

Introduction
The largest family of cell surface receptors involved in signal transduction, G protein coupled receptors (GPCRs), are one of the major targets for current drugs as well as new drug development. Ligands interacting with for e.g. adrenergic, histamine, adenosine, opioid, dopamine or serotonin receptors, constitute a large portion of currently used therapeutics. A common property of GPCRs is that upon activation (agonist binding) they transmit signals across the plasma membrane via an interaction with heterotrimeric G proteins (Stadel et al., 1997). The corresponding activated G protein subsequently interacts with an intracellular effector system, such as adenylate cyclase or phospholipase C, leading to a wide variety of distinct physiological responses.
Recent evidence suggests that GPCRs have the potential to be 'active' even in the absence of an agonist. This exhibition of spontaneous receptor activity has led to the observation that various ligands, previously considered as antagonists with no intrinsic activity, actually can inhibit this spontaneous activity, appearing to possess 'negative intrinsic activity'. This phenomenon has been termed inverse agonism and the corresponding ligands are referred to as inverse agonists.
Although intrinsic constitutive receptor activity and inverse agonism have unequivocally been demonstrated in vitro, (patho)physiological consequences are far from self-evident. Thus, in this review we should like to focus on the expression of inverse agonism under more 'physiological conditions', since it appears timely to address the physiological relevance and consequences of this new concept, both in GPCR research and drug discovery.
Historical overview
Traditional receptor theory has postulated on a single, 'quiescent' receptor state to which agonists bind inducing a conformational change of the receptor to an activated and 'functional' state. This view, initially formed in the early 1950s, was more clearly expressed by Del Castillo & Katz (1957), and stood as the foundation of receptor pharmacology for decades. It was believed that antagonists interact with the receptor, thereby preventing agonist binding, without having an effect on conformational changes of the receptor that remained in its 'quiescent' state.
The first evidence of a ligand ('antagonist') producing opposite effects to those of an agonist, and thus not merely inhibiting agonist binding, stems from the GABA-benzodiazepine field. In Braestrup et al. 1982, reported on the discovery of an agent, DMCM (methyl 6,7-dimethoxy-4-ethyl-β-carboline-e-carboxylate), which in contrast to benzodiazepines, not only was a potent convulsant in vivo, but seemed to favour binding to benzodiazepine receptors that were in the non-GABA(agonist)-stimulated conformation. For the first time, it was elaborated that GABA-benzodiazepine receptors may perhaps exist in two conformations which are in equilibrium, an open chloride channel form (activated conformation) and a closed one (inactivated conformation) for which DMCM may have a high affinity and a tendency to stabilize, thereby decreasing binding of GABA to the activated conformation.
Interestingly, the above concept of an 'agonist-independent' two-state receptor conformation, introduced initially for ion-channel-coupled receptors, soon found support from those studying the large family of GPCRs. Costa & Herz (1989) were pioneers in setting the grounds for what later would be referred to as 'inverse agonism'. They demonstrated for the first time that some antagonists of the δ opioid receptor had 'negative intrinsic activity' in vitro, in contrast to others that lacked any intrinsic activity. Moreover, such ligands diminished even further the 'basal' or constitutive activity of the receptor defined as the activity of the receptor in the absence of any ligand. It was apparent that the concept of 'negative intrinsic activity' implied a pre-existing equilibrium between (at least) two states of a receptor. These two states could easily be defined as either a G protein-bound or a free form of the receptor, the first one active and the latter inactive. In their studies, basal GTPase activity in NG108-15 cell membranes was suggested to be due to stimulated activity resulting from a spontaneous interaction between empty or free receptors and G proteins. Their data lent support to the receptor model of Wregget & De Lean (1984) which predicted that 'antagonists may be active by hindering the ability of receptors to associate spontaneously with G proteins in membranes'.
Since then, and especially over the past few years, disclosure of negative intrinsic activity has corroborated even further this two-state model of GPCR activation. Hence, a large number of publications within this decade are concerned with, and demonstrate with a variety of systems or means, the phenomenon of constitutive receptor activity and its implication for inverse agonism.
In this review we will first address some of the more seminal papers, showing that in most cases genetically engineered cell systems were pivotal for the development of this new concept. These and many more studies have been adequately and thoroughly reviewed recently by Milligan et al. (1997) and Leurs et al. (1998). We will then gradually move to more 'physiological' systems, in order to address the central issue of this review whether inverse agonism and spontaneous receptor activity are relevant phenomena in health and disease, and hence for drug discovery. It should be pointed out, however, that a classification of ligands based on their (negative) intrinsic activity is not an easy task. Due to the large influence of receptor systems and experimental conditions (whole cells versus membranes, stoichiometry of receptors/G protein, signalling proteins etc.) the same ligand may behave as an inverse agonist, a neutral antagonist or even a (partial) agonist.
