

Abstract
We have previously demonstrated that chronic and simultaneous treatment of rats with the μ-opioid receptor agonist sufentanil and the Ca2+ channel blocker nimodipine, not only prevented tolerance development, but the animals became supersensitive to the antinociceptive effect of the opioid. The focus of the present work was to determine the possible involvement of cross interactions between the adenylyl cyclase pathway and L-type voltage-sensitive Ca2+-channels, in modulating the switch from opioid tolerance into supersensitivity.
The modulatory effect of sufentanil on adenylyl cyclase activity was determined by measuring cyclic AMP production in slices from the cortex of rats rendered tolerant or supersensitive to the antinociceptive effect of the opioid. Tolerance was induced by chronic infusion of sufentanil, at a rate of 2 μg h−1, for 7 days. Supersensitivity was induced by concurrent infusion of sufentanil (2 μg h−1) and nimodipine (1 μg h−1) for 7 days. Antinociception was evaluated by the tail-flick test.
Tolerance to the analgesic effect of sufentanil was associated with a significant reduction in the response of adenylyl cyclase to forskolin. Furthermore, the effect of the opioid on forskolin-induced cyclic AMP accumulation was abolished. On the other hand, supersensitivity to the analgesic effect of the opioid was associated with an increase in both, the adenylyl cyclase response to forskolin, and the opioid inhibition of cyclic AMP production.
We suggest that sustained L-type Ca2+ channel blockade may result in changes in the adenylyl cyclase effector system triggered by μ-opioid receptor activation, leading to the switch from opioid tolerance into supersensitivity.
Keywords: Cyclic AMP, opioids, tolerance, supersensitivity, calcium channel blockers
Introduction
The most efficacious drugs employed to relieve pain are opioid analgesics. However, chronic administration of these drugs results in a progressive reduction in the antinociceptive response (tolerance). Opioid tolerance is assumed to be a pharmacodynamic process, involving neural adaptive mechanisms in some of the intracellular messenger pathways that mediate the acute actions of the agonists. Increasing evidence supports the role for adaptations in the cyclic AMP pathway, and in the function of voltage-sensitive Ca2+ channels, as important molecular mechanisms underlying opioid tolerance and dependence (see Cox, 1993; Nestler, 1996).
The search for drugs that could prevent the development of tolerance would be very useful in pain management, to improve the therapeutic benefits of opioids. In this regard, behavioural, biochemical and molecular pharmacology studies suggest that Ca2+ channel blockers may be useful in inhibiting the morphine tolerance process (Dierssen et al., 1990; Díaz et al., 1995b), and providing protection against opioid withdrawal in rodents (Bongianni et al., 1986; Baeyens et al., 1987; Pellegrini-Giampietro et al., 1988; Barrios & Baeyens, 1988; Antkiewicz-Michaluk et al., 1990; 1993; Alfaro et al., 1990; Zharkovsky et al., 1993; Krystal et al., 1996; Michaluk et al., 1998). We have demonstrated that chronic infusion of nimodipine in association with sufentanil in rats, not only prevents tolerance expression but induces supersensitivity to the antinociceptive effect of the opioid (Dierssen et al., 1990; Díaz et al., 1995b). Opioid supersensitivity was associated with an increase in the number of dihydropyridine-binding sites, and with a recovery from the μ-opioid receptor down-regulation displayed by tolerant animals (Díaz et al., 1995a,1995b). Regarding the therapeutic relevance of these experimental data, we have reported that nimodipine reduced the daily requirements of morphine to relieve pain in cancer patients who had developed dose escalation (Santillán et al., 1994; 1998).
The brain cortex is an important substrate for the opioidergic control of pain perception. Recent anatomical and physiological studies in animals, as well as functional imaging studies in humans have shown that multiple cortical areas are activated by painful stimuli. These regions include primary and secondary somatosensory cortices, anterior cingulate cortex, insular cortex, and the frontal cortex (Treede et al., 1999; Talbot et al., 1991; Bushnell et al., 1999). Moreover, opioid drugs inhibit both, the cortical structures involved in opioidergic descending inhibitory control of pain (Kharkevich & Churukanov, 1999; Burkey et al., 1996; Soto-Moyano et al., 1988), and the ascending nociceptive inputs to the somatosensory cortex (Kalliomaki et al., 1998). The aim of this study was to characterize the involvement of the adenylyl cyclase pathway in the mechanisms underlying opioid supersensitivity, by analysing the modulation of forskolin-induced cyclic AMP production by the μ-opioid agonist sufentanil, in the brain cortex of rats rendered tolerant or supersensitive to the antinociceptive effect of the opioid.
Methods
Animals
Male albino Wistar rats (Charles River, Criffa, Barcelona, Spain), weighing 250–300 g, were used. They were housed in a room kept at 22°C with a 12 : 12 h light-dark cycle. Food and water were provided ad libitum. This study was approved by the Cantabria University Institutional Laboratory Animal Care and Use Committee.
Drugs and chemicals
The μ-opioid agonist, sufentanil, and the Ca2+ channel blockers, nimodipine and furaldipine were kindly provided by Janssen Cylag, S.A. (Madrid, Spain), Química Farmacéutica Bayer, S.A. (Barcelona, Spain), and Laboratorios Alter (Madrid, Spain), respectively. The opioid antagonist, naloxone; the adenylyl cyclase activator, forskolin; and the inhibitor of cyclic AMP phosphodiesterase, isobutyl-methyl-xanthine (IBMX), were purchased from Sigma (Madrid, Spain). The chronic delivery of saline or drug-containing solution was carried out with Alzet 2001 osmotic minipumps, implanted subcutaneously, under light ether anaesthesia. These pumps deliver solutions at a constant rate of 1 μl h−1, during 7 days, and drug solutions were appropriately composed for the desired dosing regime. The acute administration of sufentanil was subcutaneously.
