

Abstract
Mechanisms of protease-activated receptor-1 (PAR1)- and PAR2-induced relaxation were investigated in pre-contracted porcine coronary artery ring preparations.
Thrombin (0.01–0.3 u ml−1) and the PAR1-activating peptide SFLLRN (0.1–10 μM) caused concentration- and endothelium-dependent relaxation. pEC50s (−log u ml−1 for enzymes, −log M for peptides) and maximum relaxations (Rmax, %) for thrombin were 1.8±0.1 and 93.5±2.8% respectively, and for SFLLRN 6.8±0.1 and 90.8±1.3%. Similar concentration- and endothelium-dependent relaxations occurred with trypsin (pEC50 2.3±0.2; Rmax 94.1±1.9%) and the PAR2-activating peptide SLIGRL (pEC50 6.5±0.2; Rmax 92.4±1.6%).
Relaxations to thrombin, SFLLRN, trypsin and SLIGRL were significantly inhibited (P<0.05) to similar extents by the nitric oxide (NO) synthase inhibitor NG-nitro-L-arginine (L-NOARG; 100 μM) and the NO scavenger oxyhaemoglobin (20 μM), both separately and in combination.
In the presence of the L-type voltage-operated calcium channel (L-VOCC) inhibitor nifedipine (0.3 μM), K+ (67 mM) abolished the L-NOARG-resistant relaxations to thrombin, SFLLRN, trypsin and SLIGRL. However, nifedipine alone significantly (P<0.05) reduced the pEC50 (1.5±0.1) and Rmax (77.5±7.0%) for thrombin but had no effect on relaxations to SFLLRN, trypsin or SLIGRL. Furthermore, L-NOARG-resistant relaxations to thrombin were abolished by nifedipine, whereas relaxations to SFLLRN, trypsin or SLIGRL were not further inhibited by combined treatment with nifedipine and L-NOARG, than they were with L-NOARG treatment alone.
Similar selective inhibition of the L-NOARG-resistant relaxation to thrombin, but not SFLLRN, occurred with verapamil (1 μM) and diltiazem (3 μM).
Our results suggest heterogeneous mechanisms in the NO-independent relaxation to thrombin and peptide activators of PAR1 in the porcine coronary artery.
Keywords: Endothelium, endothelium-derived hyperpolarizing factor, nifedipine, nitric oxide, porcine coronary artery, protease-activated receptors, thrombin, trypsin, voltage-operated calcium channels
Introduction
Thrombin is generated at sites of vascular injury and is involved in an array of responses associated with haemostasis (Fenton, 1993), including endothelium-dependent vascular relaxation in a number of species such as pig (Glusa & Markwardt, 1988; Tesfamariam et al., 1993), dog (White et al., 1984; Ku, 1986), rat (Muramatsu et al., 1992) and human (Luscher et al., 1988; Ku et al., 1992). Many of the cellular actions of thrombin are mediated by a family of protease-activated receptors (PARs: for review see Coughlin, 1994; Déry et al., 1998). PARs are seven transmembrane-spanning domain, G protein-coupled receptors activated by a unique mechanism involving site-specific proteolytic cleavage of the receptor's extracellular amino-terminal to expose an internal 'tethered' ligand sequence. Of the four cloned PARs, PAR1 and PAR3 are activated by thrombin (Vu et al., 1991; Ishihara et al., 1997), PAR2 by trypsin (Nystedt et al., 1994) and trypsin-like enzymes such as mast cell tryptase (Fox et al., 1997; Molino et al., 1997), while PAR4 appears to be activated equally well by thrombin and trypsin (Xu et al., 1998). For PAR1, PAR2 and PAR4, but surprisingly not PAR3, receptor activation can be mimicked by synthetic peptides corresponding to the tethered ligand sequences of each receptor (Vu et al., 1991; Nystedt et al., 1994; Ishihara et al., 1997; Xu et al., 1998).
Whilst all four PARs are expressed on vascular endothelial cells (Cupit et al., 1999), to date only PAR1 and PAR2 (Muramatsu et al., 1992; Hwa et al., 1996; Saifeddine et al., 1996; Hamilton et al., 1998; 1999) have been shown to mediate endothelium-dependent vascular relaxation. In pig (Hwa et al., 1996) and human (Hamilton et al., 1998) coronary arteries, such PAR-mediated responses have been shown to be due to nitric oxide (NO) and another mechanism, which may involve endothelium-dependent smooth muscle hyperpolarization (for review see Garland et al., 1995; Félétou & Vanhoutte, 1999), since thrombin, like other endothelium-dependent vasodilators, was found to cause smooth muscle hyperpolarization in pig coronary artery (Nagao & Vanhoutte, 1992).
Physiological roles for vascular PARs, particularly those on the endothelium, are unknown. However, the involvement of a number of serine proteases–including thrombin and trypsin-like enzymes such as mast cell tryptase–in inflammatory vascular diseases like atherosclerosis and ischaemia-reperfusion injury (Hatton et al., 1989; Kovanen et al., 1995; Cicala et al., 1999) suggest such enzymes may be involved in the modulation of vascular tone during inflammation. Since endothelium-dependent vasodilatation can be compromised in these conditions (Fleming & Busse, 1999), the aim of the present study was to investigate the mechanisms of PAR-mediated vascular relaxation. We report here that enzyme and peptide activators of PAR1 and PAR2 mediate endothelium - dependent relaxation of porcine isolated coronary artery via NO - dependent and -independent mechanisms, of which the latter is a K+-sensitive, endothelium - derived hyperpolarizing factor (EDHF)-like mechanism, similar to that reported for non-PAR endothelium-dependent vasodilators in this (Kilpatrick & Cocks, 1994) and other (Drummond & Cocks, 1996; Kemps & Cocks, 1997) isolated coronary artery preparations. However, a surprising outcome from our study was the observation that L-VOCC inhibitors selectively blocked the NO-independent relaxation induced by thrombin, but not that of trypsin or peptide activators of either PAR1 or PAR2.