Genetically engineered systems to facilitate detection of inverse agonism
CAMs and receptor or G protein overexpression
Inverse agonism on GPCRs is not always easily established, since basal receptor activity is generally not pronounced. Thus, various manipulations to increase basal (constitutive) receptor activity have been explored, such as construction and expression of constitutively active mutant receptors (CAM) or overexpression of either the receptor or the G protein to favourably change the receptor-G protein ratio (R : G).
CAM receptors show higher agonist-independent activity and have been reported for various receptor subtypes. Since CAM receptors have a higher basal receptor activity, the effect of inverse agonists is more readily observed. Samama et al. (1993) were the first to describe a CAM receptor of the β2-adrenoceptor (AR); replacement of four amino acids of the third intracellular loop by the corresponding residues of the α1B-AR, led to agonist-independent activation of adenylate cyclase. Previous work by Cotecchia et al. (1990) had demonstrated that substitution of residues in the third intracellular loop of the β2-AR by the corresponding residues of the α1-AR, led to chimeric receptors that were coupled to PI hydrolysis, like the native α1-AR, instead of adenylate cyclase. This provided direct evidence that the third intracellular loop is important for G protein binding and activation.
Since normal expression levels of wild type receptors do not always result in constitutive activity, numerous studies have been performed in various systems where overexpression of the wild type receptor has been induced. Examples of these are the expression of wild type β2-AR in Sf9 insect cells leading to receptor densities up to 40 pmol mg−1 protein (Chidiac et al., 1994) or the overexpression of the calcitonin receptor in HEK293 cells (Pozvek et al., 1997). In the latter case, two different clonal cell lines were selected, expressing 5×106 and 25×103 receptors/cell, respectively. Whereas the first cell line displayed an 80 fold increase in basal cyclic AMP production, the second was not constitutively active. Apparently, receptor density is positively correlated to spontaneous activity and various classes of GPCRs can display constitutive activity upon overexpression.
Overexpression of the G protein involved may also lead to increased basal levels of second messengers. High levels of Gαq cotransfected with various muscarinic receptor subtypes in NIH3T3 cells resulted in increased basal activity of the receptors (Burstein et al., 1997). This induced constitutive activity of the receptors was reversed completely by the muscarinic antagonists tested, indicating that they behaved as inverse agonists. Thus, elevation of G protein levels favours formation of the active conformation of the receptor the fraction of receptors that are coupled to the G protein and provides a more sensitive means for the detection of inverse agonism.
These genetic approaches even work in vivo, since transgenic animals overexpressing GPCRs proved another source of constitutively active receptors. Bond et al. (1995) described transgenic mice overexpressing the wild type β2-AR at various receptor levels. Baseline left atrial tension in these transgenic mice was increased 3 fold over control mice while the β2-selective ligand ICI-118,551, acting as an inverse agonist, decreased baseline tension. The inhibitory effect of ICI-118,551 was correlated with β2-AR densities, suggesting that it was a receptor-mediated event. Apart from these organ bath data, the effect of ICI-118,551 was also studied in vivo, where cardiac contractility was measured in both control and transgenic mice. ICI-118,551 decreased cardiac contractility in transgenic mice by approximately 70%, an effect that was associated with a fall both in heart rate and left ventricular systolic pressure, while the compound exhibited no effects on control hearts. Recently, the CAM β2-AR mentioned above, has also been overexpressed in mice (Samama et al., 1997). In this case, the mouse phenotype was not very different from normal, probably due to the rather modest overexpression (∼3 fold). However, long-term treatment with ICI-118,551 increased CAM β2-AR density, resulting in marked basal atrial tension and cardiac contractility. Finally, Nagaraja et al. (1999) described the effects of long term treatment of various β-adrenoceptor ligands on baseline left atrial tension in transgenic mice with modest β2-AR overexpression (50 fold compared to 200 fold β2-AR overexpression in the paper by Bond et al. (1995)). In these transgenic mice, inverse agonists such as ICI-115,881, carvedilol and propranolol increased baseline left atrial tension, whereas untreated or alprenolol-treated mice were unaffected. Baseline left atrial tension was not affected by any ligand in the hearts of wild type mice. This paper therefore showed the differential effects of a neutral (alprenolol) and inverse agonists (e.g. ICI-118,551) besides the importance of receptor density in the study of inverse agonism.