Experimental groups
Five different experimental groups were designed for the present study. Opioid tolerance and supersensitivity were induced as previously described (Dierssen et al., 1990; Díaz et al., 1995b). Group I: the control group (n=24) received 7 days chronic infusion of saline (1 μl h−1). Acute sufentanil challenging doses of 0.1 μg kg−1 (n=8), 0.5 μg kg−1 (n=8), and 1 μg kg−1 (n=8) were tested on the 7th day. Each animal received a single dose of the opioid. Group II: sufentanil tolerance was induced by subcutaneous administration of the opioid (2 μg h−1) for 7 days (n=19). Acute sufentanil challenging doses of 0.5 μg kg−1 (n=10), and 1 μg kg−1 (n=9) were tested on the 7th day, while the minipump of the opioid remained in place. Group III: sufentanil supersensitivity (n=8) was induced by chronic and simultaneous administration of the opioid (2 μg h−1) and the Ca2+ antagonist nimodipine (1 μg h−1), for 7 days. An acute sufentanil challenging dose of 0.5 μg kg−1 was tested on the 7th day of drug infusion. Group IV: nimodipine minipump was removed on the 6th day of treatment, keeping the sufentanil minipump for 7 days (n=5). A sufentanil challenging dose of 1 μg kg−1 was tested on the 7th day, 24 h after withdrawal from nimodipine. Group V: rats received nimodipine alone (1 μg kg−1 h−1) for 7 days (n=5). In all the groups, on the 7th day, 2 h after performing the analgesic test, animals were sacrificed without removing drug pumps, and the brains were processed for the cyclic AMP assay.
Evaluation of nociception
The tail-flick test (D'Armour & Smith, 1941) was used to assess nociceptive threshold. The tail-flick response was elicited by applying radiant heat to the surface of the tail. The intensity of the stimulus was adjusted so that, the control latency was within 3–5 s. A cut-off time of 10 s was set to prevent blistering. The tail-flick latency was measured before and 15 min after the injection of drugs. Analgesic end-point was defined as an increase of 100% in the individual reaction time in relation to the predrug control latency. The antinociceptive effect was expressed as the percentage of animals which reached the analgesic end-point.
Cyclic AMP assay
Rats were decapitated under light ether anaesthesia and the cerebral cortices were dissected on ice. The tissue was cut (0.35×0.35 mm) using a chopper. The cortical slices were rinsed in 10 ml per brain of an oxygenated (95% O2-5% CO2) Krebs-Ringer-bicarbonate buffer (KRB) in mM: NaCl 108, KCl 4.7, KH2PO4 0.9, MgSO4 1.2, CaCl2 2.5, NaHCO3 25, glucose 10, pH 7.4, for 60 min on ice. The buffer was renovated every 15 min. Cortical slices were incubated for 20 min at 37°C in the oxygenated KRB and then washed twice with 10 ml of KRB and resuspended in 10 ml per cortex of oxygenated KRB. Cortical slices (400 μl) were incubated in IBMX (1 mM) for 10 min at 37°C in the absence (basal) or in the presence of forskolin (10 μM) (stimulated). The ability of sufentanil to inhibit adenylyl cyclase activity was evaluated by incubating slices with increasing concentrations of the opioid in the presence of forskolin (10 μM) and IBMX (1 mM). After the 10 min incubation the reaction was stopped by heating in boiling water for 10 min. The mixture was homogenized and centrifuged at 13,000×g for 5 min. Aliquots (50 μl) of the supernatant were assayed in triplicate for cyclic AMP content, using an isotopic displacement commercial assay Kit (TRK 432, Amersham International PLC, Amersham, U.K.). Protein content was determined by the Lowry method (Lowry, 1951). Adenylyl cyclase activity was expressed as pmol of cyclic AMP produced per mg protein in 10 min.
Statistical analysis
In the cyclic AMP assay, data are expressed as the mean±s.e.mean of at least four different experiments. The percentage of cyclic AMP variation in presence of forskolin was calculated as [100×(forskolin-stimulated value−basal value)×basal value−1]. Within-group analysis was made by one-way ANOVA and post hoc Bonferroni test. Between-groups analysis was made by four-factor ANOVA and post hoc Bonferroni procedure. Comparison between two groups was made by Student t-test. The antinociceptive effect was expressed as the percentage of animals which reached the analgesic end-point. To compare the antinociceptive effect of each given acute sufentanil dose with the equivalent dose of the control group, the Mann-Whitney U-test was used. A P value <0.05 was considered to be statistically significant.