Methods
Tissue preparation
Right coronary arteries were dissected from the hearts of Large White pigs (30–50 kg, either sex) which were obtained from a local abattoir and transported to the laboratory in ice-cold Krebs solution (composition in mM: Na+ 143.1, Cl− 127.8, HCO3− 25.0, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, H2PO4− 1.2, SO42− 1.2 and glucose 11; pH 7.4). Artery ring segments ∼3 mm long were mounted between two parallel wire hooks and immersed in 10 ml organ baths containing Krebs solution maintained at 37°C and continuously bubbled with 95% O2, 5% CO2 to keep the pH at 7.4. One wire hook was attached to a micrometer-adjustable support leg and the other to a force transducer (model FT03C, Grass Instruments, MA, U.S.A.) to record changes in isometric, circumferential force which were amplified and displayed on flat bed chart recorders (W&W Scientific Instruments, Basel, Switzerland).
Tissue equilibration
Following a 60 min equilibration period, artery ring preparations were stretched to 5 g passive force and allowed to recover for 30 min before again being stretched to 5 g. After a further 30 min, tissues were exposed to an isotonic, high potassium Krebs solution (KPSS: composition in mM: K+ 124.9, Cl− 128.7, Na+ 25.0, HCO3− 25.0, Ca2+ 2.5, Mg2+ 1.2, SO42− 1.2, H2PO4− 1.2, glucose 6.1) to obtain a maximum contraction for each artery ring (KPSSmax: (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996)). The KPSS was then replaced with normal Krebs solution and the tissues allowed to return to their optimal passive force level over 30–60 min.
Responses to PAR activators
Tissues were contracted to ∼50% KPSSmax with titrated concentrations of the thromboxane A2 mimetic, U46619 (1–100 nM). Once the U46610-induced contraction had reached a stable plateau, cumulative concentrations of thrombin or trypsin (0.0001–1 u ml−1), or the synthetic PAR1 (SFLLRN or TFLLR) or PAR2 (SLIGRL) tethered ligand sequences (0.01–30 μM), were added to the organ bath. The maximum endothelium-dependent and -independent relaxation of each ring preparation was then determined with the addition of bradykinin (0.3 μM) and isoprenaline (1 μM), respectively.
Effect of NO inhibitors
The contribution of NO to PAR-mediated relaxation was determined in coronary artery ring segments treated with the NO synthase inhibitor NG-nitro-L-arginine (L-NOARG; 100 μM) and the NO scavenger oxyhaemoglobin (HbO; 20 μM), either separately or in combination, 30 min before the U46619-induced contraction. To minimise the possibility of HbO denaturation, 10 μM HbO was added prior to, and a further 10 μM after, the U46619-induced contraction (although the final bath concentration of HbO was taken as 20 μM).
Effect of high extracellular K+
To examine the contribution of K+ channels to PAR-mediated relaxations, high extracellular K+ (67 mM KCl, isotonic) was used to inhibit K+ channel activity (Chen & Suzuki, 1989) and the subsequent tissue hyperpolarization (Nagao & Vanhoutte, 1992) and smooth muscle relaxation (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996). All tissues exposed to high K+ were treated with nifedipine (0.3 μM) to inhibit K+-induced contractions (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996). Therefore, to provide appropriate controls in this series of experiments, tissues were either left untreated or were treated with nifedipine, nifedipine and K+, nifedipine and L-NOARG (100 μM) or nifedipine plus K+ and L-NOARG.
Effect of L-VOCC inhibitors
In this group of experiments, tissues were left untreated or were treated with nifedipine (0.3–3 μM), L-NOARG (100 μM), or a combination of nifedipine and L-NOARG. Cumulative concentrations of thrombin, SFLLRN or TFLLR were then added to the organ bath. To confirm the involvement of L-VOCCs, this series of experiments was repeated with verapamil (1 μM) or diltiazem (3 μM). In addition, responses to a single concentration of thrombin (1 u ml−1) were obtained in the absence and presence of the same set of inhibitors as described for the cumulative responses to thrombin.
Specificity of L-VOCC inhibitors and desensitization to SFLLRN
To investigate the possibility of a direct interaction between nifedipine and thrombin, artery rings were left untreated or were treated with thrombin (1 u ml−1) before being contracted to ∼50% KPSSmax with KCl (10–18 mM) and then relaxed with cumulative additions of nifedipine (0.1 nM–1 μM). In desensitization experiments, untreated tissues were contracted to ∼50% KPSSmax with U46619 and exposed to the concentration of SFLLRN required to elicit maximum endothelium-dependent relaxation (10 μM). When the SFLLRN-induced relaxation had recovered to the initial precontraction level of tone, SFLLRN (10 μM) was again added to the tissue and this procedure repeated until no further response was observed. When the tissue was desensitized to SFLLRN, responses to the concentration of thrombin required to elicit maximum endothelium-dependent relaxation (1 u ml−1) were examined.