Thus, constitutive receptor activity has been demonstrated for several GPCRs after some form of genetic manipulation. We will now review other types of studies that may have a more direct link to in vivo pharmacology and physiology. These studies include cell systems with 'normal' levels of receptor expression and tissue or organ bath preparations. The issue of 'endogenous' inverse agonists will also be discussed.
Inverse agonism at wild type receptors
Various authors have described inverse agonism and constitutive activity on wild type receptors expressed in artificial cell lines but at more or less 'physiological' levels of expression. Examples of such studies are listed in Table 1 and discussed below.
Table 1.
Examples of constitutively active wild type receptors expressed at more or less ‘physiological' receptor levels

The rat histamine H2 receptor stably transfected in CHO cells was studied by Smit et al. (1996). This receptor had pronounced basal activity, as indicated by an increase in basal cyclic AMP production. Although this basal cyclic AMP was further increased by the endogenous agonist histamine, the H2 blockers cimetidine and ranitidine were shown to decrease basal cyclic AMP production, therefore expressing inverse agonism in this system. Burimamide, on the other hand, did not alter basal cyclic AMP levels but was able to block both the histamine-induced increase and the cimetidine-induced decrease of basal cyclic AMP production. Hence, burimamide behaved as a neutral antagonist. Similar results were obtained for the human histamine H2 receptor (Alewijnse et al., 1998), although at this receptor burimamide behaved as a weak partial agonist, increasing basal cyclic AMP production by 16%, compared to the maximal response induced by histamine.
Newman-Tancredi et al. (1997) studied the 5-HT1A subtype of the serotonin receptor expressed in CHO cells at a receptor density of 1.6 pmol mg−1 protein. Modulation of [35S]-GTPγS binding in a membrane preparation was used to discriminate between the various ligands tested. 5-Carboxamidotryptamine (5-CT) was classified as a full agonist increasing [35S]-GTPγS binding to the same extent as serotonin (5-HT), whereas spiperone was identified as an inverse agonist since it decreased basal [35S]-GTPγS binding by 30%. Meanwhile, WAY100,635 behaved as a neutral antagonist. It showed no effect on basal [35S]-GTPγS binding by itself, but was able to block both 5-CT-induced stimulation and spiperone-induced inhibition of basal [35S]-GTPγS binding. The effect of spiperone could not be explained by a simple displacement of endogenous 5-HT; not only had the membranes been extensively washed, but if basal activity had been due to 5-HT1A receptor activation by endogenous 5-HT, the antagonist WAY100,635 should have also blocked this activation.
The behaviour of both subtypes of the human cannabinoid (CB1 and CB2) receptor was analysed by Bouaboula et al. (1997; 1999). CHO cells, stably expressing either the CB1 or the CB2 receptor, showed higher basal MAPK activity compared to untransfected CHO cells. In both transfected cell lines CP-55940, a non-selective cannabinoid agonist, further increased basal MAPK activity. The CB1-selective compound SR141,716A decreased basal MAPK activity in CHO cells expressing the CB1 receptor, thus behaving as an inverse agonist for this receptor (Bouaboula et al., 1997). SR141,716A also displayed effects opposite to agonists in a cyclic AMP-related luciferase assay. Instead of a decrease in luciferase activity induced by cannabinoid agonists, SR141,716A elicited an increase. Apparently, SR141,716A acted as an inverse agonist in two different signal transduction pathways, i.e. Gβγ-mediated MAPK-activity and Gα,i-mediated adenylate cyclase inhibition. Similar results were obtained with SR144,528, a CB2-selective ligand that behaved as an inverse agonist, decreasing both basal MAPK activity and [35S]-GTPγS binding on CHO cells expressing the CB2 receptor (Bouaboula et al., 1999). Furthermore, Landsman et al. (1998) reported on another inverse agonist for the human CB1 receptor, AM630, which decreased basal [35S]-GTPγS binding, in contrast to the cannabinoid agonist WIN55,212-2.
Inverse agonistic effects were also shown at various dopamine receptor subtypes. Tiberi & Caron (1994) reported basal receptor activity of the dopamine D1A and D1B receptor, the human D1B receptor being linked to higher intracellular basal cyclic AMP levels, compared to the D1A receptor. Similar results were obtained for the corresponding rat receptors. Two dopamine antagonists, (+)-butaclamol and flupentixol, were able to decrease basal cyclic AMP levels; the inhibitory effect of these compounds, acting as inverse agonists, was more pronounced at the human D1B receptor, since the basal cyclic AMP level was higher. More recently, Griffon et al. (1996) showed that various antipsychotics inhibited [3H]-thymidine incorporation in NG108-15 cells expressing the recombinant human dopamine D3 receptor. Since dopamine agonists enhanced [3H]-thymidine incorporation, the antipsychotics tested (haloperidol, fluphenazine and chlorpromazine), behaved as inverse agonists. Nafadotride, a D3 receptor-preferring antagonist, had no effect of its own on [3H]-thymidine incorporation, therefore behaving as a neutral antagonist.