Results
Antinociceptive response induced by sufentanil
Before performing the biochemical component of the study, we assessed the efficacy of the different treatment protocols, which have been shown previously to induce tolerance and supersensitivity to the antinociceptive effect of sufentanil (Díaz et al., 1995b). The results obtained in the analgesic assay are summarized in Table 1. Briefly, acute administration of sufentanil (0.1, 0.5 and 1 μg kg−1) in the control group (n=24) produced a dose-dependent antinociceptive effect. Tolerance was assessed in a group of rats (n=19) chronically treated with the opioid (2 μg h−1) for 7 days. The antinociceptive response induced by challenging doses of sufentanil (0.5 and 1 μg kg−1) administered on the 7th day of opioid infusion, was significantly reduced. After 7 days of chronic and simultaneous treatment with sufentanil (2 μg h−1) and nimodipine (1 μg h−1) (n=8), a challenging dose of the opioid (0.1 μg kg−1), administered on the 7th day of drug infusion, reached the antinociceptive threshold in 75% of animals. However, when the nimodipine minipump was removed on the 6th day of infusion (n=5), and the opioid infusion maintained, no analgesic response was elicited, on the 7th day, by the same challenging dose of sufentanil (0.1 μg kg−1). Chronic nimodipine (1 μg h−1) administered alone for 7 days, failed to modify either the tail-flick basal reaction latencies or the antinociceptive response induced by a challenging dose of sufentanil (0.5 μg kg−1) (data not shown).
Table 1.
Antinociceptive effect of sufentanil in the different experimental groups

Basal and forskolin-induced accumulation of cyclic AMP in the different experimental groups
The basal cyclic AMP production was similar in all groups (Figure 1). However, the ability of adenylyl cyclase to increase cyclic AMP production in response to direct stimulation by forskolin (10 μM) was differently affected, depending on the experimental condition (Figure 1). In control animals chronically treated with saline (1 μl h−1, 7 days) (n=24), forskolin induced a 181±31% increase in cyclic AMP production. In animals rendered tolerant to the antinociceptive effect of the opioid by chronic treatment with sufentanil (2 μg h−1, 7 days) (n=19), the cyclic AMP accumulation was only increased 58±16% by forskolin. In order to assess if this reduction in the response to forskolin was a consequence of residual sufentanil present in the preparation, we compared the effects induced by adding naloxone (1 μM) to the slices of untreated (n=4) and chronically treated with sufentanil animals (n=4). In the control group, naloxone did not modify both basal and forskolin-induced accumulation of cyclic AMP. In the tolerant group, the reduced response to forskolin was not prevented by naloxone (data not shown). In animals rendered supersensitive to the analgesic effect of the opioid by 7 days concurrent administration of sufentanil (2 μg h−1) and nimodipine (1 μg h−1) (n=9), the response to forskolin was 258±60% of the cyclic AMP basal value. In the group where the nimodipine pump was removed 24 h before performing the cyclic AMP production assay, the stimulatory effect of forskolin was completely abolished.
Figure 1.
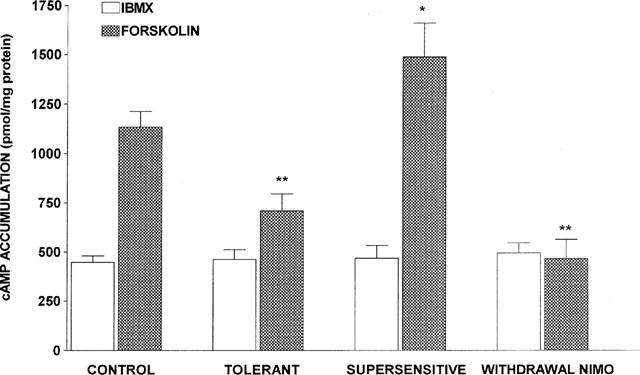
Basal and forskolin-stimulated cyclic AMP production in the different experimental groups. Cyclic AMP was determined in brain cortex slices incubated for 10 min in the absence (IBMX) or in the presence of 10 μM forskolin. Control animals (n=24) were treated with saline (1 μl h−1) for 7 days. Tolerant animals (n=19) received sufentanil (2 μg h−1) for 7 days. Supersensitive animals (n=8) were concurrently treated with sufentanil (2 μg h−1) and nimodipine (1 μg h−1) for 7 days. In the nimodipine withdrawal group (n=5), nimodipine minipump was removed on the 6th day of infusion, while sufentanil minipump remained in place for 7 days. Data are expressed as mean±s.e.mean. F=13.43, P<0.001 (ANOVA); **P<0.01, *P<0.05 vs control forskolin (Bonferroni test).
Effects of sufentanil on forskolin-induced cyclic AMP accumulation in the different experimental groups
In the cortex of control rats (n=24), incubation of the slices with increasing concentrations of sufentanil (0.1–5 μM), produced a concentration-dependent inhibition of forskolin-stimulated adenylyl cyclase activity (Figure 2A). At a concentration of 5 μM, sufentanil produced a maximum inhibition of 58±5%. In tolerant animals, chronically treated with sufentanil (2 μg h−1, 7 days) (n=19), the ability of the opioid (1–5 μM) to inhibit forskolin-induced cyclic AMP accumulation was abolished (Figure 2B). In supersensitive animals, chronically treated with sufentanil (2 μg h−1) and nimodipine (1 μg h−1) for 7 days (n=9), the ability of sufentanil (0.1–2.5 μM) to inhibit forskolin-induced cyclic AMP accumulation, not only recovered to control values, but was significantly increased when compared with the control and with the tolerant groups (Figure 2C). Chronic treatment with nimodipine alone (2 μg h−1, 7 days) (n=5), did not significantly modify the inhibitory effect of sufentanil (0.1, 2.5 and 5 μM) on forskolin-induced cyclic AMP accumulation when compared with the control group (data not shown). In animals chronically treated with sufentanil and nimodipine, withdrawal from nimodipine 24 h before performing the experiment on the 7th day, resulted in a complete loss of the ability of sufentanil (0.1–5 μM) to inhibit forskolin-induced cyclic AMP accumulation (Figure 2D), as occurred in tolerant animals.