Materials
Bradykinin triacetate, (−)-isoprenaline, NG-nitro-L-arginine (L-NOARG) and α-thrombin (bovine serum) were obtained from Sigma (MO, U.S.A.). U46619 (9,11-dideoxy-9α,11α-methanoepoxy-prostaglandin F2α), nifedipine, verapamil hydrochloride and diltiazem hydrochloride were purchased from Sapphire Bioscience (Sydney, Australia). Trypsin (bovine pancreas) was from Worthington Biochem (NJ, U.S.A.) and the amidated synthetic PAR1 (SFLLRN-NH2 and TFLLR-NH2) and PAR2 (SLIGRL-NH2) tethered ligand sequences were from Auspep (Parkville, Australia). Stock solutions of L-NOARG (100 mM) were prepared in 1 M NaHCO3 while those for U46619 (1 mM) and nifedipine (10 mM) were in absolute ethanol. Further dilutions of these stock solutions and preparation of all other stock solutions were in distilled water. Neither the ethanol (0.003% v v−1) nor the NaHCO3 (1 mM) vehicles had any effect on tissue function.
Statistical analyses
All relaxations were normalized as a per cent of each tissue's response to 1 μM isoprenaline and all data are expressed as mean±s.e.mean. Normalized concentration-response curves were computer fitted to a sigmoidal regression curve (Graphpad Prism, Graphpad Software Inc.) to generate values for sensitivity (pEC50). Differences in mean pEC50s and maximum responses (Rmax) were tested for significance either by an unpaired Student's t-test or a one-way analysis of variance (ANOVA) with a Tukey-Kramer modified t statistic for multiple comparisons. In all cases, differences were considered significant when P<0.05.
Results
Responses to PAR activators
The proteolytic and non-proteolytic activators of PAR1, thrombin and SFLLRN respectively, and of PAR2, trypsin and SLIGRL respectively, each caused concentration-dependent relaxations of U46619-contracted coronary artery preparations, which in all cases were abolished by removal of the endothelium (data not shown). pEC50 (−log u ml−1 for enzymes and −log M for peptides) and Rmax values for each agonist were, respectively: thrombin 1.8±0.1 and 93.5±2.8%; SFLLRN 6.8±0.1 and 90.8±1.3%; trypsin 2.3±0.2 and 94.1±1.9%; SLIGRL 6.5±0.2 and 92.4±1.6% (Figure 1).
Figure 1.
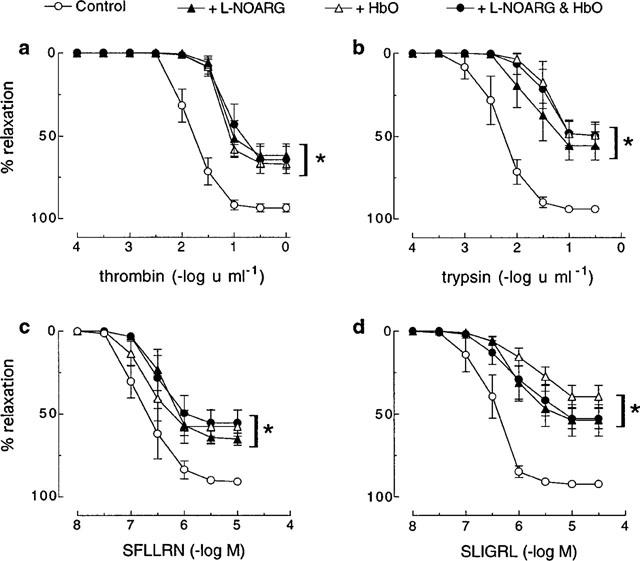
Cumulative concentration-response curves to (a) thrombin, (b) trypsin, (c) SFLLRN and (d) SLIGRL in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with U46619 in the absence (control) and presence of L-NOARG (100 μM), HbO (20 μM) and a combination of L-NOARG and HbO. Each point represents the mean±s.e.mean of five experiments. *Denotes Rmax and pEC50 values significantly different from control (P<0.05).
Effect of NO inhibitors
The NO synthase inhibitor, L-NOARG (100 μM), significantly (P<0.05) decreased both the sensitivity and maximum relaxation to thrombin (pEC50 1.2±0.1, Rmax 62.0±8.0%), SFLLRN (pEC50 6.4±0.1, Rmax 65.2±4.0%), trypsin (pEC50 1.6±0.2, Rmax 55.8±9.7%) and SLIGRL (pEC50 6.0±0.1, Rmax 53.9±10.6%) (Figure 1). For all activators, the inhibition by L-NOARG was not significantly different to that observed with HbO (20 μM) either alone or in combination with L-NOARG (Figure 1).
Effect of high extracellular K+
In the presence of nifedipine (0.3 μM), high extracellular K+ (67 mM) had no effect on responses to trypsin, SFLLRN or SLIGRL but abolished the L-NOARG-resistant relaxations to these agonists (Figure 2). In contrast, both the sensitivity and maximum relaxation to thrombin were slightly, but significantly (P<0.05), inhibited by nifedipine alone (pEC50 1.5±0.1; Rmax 77.5±7.0%) (Figure 2). No further inhibition of the thrombin-induced response occurred with higher concentrations (1 and 3 μM) of nifedipine (data not shown). The inhibition of the thrombin-induced relaxation by nifedipine alone was no different to that observed in the presence of nifedipine in combination with high extracellular K+ (pEC50 1.5±0.1; Rmax 71.5±10.0%) (Figure 2). In addition, in the presence of nifedipine, L-NOARG now abolished relaxations to thrombin, whereas relaxations to SFLLRN, trypsin and SLIGRL were not further inhibited in the presence of nifedipine in combination with L-NOARG than they were with L-NOARG treatment alone (Figure 1 vs Figure 2). Importantly, tissues relaxed to approximately baseline levels of force with the addition of isoprenaline (1 μM) irrespective of their treatment (data not shown).
Figure 2.
