-specific effects on cells containing human papillomavirus type 16. Nucleic Acids Res. 1991;19:4109–4114. doi: 10.1093/nar/19.15.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAHLESTEDT C. Antisense oligonucleotide strategies in neuropharmacology. Trends Pharmacol. Sci. 1994;15:42–46. doi: 10.1016/0165-6147(94)90107-4. [DOI] [PubMed] [Google Scholar]
- WHITESELL L., GESELOWITZ D., CHAVANY C., FAHMY B., WALBRIDGE S., ALGER J., NECKERS L.M. Stability, clearance, and disposition of intraventricularly administered oligodeoxynucleotides: Implications for therapeutic application within the central nervous system. Proc. Natl. Acad. Sci. U.S.A. 1993;90:4665–4669. doi: 10.1073/pnas.90.10.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAKSH T.L., DIRKSEN R., HARTY G. Antinociceptive effects of intrathecally injected cholinomimetic drugs in the rat and cat. Zur. J. Pharmacol. 1985;117:81–88. doi: 10.1016/0014-2999(85)90474-1. [DOI] [PubMed] [Google Scholar]
- ZANG Z., FLORIJN F., CREESE I. Reduction in muscarinic receptors by antisense oligodeoxynucleotide. Biochem. Pharmacol. 1994;48:225–228. doi: 10.1016/0006-2952(94)90090-6. [DOI] [PubMed] [Google Scholar]
- ZHUO M., GEBHART G.F. Tonic cholinergic inhibition of spinal mechanical transmission. Pain. 1991;46:211–222. doi: 10.1016/0304-3959(91)90078-C. [DOI] [PubMed] [Google Scholar]