Figure 2.
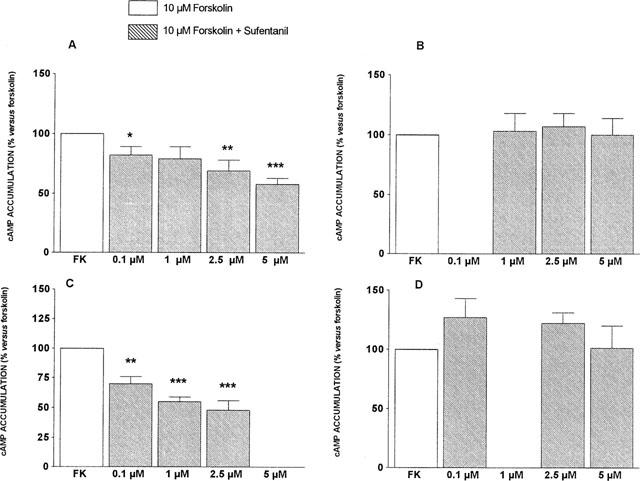
Effects of sufentanil on forskolin induced cyclic AMP accumulation in the different experimental groups. Cyclic AMP was determined in brain cortex slices incubated for 10 min in the presence of 10 μM forskolin alone or with increasing concentrations of sufentanil (0.1–5 μM). (A) Control animals (n=24) were treated with saline (1 μl h−1) for 7 days. (B) Tolerant animals (n=19) received sufentanil (2 μg h−1) for 7 days. (C) Supersensitive animals (n=8) were concurrently treated with sufentanil (2 μg h−1) and nimodipine (1 μg h−1) for 7 days. (D) In the nimodipine withdrawal group (n=5), nimodipine minipump was removed on the 6th day of infusion, while sufentanil minipump remained in place for 7 days. Data are expressed as mean±s.e.mean. Within-group comparisons were performed by one-way ANOVA (Group A: F=7.43, P<0.001; Group B: F=0.15, NS; Group C: F=17.11, P<0.001; Group D: F=1.18, NS). ***P<0.001, **P<0.01,*P<0.05 vs forskolin (Bonferroni test). Four-factor ANOVA was used for comparing sufentanil effects between groups (F=3.31, P=0.001). Bonferroni Post hoc test showed significant differences between control group and tolerant (P<0.001), supersensitive (P<0.01), and nimodipine withdrawal (P<0.001) groups; between supersensitive group and tolerant (P<0.001) and nimodipine withdrawal (P<0.001) groups, but not between tolerant and nimodipine withdrawal groups.
Effects in vitro addition of furaldipine to the slices on forskolin-induced cyclic AMP accumulation
In the cortex of control rats (n=4), incubation of the slices with the Ca2+ channel blocker furaldipine (1 μM), did not modify basal or forskolin-stimulated adenylyl cyclase activity (Figure 3). In cortices of tolerant animals, Ca2+ channel blockade enhanced forskolin-induced cyclic AMP accumulation up to the values of the supersensitive group (Figure 3). In cortices of supersensitive animals, addition of Ca2+ channel blockers to the assay did not further increase the stimulatory effect of forskolin on cyclic AMP accumulation (Figure 3).
Figure 3.
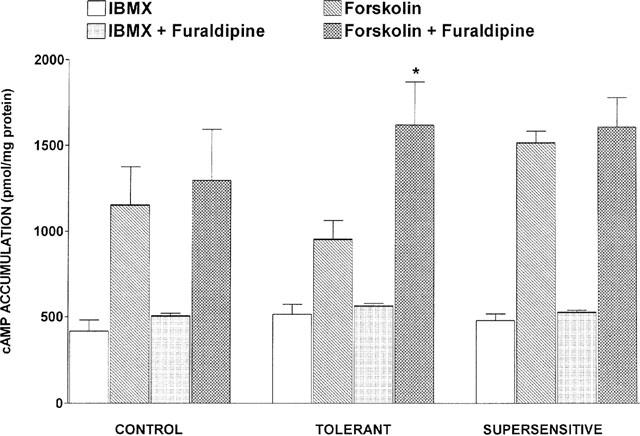
Effects of in vitro furaldipine on basal (IBMX) and forskolin-stimulated cyclic AMP production in the different experimental groups. Cyclic AMP was determined in brain cortex slices incubated for 10 min with 10 μM forskolin alone or with 1 μM furaldipine. Control animals were treated with saline (1 μl h−1) for 7 days. Tolerant animals received sufentanil (2 μg h−1) for 7 days. Supersensitive animals were concurrently treated with sufentanil (2 μg h−1) and nimodipine (1 μg h−1) for 7 days. Data are expressed as mean±s.e.mean. *P<0.05 vs forskolin (Student t-test).
Discussion
Previous studies, performed in our laboratory, demonstrated that chronic and simultaneous treatment of rats with the opioid sufentanil and the Ca2+ channel blocker, nimodipine, not only prevented tolerance development, but the animals became supersensitive to the antinociceptive effect of the opioid (Dierssen et al., 1990; Díaz et al., 1995b). The focus of the present work was to determine the possible involvement of cross interactions between the adenylyl cyclase pathway and L-type voltage-sensitive Ca2+-channels, in modulating the switch from opioid tolerance into supersensitivity. For this purpose, we have analysed the modulation by sufentanil of forskolin-induced cyclic AMP production in the cortex of rats rendered tolerant or supersensitive to the antinociceptive effect of the opioid.