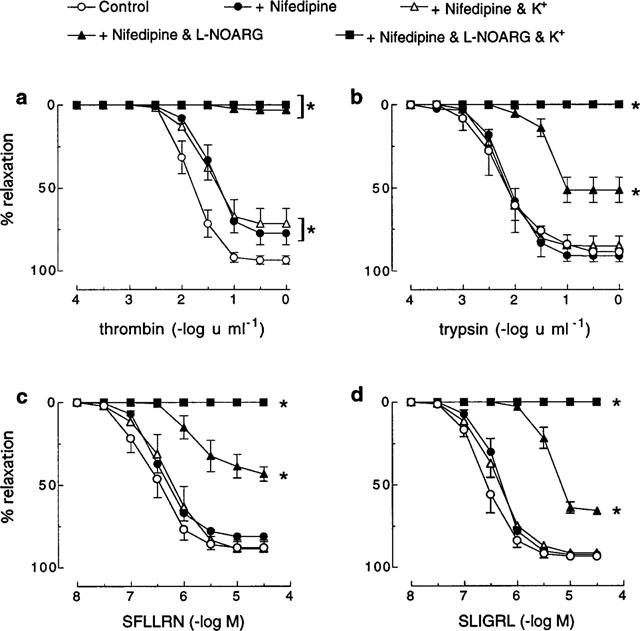
Cumulative concentration-response curves to (a) thrombin, (b) trypsin, (c) SFLLRN and (d) SLIGRL in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with U46619 in the absence (control) and presence of nifedipine (0.3 μM), nifedipine and K+ (67 mM), nifedepine and L-NOARG (100 μM) or nifedipine plus K+ and L-NOARG. Each point represents the mean±s.e.mean of six (a, b, c) and three (d) experiments. *Denotes Rmax and pEC50 values significantly different from control (P<0.05).
Effect of L-VOCC inhibitors
As for nifedipine, both verapamil (1 μM) and diltiazem (3 μM) slightly, but significantly, decreased the Rmax for thrombin (control=92.6±1.0% vs verapamil=83.4±2.1%; control=91.2±1.8% vs diltiazem=84.6±2.7%; for both P<0.05), but had no effect on relaxations induced by SFLLRN (control=91.8±3.6% vs verapamil=91.7±1.5%; control=92.6±1.7% vs diltiazem=90.7±2.2%) (Figure 3). Also, a significant (P<0.05) further inhibition of the relaxation to thrombin was observed in the presence of verapamil or diltiazem in combination with L-NOARG (100 μM) than was observed in the presence of L-NOARG alone (Figure 1 vs Figure 3). By contrast, L-NOARG did not further inhibit the SFLLRN-induced relaxation when applied in combination with either verapamil or diltiazem (Figure 1 vs Figure 3).
Figure 3.
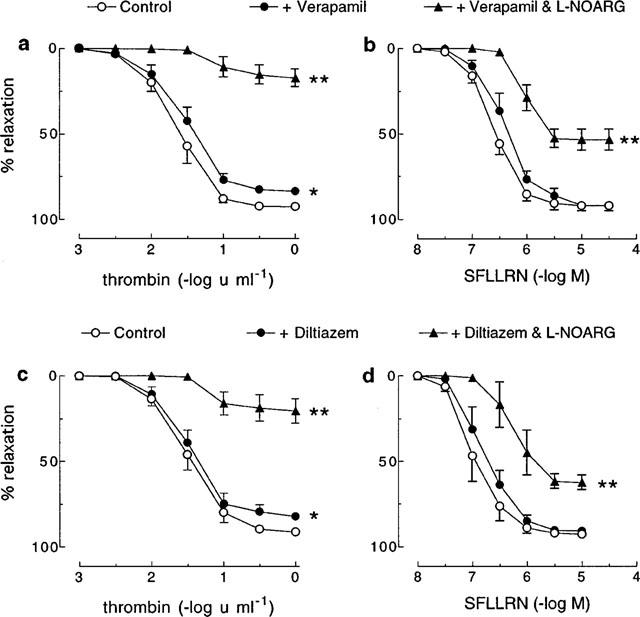
Cumulative concentration-response curves to (a, c) thrombin and (b, d) SFLLRN in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with U46619 in the absence (control) and presence of (a, b) verapamil (1 μM) or (c, d) diltiazem (3 μM) alone and in combination with L-NOARG (100 μM). Each point represents the mean±s.e.mean of 4–8 experiments. *Denotes Rmax value significantly different from control (P<0.05). **Denotes Rmax and pEC50 values significantly different from verapamil/diltiazem control (P<0.05).
Specificity of L-VOCC inhibitors and receptor desensitization
In artery rings contracted with KCl (10–18 mM), relaxations induced by nifedipine (1 nM–1 μM) were not affected by pretreatment with thrombin (1 u ml−1) (Figure 4). Also, as was observed with SFLLRN, nifedipine had no effect on relaxations to the more selective PAR1-activating peptide TFLLR (Kawabata et al., 1999) in the absence or presence of L-NOARG (Figure 5). Furthermore, desensitization to SFLLRN abolished relaxations to thrombin (Figure 6), which were restored following washout and recovery of the tissue (not shown). By contrast, desensitization to thrombin did not affect the response to SFLLRN (data not shown).
Figure 4.