Tolerance to the opioid antinociceptive effect was effectively induced, following the experimental protocol previously described, by chronic infusion of the μ-opioid agonist, sufentanil, at a rate of 2 μg h−1, for 7 days (Dierssen et al., 1990; Díaz et al., 1995b). The present data show that the loss of analgesic potency is associated with a reduction in the inhibitory effect of sufentanil on forskolin-induced cyclic AMP accumulation in the cerebral cortex of tolerant rats. The effects of chronic opioid drug treatment on opioid-inhibited adenylyl cyclase represent one of the most thoroughly studied models of biochemical analogy to opioid tolerance (for reviews see Cox, 1993; Childers, 1993; Nestler, 1996). Receptor desensitization, characterized as the loss in the ability of the opioid agonist to inhibit adenylyl cyclase activity, and receptor down-regulation, appear to be important factors contributing to the reduced sensitivity upon chronic opioid treatment (Bot et al., 1998; Puttfarcken et al., 1988; Chakrabarti et al., 1995). Employing the same experimental protocol as in the present report (Díaz et al., 1995a), we have previously shown that there is a decrease in the number of opioid binding sites.
In control animals, forskolin produced an increase in cyclic AMP accumulation of 181±31%. However, after tolerance development, the forskolin response was reduced to 58±16%. This effect does not appear to be due to residual sufentanil retained in the preparation, as it was not observed in the animals concurrently treated with sufentanil and nimodipine. Furthermore, the effect was not reversed by the inclusion of naloxone in the assay. Considering that the dose of forskolin used directly activates adenylyl cyclase enzymatic activity (Sutkovski et al., 1996), these data indicate that the catalytic function of the adenylyl cyclase was decreased in the cortex of rats chronically treated with sufentanil. Similarly, it has been reported that the adenylyl cyclase responsiveness to forskolin stimulation was markedly reduced in cultures from the rat cortex after long term opioid treatment (Eriksson et al., 1992), and in HEK-293 cells stably transfected with the μ-opioid receptor, chronically treated with sufentanil (Bot et al., 1998). Conversely, an increase in adenylyl cyclase and PKA activities occurs after withdrawal from chronic morphine administration in some brain regions, such as the locus coeruleus and the nucleus accumbens. These changes have been implicated in the behavioural expression opioid abstinence (Duman et al., 1988; Terwilliger et al., 1991; Maldonado et al., 1995; Nestler, 1996; Valverde et al., 1996).
Our results indicate that in animals chronically treated with sufentanil, the cyclic AMP-elevating effect of forskolin was enhanced by blocking Ca2+ entry through L-type channels either in vitro, by adding furaldipine to the cortical slices in the tolerant group, or in vivo, by simultaneous treatment with sufentanil and nimodipine, as observed in the supersensitive group. These data suggest the involvement of Ca2+-inhibitable forms of adenylyl cyclase in the adaptive mechanisms brought about by chronic opioid treatment. Adenylyl cyclases are important targets for modulatory effects of Ca2+ (Cooper et al., 1995; Sunahara et al., 1996), and it has been suggested that entry of Ca2+ through L-type channels mediates the inhibitory effect of Ca2+ on type V and VI adenylyl cyclase (Yu & Green, 1993; Cooper et al., 1995). Considering that chronic exposure to opioid drugs, including sufentanil, produced L-type Ca2+ channel up-regulation in rat brain (Ramkumar & El-Fakahany, 1988; Antkiewickz-Michaluk et al., 1990; Ohnishi et al., 1990; Zharkovsky et al., 1993; Díaz et al., 1995b, Michaluk et al., 1998), it is possible that an increased influx of Ca2+ through these channels could account for the reduction in the adenylyl cyclase enzymatic activity, and might prevent the enzyme from responding any further to the regulatory effect derived from μ-opioid receptor activation.
There is general agreement that L-type Ca2+ channel blockers reduce the expression of opioid tolerance in humans (Santillán et al., 1994; 1998), and in rats (Dierssen et al., 1990; Díaz et al., 1995b; Michaluk et al., 1998), and provide protection against opioid withdrawal (Alfaro et al., 1990; Antkiewicz-Michaluk et al., 1990; Baeyens et al., 1987; Barrios & Baeyens, 1988; Bongianni et al., 1986; Pellegrini-Giampietro et al., 1988; Zharkovsky et al., 1993; Krystal et al., 1996; Michaluk et al., 1998). When nimodipine was chronically infused in conjunction with sufentanil, the antinociceptive effect of the opioid was strongly potentiated, confirming that these experimental conditions lead to a switch from opioid tolerance into supersensitivity (Dierssen et al., 1990; Díaz et al., 1995b). Interestingly, the ability of forskolin to stimulate cyclic AMP accumulation in the cortex of the animals treated with sufentanil and nimodipine not only was recovered, but an increased response was observed when compared with the control group. Furthermore, the inhibitory effect of sufentanil on forskolin-stimulated cyclic AMP production was also significantly increased. Thus, contrary to what occurs in the tolerant condition, the presence of nimodipine during chronic sufentanil treatment induced opioid antinociceptive supersensitivity associated with a recovery of the adenylyl cyclase enzymatic activity, and with supersensitivity of the enzyme to the inhibitory effect of the μ-opioid agonist. On the other hand, chronic infusion of nimodipine alone produced no significant changes in the opioid effects. These results indicate that the concurrent presence of both sufentanil and nimodipine, but not each drug individually, induced changes in the adenylyl cyclase effector system linked to the μ-opioid receptor, thus leading to a state of supersensitivity to the activation of the receptor by agonists. The functional correlate of this biochemical adaptation was an increased opioid antinociceptive potency. The sustained blockade of the Ca2+ channel was essential to maintain the increased response to sufentanil, because withdrawal of nimodipine returned both the opioid antinociceptive response, and the inhibitory effect on cyclic AMP accumulation to the values of tolerant animals (present data and Zharkovsky et al., 1999).
We have previously demonstrated that chronic association of nimodipine with sufentanil prevented the down-regulation of μ-opioid receptors observed in tolerant animals in areas related to pain transmission, such as the somatosensory cortex, the central grey, and the dorsal horn of the spinal cord (Díaz et al., 1995a and unpublished observations). These data, taken together with the present results, indicate that chronic nimodipine administered during sufentanil tolerance development, was able to prevent the uncoupling of μ-opioid receptors from their effector systems, their subsequent down-regulation, and the functional expression of antinociceptive tolerance.
Regarding the mechanism underlying opioid supersensitivity, we suggest that chronic blockade of L-type Ca2+ channels concurrently with μ-opioid receptor activation, could lead to a chronic cross-talk which results in the amplification of the cellular and behavioural responses to μ-opioid receptor activation. Possibly there are common gene expression control features between these two systems. In this regard, the transcription factor CREB (cyclic-AMP-response-element binding protein) is critical for the adaptive responses triggered by chronic opioid treatment (Maldonado et al., 1996; Blendy & Maldonado, 1998), and can function as a Ca2+ sensitive transcription factor (Hardinghan et al., 1997; Hu et al., 1999).
In conclusion, the results of the present study indicate that tolerance and supersensitivity to the antinociceptive effect of sufentanil are matched by parallel changes in the ability of the opioid to inhibit the cyclic AMP production system. We suggest that association of the Ca2+ channel blocker, nimodipine, to the chronic treatment with opioid analgesic drugs, could be useful in preventing tolerance development by avoiding the uncoupling of μ-opioid receptors from their effector systems.
Acknowledgments
This work was supported by Ministerio de Educación y Cultura (CICYT SAF96-0284), and Fundación Marqués de Valdecilla (14-99) grants. E.M. Valdizán is supported by the Fundación Marcelino Botín. I. Goirigolzarri is recipient of a predoctoral fellowship from the Fundación Upsamédica. The authors wish to thank Ms Nieves García-Iglesias and Mr Jaime Perez-Casar for their excellent technical assistance. We thank Dr J. Flórez for his helpful comments. The generous gift of nimodipine by Química Farmáceutica Bayer (Spain), furaldipine by Laboratorios Alter (Spain), and sufentanil by Janssen-Cylag (Spain) are gratefully acknowledged.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- CREB
cyclic-AMP-response-element binding protein
- FK
forskolin
- IBMX
isobutyl-methyl-xanthine
- KRB
Krebs-Ringer-bicarbonate buffer
References
- ALFARO M.J., COLADO M.I., LÓPEZ F., MARTÍN M.I. Effect of clonidine, nimodipine and diltiazem on the in vitro opioid withdrawal response in the guinea-pig ileum. Br. J. Pharmacol. 1990;101:958–960. doi: 10.1111/j.1476-5381.1990.tb14187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANTKIEWICZ-MICHALUK L., MICHALUK J., ROMANSKA I., VETULANI J. Cortical dihydropyridine binding sites and behavioural syndrome in morphine-abstinent rats. Eur. J. Pharmacol. 1990;180:129–135. doi: 10.1016/0014-2999(90)90600-b. [DOI] [PubMed] [Google Scholar]
- ANTKIEWICZ-MICHALUK L., MICHALUK J., ROMÁNSKA I., VENTULANI J. Reduction of morphine dependence and potentiation of analgesia by chronic co-administration of nifedipine. Psychopharmacol. 1993;111:457–464. doi: 10.1007/BF02253536. [DOI] [PubMed] [Google Scholar]
- BAEYENS J.M., ESPOSITO E., OSSOWSKA G., SAMANIN R. Effects of peripheral and central administration of Ca2+ channel blockers in the naloxone-precipitated abstinence syndrome in morphine-dependent rats. Eur. J. Pharmacol. 1987;137:9–13. doi: 10.1016/0014-2999(87)90176-2. [DOI] [PubMed] [Google Scholar]
- BARRIOS M., BAEYENS J.M. Differential effects of Ca2+ channel blockers and stimulants on morphine withdrawal in vitro. Eur. J. Pharmacol. 1988;152:175–178. doi: 10.1016/0014-2999(88)90852-7. [DOI] [PubMed] [Google Scholar]
- BLENDY J.A., MALDONADO R. Genetic analysis of drug addiction: the role of cAMP response element binding protein. J. Mol. Med. 1998;76:104–110. doi: 10.1007/s001090050197. [DOI] [PubMed] [Google Scholar]
- BONGIANI F., CARLA V., MORONI F., PELLEGRINI-GIAMPIETRO D.E. Calcium channel inhibitors suppress the morphine-withdrawal syndrome in rats. Br. J. Pharmacol. 1986;88:561–567. doi: 10.1111/j.1476-5381.1986.tb10236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOT G., BLAKE A.D., LI S., REISINE T. Fentanyl and its analogs sesensitize the cloned mu opioid receptor. J. Pharmacol. Exp. Ther. 1998;285:1207–1218. [PubMed] [Google Scholar]