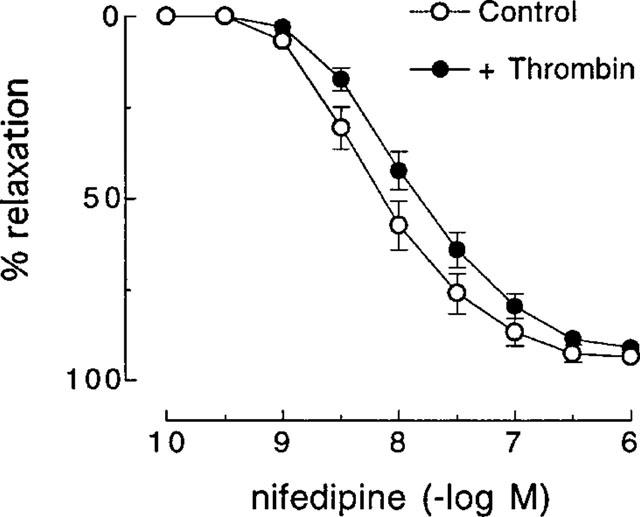
Cumulative concentration-response curves to nifedipine in the absence (control) and presence of thrombin (1 u ml−1) in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with KCl. Each point represents the mean±s.e.mean of six experiments.
Figure 5.
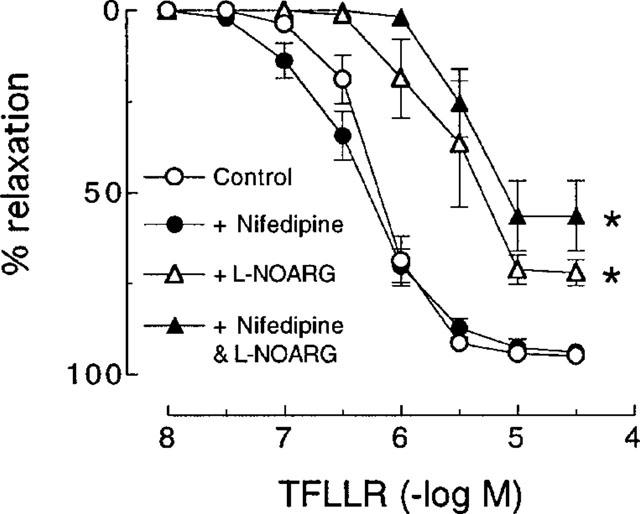
Cumulative concentration-response curves to TFLLR in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with U46619 in the absence (control) and presence of nifedipine (0.3 μM), L-NOARG (100 μM) and a combination of nifedipine and L-NOARG. Each point represents the mean±s.e.mean of five experiments. *Denotes Rmax and pEC50 values significantly different from control (P<0.05).
Figure 6.
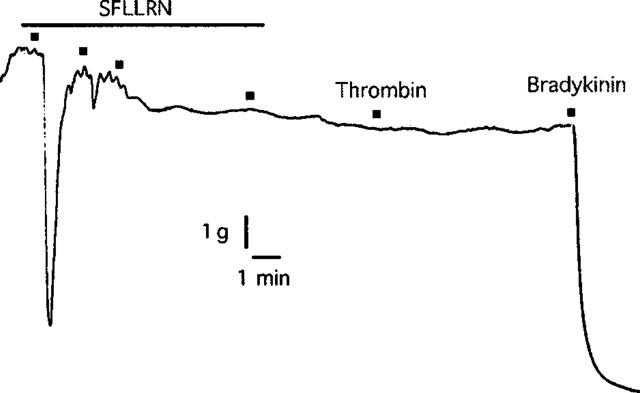
Digitized trace of an original chart recording showing the response to thrombin (1 u ml−1) following desensitization to SFLLRN (10 μM) in pig coronary artery ring preparations contracted to ∼50% of their maximum contraction with U46619. This trace is representative of four experiments.
Overcoming the inhibition by nifedipine of thrombin-induced relaxation
As described above, when thrombin was added cumulatively to the organ bath (0.001–1 u ml−1), nifedipine (0.3 μM) inhibited the maximum relaxation induced by thrombin and abolished the response in the presence of L-NOARG (100 μM) (Figure 2). By contrast, if thrombin was added as a single, high concentration (1 u ml−1) rather than in cumulatively increasing concentrations, nifedipine had no effect on thrombin-induced relaxations in tissues, with or without L-NOARG pretreatment (Figure 7).
Figure 7.
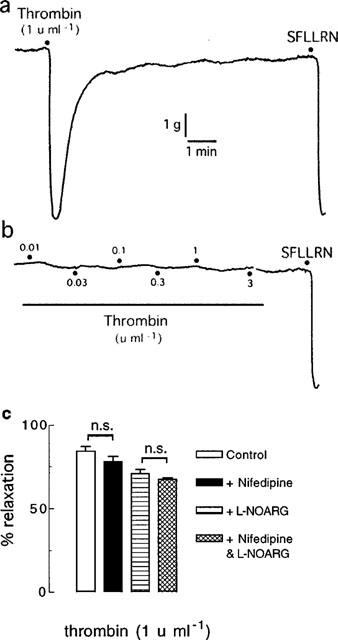
Digitized traces of original chart recordings showing responses to thrombin added (a) as a single, high concentration (1 u ml−1) and (b) cumulatively (0.01–3 u ml−1) to artery rings treated with nifedipine (0.3 μM) and L-NOARG (100 μM) and contracted to ∼50% of their maximum contraction with U46619. These traces are representative of six experiments. (c) Group data from similar experiments (each n=6) as depicted in (a). n.s.=not significant.
Discussion and conclusions
The main finding of this study was that the endothelium-dependent, NO-independent component of the relaxation to thrombin, but not to PAR1-activating peptides, was blocked by L-VOCC inhibitors. The involvement of a NO-independent vascular relaxation mechanism in the responses to both enzyme and peptide activators of PAR1 and PAR2 was evident by the similar degree of inhibition of each response by the NO synthase inhibitor, L-NOARG and/or the NO scavenger, HbO. These findings indicate that the only source of NO that contributed to the relaxations to each PAR activator was likely to be endothelial NO synthase and that its activity was abolished by L-NOARG, as reported for bradykinin in cow (Drummond & Cocks, 1996) and human (Kemp & Cocks, 1997) coronary arteries. Also, as for bradykinin in human and cow arteries (Drummond & Cocks, 1996; Kemp & Cocks, 1997) and the pig coronary artery (Kilpatrick & Cocks, 1994), the NO-independent component of the PAR1- and PAR2-mediated relaxations observed here were likely due to EDHF-like mechanisms (for review see Garland et al., 1995; Félétou & Vanhoutte, 1999), since they were abolished by high extracellular K+, which inhibits K+ channel activity (Chen & Suzuki, 1989) and the subsequent endothelial and smooth muscle hyperpolarization (Nagao & Vanhoutte, 1992; Corriu et al., 1996) and vascular relaxation (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996; Kemp & Cocks, 1997).
Endothelium-dependent hyperpolarization of vascular smooth muscle is generally thought to cause smooth muscle relaxation via closure of L-VOCCs (Garland et al., 1995). However, L-VOCC-independent mechanisms can also account for non-NO, K+-sensitive relaxations to endothelium-dependent vasodilators, since relaxations to bradykinin and substance P in pig (Kilpatrick & Cocks, 1994) and cow (Drummond & Cocks, 1996) coronary arteries were unaffected by L-VOCC inhibitors. While we observed a similar L-VOCC-independent, EDHF-like relaxation to trypsin and peptide activators of PAR1 and PAR2, intriguingly, we found that under similar assay conditions L-VOCC inhibitors selectively blocked the non-NO, K+-sensitive relaxation to thrombin.
The most likely explanation for the apparent selective involvement of L-VOCCs in thrombin-induced relaxations is that thrombin activated a different receptor to that activated by SFLLRN and TFLLR in the pig coronary artery. PAR3 and PAR4 are the most obvious candidates, since thrombin activates these receptors in addition to PAR1 (Ishihara et al., 1997; Xu et al., 1998). However, the contribution of PAR3 and/or PAR4 in the thrombin-induced endothelium-dependent relaxation in pig coronary artery appears unlikely since desensitization of tissues with SFLLRN, which is incapable of activating PAR3 and PAR4 (Ishihara et al., 1997; Xu et al., 1998), abolished the response to thrombin, suggesting that a common receptor mediated the response to each agonist. In addition, PAR4 was unlikely to have contributed to the endothelium-dependent relaxation induced by thrombin in the pig coronary artery since peptide activators of PAR4 are inactive in this preparation (Hamilton & Cocks, unpublished observations). While similar studies using PAR3-activating peptides cannot determine the presence or absence of functional PAR3 in tissues due to the inability of such peptides to activate the receptor (Ishihara et al., 1997), the fact that thrombin did not stimulate endothelium-dependent relaxation following desensitization with the PAR1-activating peptides points to a lack of involvement of PAR3 in the response to thrombin in the pig coronary artery. While desensitization to thrombin did not affect responses to SFLLRN, this is not unusual, since thrombin-activated PAR1 recycle back to the plasma membrane in a state in which they can be activated by synthetic tethered ligands such as SFLLRN but not by enzymes (Hamilton et al., 1998; Hammes & Coughlin, 1999). Whilst high concentrations of SFLLRN can activate PAR2 in some systems (Blackhart et al., 1996; Damiano et al., 1999; Kawabata et al., 1999), a similar lack of selectivity for SFLLRN does not appear to occur for endothelial PAR1 and PAR2 in the pig coronary artery (Hwa et al., 1996; Hamilton and Cocks, unpublished observations). Furthermore, the failure of nifedipine to block the non-NO, K+-sensitive relaxation to SFLLRN was unlikely to be due to activation of a different receptor (e.g. PAR2), since similar relaxations to the more selective PAR1 agonist TFLLR (Kawabata et al., 1999) were also unaffected by nifedipine. However, while it is unlikely that both thrombin and the PAR1-activating peptides, SFLLRN and TFLLR, acted on PAR3 or PAR4, we cannot rule out the possibility that either type of agonist acted at another, unknown receptor (protease-activated or other).
The effect of nifedipine on the non-NO, K+-sensitive relaxation to thrombin was most likely due to L-VOCC inhibition since similar selective effects on thrombin were observed with the chemically distinct L-VOCC inhibitors, verapamil and diltiazem. Importantly, the inhibition by each L-VOCC inhibitor was observed using the concentration required to abolish K+-induced contractions (and therefore, presumably, L-VOCC activity) in this tissue. Nevertheless, it remains possible that all three classes of L-VOCC inhibitors blocked the non-NO-mediated relaxation to thrombin independently of L-VOCC inhibition. For example, such non-selectivity has been reported for nimodipine, verapamil and diltiazem as inhibitors of 5-hydroxytryptamine3 receptors (Hargreaves et al., 1996). However, our finding that pretreatment of artery rings with thrombin had no effect on nifedipine-induced relaxations, does not support a direct interaction between L-VOCC inhibitors and thrombin. Also, since no further inhibition of thrombin-induced relaxations occurred when the concentration of nifedipine was increased from 0.3 μM to 1 and 3 μM, an interaction between nifedipine and PAR1 appears equally unlikely.