- BURKEY A.R., CARSTENS E., WENNIGER J.J., TANG J., JASMIN L. An opioidergic cortical antinociception triggering site in the agranular insular cortex of the rat contributes to morphine antinociception. J. Neurosci. 1996;16:6612–6623. doi: 10.1523/JNEUROSCI.16-20-06612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSHNELL M.C., DUNCAN G.H., HOFBAUER R.K., HA B., CHEN J.I., CARRIER B. Pain perception: is there a role for primary somatosensory cortex. Proc. Natl., Acad. Sci. U.S.A. 1999;96:7705–7709. doi: 10.1073/pnas.96.14.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOPER D.M.F., MONS N., KARPEN J.W. Adenylyl cyclases and the interaction between calcium and cyclic AMP signalling. Nature. 1995;374:421–424. doi: 10.1038/374421a0. [DOI] [PubMed] [Google Scholar]
- COX B.M.Opioid receptor-G protein interactions: acute and chronic effects of opioids Hdb. Exp. Pharmacol.: Opioids 1993Berlin: Springer-Verlag; 145–188.ed. Herz, A., Akil, H., Simon, E. pp [Google Scholar]
- CHAKRABARTI S., LAW P-Y, LOH H.H. Neuroblastoma Neuro 2A cells stably expressing a cloned μ-opioid receptor: a specific cellular model to study acute and chronic effects of morphine. Mol. Brain Res. 1995;30:269–278. doi: 10.1016/0169-328x(95)00014-j. [DOI] [PubMed] [Google Scholar]
- CHILDERS S.R.Opioid receptor coupled second messenger systems Hdb. Exp. Pharmacol.: Opioids 1993Berlin: Springer-Verlag; 145–188.ed.: Herz, A., Akil, H. & Simon, E. pp [Google Scholar]
- D'ARMOUR F.E., SMITH D.L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- DÍAZ A., RUIZ F., FLÓREZ J., HURLÉ M.A., PAZOS A. Mu-opioid receptor regulation during opioid tolerance and supersensitivity in rat central nervous system. J. Pharmacol. Exp. Ther. 1995a;274:1552–1561. [PubMed] [Google Scholar]
- DÍAZ A., RUIZ F., FLÓREZ J., PAZOS A., HURLÉ M.A. Regulation of dihydropyridine-sensitive Ca++ channels during opioid tolerance and supersensitivity in rats. J. Pharmacol. Exp. Ther. 1995b;274:1545–1551. [PubMed] [Google Scholar]
- DIERSSEN M., FLÓREZ J., HURLÉ M.A. Calcium channel modulation by dihydropyridines modifies sufentanil-induced antinociception in acute and tolerant conditions. Naunyn-Schmidebergs's Arch. Pharmacol. 1990;342:559–565. doi: 10.1007/BF00169046. [DOI] [PubMed] [Google Scholar]
- DUMAN R.S., TALLMAN J.F., NESTLER E.J. Acute and chronic opiate-regulation of adenylyl cyclase in brain: specific effects in locus coeruleus. J. Pharmacol. Exp. Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- ERIKSSON P.S., CARLSSON B., ISAKSSON E., RONNBACK L. Altered amounts of G-protein mRNA and cyclic AMP accumulation after long-term opioid receptor stimulation of neurones in primary culture from the rat cerebral cortex. Brain Res. Mol. Brain Res. 1992;14:317–325. doi: 10.1016/0169-328x(92)90099-w. [DOI] [PubMed] [Google Scholar]
- HARDINGHAM G.E., CHAWLA S., JOHNSON C.M., BADING H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- HU S.C., CHRIVIA J., GHOSH A. Regulation of CBP-mediated transcription by neural calcium signalling. Neuron. 1999;22:799–808. doi: 10.1016/s0896-6273(00)80738-2. [DOI] [PubMed] [Google Scholar]
- KALLIOMAKI J., LUO X.L., YU Y.B., SCHOUENBORG J. Intrathecally applied morphine inhibits nociceptive C fibre input to the primary somatosensory cortex (SI) of the rat. Pain. 1998;77:323–329. doi: 10.1016/S0304-3959(98)00115-8. [DOI] [PubMed] [Google Scholar]
- KHARKEVICH D.A. &, CHURUKANOV V.V. Pharmacological regulation of descending cortical control of the nociceptive processing. Eur. J. Pharmacol. 1999;375:121–131. doi: 10.1016/s0014-2999(99)00264-2. [DOI] [PubMed] [Google Scholar]
- KRYSTAL J.H., COMPERE S., NESTLER E.J., RASMUSSEN K. Nimodipine can reduce naloxone-precipitated locus coeruleus activation and abstinence behaviour in morphine-dependent rats. Physiol. Behav. 1996;59:863–866. doi: 10.1016/0031-9384(95)02206-6. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MALDONADO R., BLENDY J.A., TZAVARA E., GASS P., ROQUES B.P., HANOUNE J. Reduction of morphine abstinence in mice with a mutation of the gene encoding CREB. Science. 1996;273:657–659. doi: 10.1126/science.273.5275.657. [DOI] [PubMed] [Google Scholar]
- MALDONADO R., VALVERDE O., GARBAY C., ROQUES B.P. Protein kinases in the locus coeruleus and periaqueductal gray matter are involved in the expression of opiate withdrawal. Naunyn-Schmidebergs's Arch. Pharmacol. 1995;352:565–575. doi: 10.1007/BF00169392. [DOI] [PubMed] [Google Scholar]