Therefore, on the assumption that the endothelium-dependent relaxation induced by thrombin and SFLLRN/TFLLR in the pig coronary artery is due solely to activation of PAR1, our data suggest that the non-NO, K+-sensitive relaxation to PAR1 in the pig coronary artery involves L-VOCCs, but only when the receptor is activated proteolytically with thrombin. If this is the case, then it is difficult to explain the mechanism by which such heterogeneous signalling by PAR1 occurs, following the different modes of receptor activation. Perhaps a clue can be found in the observations that PAR3, the second thrombin-activated receptor, cannot be activated by its synthetic tethered ligand sequence (Ishihara et al., 1997) and that PAR4, also regarded as a thrombin receptor, is only activated by high concentrations of its tethered ligand sequence (Xu et al., 1998). These discrepancies between the sensitivities of enzyme and synthetic tethered ligand PAR activators may indicate that the different modes of receptor activation result in dissimilar cellular events for PAR3 and PAR4. If this is the case, then similar differences may extend to PAR1 (and PAR2). However, this remains to be tested.
Finally, the ability of a single, high concentration of thrombin to overcome the L-VOCC-sensitivity of relaxations observed with the cumulative addition of thrombin supports the finding of Ishii et al. (1993) that the rate of PAR1 activation by thrombin determines the level of cellular response. Thus, in rat fibroblasts transfected with cloned human PAR1, concentrations of thrombin that did not cause any cell signalling were shown to eventually cleave and disable the entire PAR1 population from subsequent enzymic activation (Ishii et al., 1993). This contrasts with more classical free ligand-activated receptors (including activation of PARs with synthetic tethered ligand sequences), where downstream cellular responses are dependent on the level of ligand occupation of the receptor. Therefore, in our experiments, cumulative thrombin addition beginning with a concentration of thrombin subthreshold for generation of endothelium-dependent relaxation would be expected to activate some cell surface PAR1. By the time the concentration of thrombin required to induce a response was reached, however, a proportion of receptors would be cleaved and thus unresponsive to further addition of thrombin. By contrast, addition of a single, high concentration of thrombin would enable the enzyme to activate enough PAR1 to cause cell signalling and a response.
In conclusion, our study has shown that clinically used L-VOCC inhibitors like nifedipine selectively block the non-NO component of endothelium-dependent relaxation to physiological concentrations of thrombin in pig coronary arteries. The involvement of L-VOCCs in endothelium-dependent relaxation of large arteries is apparently specific for thrombin since it was not observed following activation of PAR1 with PAR1-activating peptides or PAR2 with trypsin or PAR2-activating peptide and does not occur with other, non-PAR, endothelium-dependent vasodilators such as bradykinin and substance P (Kilpatrick & Cocks, 1994; Drummond & Cocks, 1996). If similar selective effects of L-VOCC inhibitors occur in human coronary arteries, then our findings have important implications for thrombogenesis in coronary arteries in which the NO-dependent vasodilator mechanisms are compromised by disease states such as atherosclerosis.
Acknowledgments
This work was supported by a grant from the National Health & Medical Research Council (Australia). The authors wish to thank Mr Stavros Selemidis and Dr Christopher Sobey for helpful discussions.
Abbreviations
- EDHF
endothelium-derived hyperpolarizing factor
- HbO
oxyhaemoglobin
- KPSS
high potassium physiological Krebs solution
- PAR
protease-activated receptor
- Rmax
maximum response
References
- BLACKHART B.D., EMILSSON K., NGUYEN D., TENG W., MARTELLI A.J., NYSTEDT S., SUNDELIN J., SCARBOROUGH R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. J. Physiol. 1989;410:91–106. doi: 10.1113/jphysiol.1989.sp017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CICALA C., PINTO A., BUCCI M., SORRENTINO R., WALKER B., HARRIOT P., CRUCHLEY A., KAPAS S., HOWELLS G.L., CIRINO G. Protease-activated receptor-2 involvement in hypotension in normal and endotoxemic rats in vivo. Circulation. 1999;99:2590–2597. doi: 10.1161/01.cir.99.19.2590. [DOI] [PubMed] [Google Scholar]
- CORRIU C., FÉLÉTOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUGHLIN S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUPIT L.D., SCHMIDT V.A., BAHOU W.F. Proteolytically activated receptor-3: a member of an emerging gene family of protease-activated receptors expressed on vascular endothelial cells and platelets. Trends Cardiovasc. Med. 1999;9:42–48. doi: 10.1016/s1050-1738(99)00005-5. [DOI] [PubMed] [Google Scholar]
- DIAMANO B. P., CHEUNG W.-M., SANTULLI R.J., FUNG-LEUNG W.-P., NGO K., YE R.D., DARROW A.L., DERIAN C. K., DE GARAVILLA L., ANDREA-GORDON P. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J. Pharmacol. Exp. Ther. 1999;288:671–678. [PubMed] [Google Scholar]
- DÉRY O., CORVERA C.U., STEINHOFF M., BUNNET N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Evidence for mediation by endothelium-derived hyperpolarizing factor of relaxation to bradykinin in the bovine isolated coronary artery independently of voltage-operated Ca2+ channels. Br. J. Pharmacol. 1996;117:1035–1040. doi: 10.1111/j.1476-5381.1996.tb16693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FÉLÉTOU M., VANHOUTTE P.M. Endothelial dysfunction: a novel therapeutic target. The alternative: EDHF. J. Mol. Cell Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FENTON J.W., , II Understanding thrombin and hemostasis. Hematol. Oncol. Clin. North. Am. 1993;7:1107–1119. [PubMed] [Google Scholar]
- FLEMING I., BUSSE R. Endothelial dysfunction: a novel therapeutic target. NO: the primary EDRF. J. Mol. Cell Cardiol. 1999;31:5–14. doi: 10.1006/jmcc.1998.0839. [DOI] [PubMed] [Google Scholar]
- FOX M.T., HARRIOTT P., WALKER B., STONE S.R. Identification of potential activators of proteinase-activated receptor-2. FEBS Lett. 1997;417:267–269. doi: 10.1016/s0014-5793(97)01298-2. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GLUSA E., MARKWARDT F. Studies on thrombin-induced endothelium-dependent vascular effects. Biomed. Biochem. Acta. 1988;47:623–630. [PubMed] [Google Scholar]
- HAMILTON J.R., CHOW J.M., COCKS T.M. Protease-activated receptor-2 turnover stimulated independently of receptor activation in porcine coronary endothelial cells. Br. J. Pharmacol. 1999;127:617–622. doi: 10.1038/sj.bjp.0702583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON J.R., NGUYEN P.B., COCKS T.M. Atypical protease-activated receptor mediates endothelium-dependent relaxation of human coronary arteries. Circ. Res. 1998;82:1306–1312. doi: 10.1161/01.res.82.12.1306. [DOI] [PubMed] [Google Scholar]
- HAMMES S.R., COUGHLIN S.R. Protease-activated receptor-1 can mediate responses to SFLLRN in thrombin-desensitized cells: evidence for a novel mechanism for preventing or terminating signaling by PAR1's tethered ligand. Biochemistry. 1999;38:2486–2493. doi: 10.1021/bi982527i. [DOI] [PubMed] [Google Scholar]
- HARGREAVES A.C., GUNTHORPE M.J., TAYLOR C.W., LUMMIS S.C. Direct inhibition of 5-hydroxytryptamine3 receptors by antagonists of L-type Ca2+ channels. Mol. Pharmacol. 1996;50:1284–1294. [PubMed] [Google Scholar]
- HATTON M.W.C., MOAR S.L., RICHARDSON M. Deendothelialization in vivo initiates a thrombogenic reaction at the rabbit aorta surface: correlation of uptake of fibrinogen and antithrombin III with thrombin generation by the exposed endothelium. Am. J. Path. 1989;135:499–508. [PMC free article] [PubMed] [Google Scholar]
- HWA J.J., GHIBAUDI L., WILLIAMS P., CHINTALA M., ZHANG R., CHATTERJEE M., SYBERTZ E. Evidence for the presence of a proteinase-activated receptor district from the thrombin receptor in vascular endothelial cells. Circ. Res. 1996;78:581–588. doi: 10.1161/01.res.78.4.581. [DOI] [PubMed] [Google Scholar]
- ISHIHARA H., CONNOLLY A.J., ZENG D., KAHN M.L., ZENG Y.W., TIMMONS C., TRAM T., COUGHLIN S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- ISHII K., HEIN L., KOBILKA B., COUGHLIN S.R. Kinetics of thrombin receptor cleavage on intact cells: relation to signaling. J. Biol. Chem. 1993;268:9780–9786. [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL-ANI B., LEBLOND L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J. Phrmacol. Exp. Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- KEMPS B.K., COCKS T.M. Evidence that mechanisms dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary arteries. Br. J. Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOVANEN P.T., KAARTINEN M., PAAVONEN T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion of rupture in myocardial infarction. Circulation. 1995;92:1084–1088. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- KU D.D. Mechanism of thrombin-induced endothelium-dependent coronary vasodilation in dogs: role of its proteolytic activity. J. Cardiovasc. Res. 1986;8:29–36. doi: 10.1097/00005344-198601000-00005. [DOI] [PubMed] [Google Scholar]
- KU D.D., WINN M.J., GRIGSBY T., CAUFIELD J.B. Human coronary vascular smooth muscle and endothelium-dependent responses after storage at −75°C. Cryobiology. 1992;29:199–209. doi: 10.1016/0011-2240(92)90020-3. [DOI] [PubMed] [Google Scholar]
- LÜSCHER T.F., DIEDERICH D., SIEBENMANN R., LEHMANN K., STULZ P., VON SEGESSER L., YANG Z., TURINA M., GRÄDEL E., WEBER E., BÜHLER F.R. Difference between endothelium-dependent relaxation in arterial and in venous coronary bypass grafts. N. Engl. J. Med. 1988;319:462–467. doi: 10.1056/NEJM198808253190802. [DOI] [PubMed] [Google Scholar]
- MOLINO M., BARNATHAN E.S., NUMEROF R., CLARK J., DREYER M., CUMASHI A., HOXIE J.A., SCHECHTER N., WOOLKALIS M., BRASS L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- MURAMATSU I., LANIYONU A., MOORE G.J., HOLLENBERG M.D. Vascular actions of thrombin receptor peptide. Can. J. Physiol. Pharmacol. 1992;70:996–1003. doi: 10.1139/y92-137. [DOI] [PubMed] [Google Scholar]
- NAGAO T., VANHOUTTE P.M. Hyperpolarization as a mechanism for endothelium-dependent relaxations in the porcine coronary artery. J. Physiol. 1992;445:355–367. doi: 10.1113/jphysiol.1992.sp018928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTEDT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., ALLEN G.T., NORMANDIN D., ANTONACCIO M.J. Involvement of the 'tethered ligand' receptor in thrombin-induced endothelium-mediated relaxations. Am. J. Physiol. 1993;265:H1744–H1749. doi: 10.1152/ajpheart.1993.265.5.H1744. [DOI] [PubMed] [Google Scholar]
- VU T.-K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WHITE R.P., SHIMAZAKI Y., ROBERTSON J.T. Comparison of responses elicited by alpha-thrombin in isolated canine basilar, coronary, mesenteric and renal arteries. Blood Vessels. 1984;21:12–22. doi: 10.1159/000158490. [DOI] [PubMed] [Google Scholar]
- XU W., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]