- MICHALUK J., KAROLEWICZ B., ANTKIEWICZ-MICHALUK L., VENTULANI J. Effects of various Ca2+ channel antagonists on morphine analgesia, tolerance and dependence, and on blood pressure in the rat. Eur. J. Pharmacol. 1998;352:189–197. doi: 10.1016/s0014-2999(98)00373-2. [DOI] [PubMed] [Google Scholar]
- NESTLER E.J. Under siege: The brain on opiates. Neuron. 1996;16:897–900. doi: 10.1016/s0896-6273(00)80110-5. [DOI] [PubMed] [Google Scholar]
- OHNISHI T., SAITO K., MAEDA S., MATSUMOTO K., SAKUDA M., INOKI R. Intracerebroventricular treatment of mice with pertussis toxin induces hyperalgesia and enhances 3H-nitrendipine binding to synaptic membranes: Similarity with morphine tolerance. Naunyn-Schmidebergs's Arch. Pharmacol. 1990;341:123–127. doi: 10.1007/BF00195068. [DOI] [PubMed] [Google Scholar]
- PELLEGRINI-GIAMPETRO D.E. , BACCIOTTIN L., MORONI F. Morphine withdrawal in cortical slices: Suppression by Ca++ channel inhibitors of abstinence-induced 3H-noradrenaline release. Br. J. Pharmacol. 1988;93:535–540. doi: 10.1111/j.1476-5381.1988.tb10308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTTFARCKEN P.S., WERLING L.L., COX B.M. Effects of chronic morphine exposure on opioid inhibition of adenylyl cyclase in 7315c cell membranes: A useful model for the study of tolerance at μ opioid receptors. Mol. Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- RAMKUMAR V., EL-FAKAHANY E.E. Prolonged morphine treatment increases rat brain dihydropyridine binding sites: Possible involvement in development of morphine dependence. Eur. J. Pharmacol. 1988;146:73–83. doi: 10.1016/0014-2999(88)90488-8. [DOI] [PubMed] [Google Scholar]
- SANTILLÁN R., HURLÉ M.A., ARMIJO J.A., DE LOS MOZOS R., FLÓREZ J. Nimodipine-enhanced opiate analgesia in cancer patients requiring morphine dose escalation: A double blind, placebo-controlled study. Pain. 1998;76:17–26. doi: 10.1016/s0304-3959(98)00019-0. [DOI] [PubMed] [Google Scholar]
- SANTILLÁN R., MAESTRE J.M., HURLÉ M.A., FLÓREZ J. Enhancement of opiate analgesia by nimodipine in cancer patients chronically treated with morphine A preliminary report. Pain. 1994;58:129–132. doi: 10.1016/0304-3959(94)90192-9. [DOI] [PubMed] [Google Scholar]
- SOTO-MOYANO R., GALVEZ J., VALLEJOS C., HERNANDEZ A. Topical application of morphine to the rat somatosensory cortex produces analgesia to tonic pain. J. Neurosci. Res. 1988;19:511–514. doi: 10.1002/jnr.490190416. [DOI] [PubMed] [Google Scholar]
- SUNAHARA R.K., DESSAUER C., GILMAN A.G. Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- SUTKOWSKI E.M., ROBBINS J.D., TANG W-J., SEAMON K.B. Irreversible inhibition of forskolin interactions with type I adenylyl cyclase by a 6-isothiocyanate derivative of forskolin. Mol. Pharmacol. 1996;50:299–305. [PubMed] [Google Scholar]
- TALBOT J.D., MARRET S., EVANS A.C., MEYER E., BUSHNELL M.C., DUNCAN G.H. Multiple representations of pain in human cerebral cortex. Science. 1991;251:1355–1358. doi: 10.1126/science.2003220. [DOI] [PubMed] [Google Scholar]
- TERWILLIGER R.Z., BEITNER-JOHNSON D., SEVARINO K.A., CRAIN S.M., NESTLER E.J. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- TREEDE R-D., KENSHALO D.R., GRACELY R.H., JONES A.K.P. The cortical representation of pain. Pain. 1999;79:105–111. doi: 10.1016/s0304-3959(98)00184-5. [DOI] [PubMed] [Google Scholar]
- VALVERDE O., TZAVARA E., HANOUNE J., ROQUES B.P., MALDONADO R. Protein kinases in the rat nucleus accumbens are involved in the aversive component of opiate withdrawal. Eur. J. Neurosci. 1996;8:2671–2678. doi: 10.1111/j.1460-9568.1996.tb01562.x. [DOI] [PubMed] [Google Scholar]
- YU H.J., MA H., GREEN R.D. Calcium entry via L-type calcium channels acts as a negative regulator of adenylyl cyclase activity and cyclic AMP levels in cardiac myocytes. Mol. Pharmacol. 1993;44:689–693. [PubMed] [Google Scholar]
- ZHARKOVSKY A., KATAJAMAKI J., SEPPALA T., AHTEE L. Morphine-induced analgesia in rats withdrawn from concurrent nimodipine and morphine treatment. Pain. 1999;79:217–222. doi: 10.1016/s0304-3959(98)00176-6. [DOI] [PubMed] [Google Scholar]
- ZHARKOVSKY A., TÖTTERMAN A.M., MOISIO J., ATHEE L. Concurrent nimodipine attenuates the withdrawal signs and the increase of dihydropyridine binding after chronic morphine treatment in rats. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;347:483–486. doi: 10.1007/BF00166739. [DOI] [PubMed] [Google Scholar]