The two isoforms of the human calcitonin receptor (referred to as hCTR-1 and hCTR-2) activate adenylate cyclase, while in addition one of them (hCTR-2) is also able to activate phospholipase C to generate inositol phosphates (IP). Cohen et al. (1997) described constitutive receptor activity of human calcitonin receptors. Both hCTR-1 and hCTR-2 receptors expressed in COS-1 cells were constitutively active as shown by an increase in basal cyclic AMP production, although to a different extent. However, the hCTR-2 receptor did not show an increase in basal IP production. Apparently, spontaneous activity of this receptor was more readily observed for activation of adenylate cyclase. Addition of salmon calcitonin (sCT), a calcitonin receptor agonist, increased cyclic AMP production further, whereas the analogue Nα-acetyl-sCT-(8-32)amide did not elicit an effect. The paucity of available ligands prevented a further demonstration of inverse agonism.
A last example is the human formyl peptide (FP) receptor, a chemoattractant GPCR, studied by Wenzel-Seifert et al. (1998). This receptor was expressed in Sf9 and HEK293 cells at receptor densities of approximately 1 pmol mg−1 protein. These receptor levels are in the same range as the receptor density in HL-60 cells that endogenously express the FP receptor. It was shown that basal [35S]-GTPγS binding increased in the presence of N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP), a FP receptor agonist. On the contrary cyclosporin H decreased basal [35S]-GTPγS binding, thereby behaving as an inverse agonist. It seems, therefore, that at physiological receptor levels and expressed in different cell lines, the human FP receptor is constitutively active.
Inverse agonism in 'physiological' (or non-engineered) systems
A prerequisite for considering the physiological relevance of inverse agonism, is its study in experimental conditions that are as close to physiological as possible. Data related to inverse agonism obtained in intact animals (in vivo) are in most cases difficult to acquire, unless animals are genetically altered (e.g. transgenic mice) as described previously. Thus, apart from potential 'in vivo' data, other more or less 'physiological studies' may include data from cell lines endogenously expressing the receptor of interest, as for example the previously mentioned NG108-15 cells expressing δ opioid receptors (Costa & Herz 1989), or from tissue preparations such as cardiac or brain cortex membranes. Both latter methods may provide ex vivo data of wild type receptors at physiological or pathophysiological levels. Various examples of such studies in which potential inverse agonism was detected are summarized in Table 2.
Table 2.
Inverse agonism in ‘physiological' studies

Hilf & Jakobs (1992) described a decrease in G protein activation by antagonists of the muscarinic receptor in porcine atrial membranes. With this membrane preparation, enriched by sucrose density gradient centrifugation to contain ∼1.4 pmol receptors per mg protein, inhibition of both basal and carbachol-induced [35S]-GTPγS binding by atropine was shown. The presence of endogenous acetylcholine (ACh) was ruled out in this study by pretreatment of the membranes with 10 μM atropine or 100 μM GDP and subsequent washes, both compounds displacing all ACh possibly present. Jakubik et al. (1995) showed inverse agonism in rat cardiomyocytes expressing the M2 receptor. Atropine and QNB, both muscarinic antagonists, increased basal as well as forskolin-induced cyclic AMP production, an effect that was opposite to that of agonists. Similar results were obtained for three other subtypes of the muscarinic receptors, M1, M3 and M4, albeit in a 'non-physiological' setting, i.e. in CHO cells stably transfected with the human receptor gene.
To detect inverse agonism in H.E.L. 92.1.7 cells endogenously expressing the human α2A-AR, Jansson et al. (1998) used two different assays. Effects of various α2-adrenoceptor ligands on both intracellular levels of Ca2+ ([Ca2+]i) and forskolin-stimulated cyclic AMP production were investigated. The ligands used were thus classified from full agonists to inverse agonists. Both assays gave similar indications of intrinsic activities, showing that different assays and/or signalling pathways can sometimes be used to detect and classify ligands as inverse agonists. Quite remarkable in these studies were the opposite effects of the enantiomers of medetomidine; while dexmedetomidine acted as a partial agonist, increasing [Ca2+]i and decreasing forskolin-stimulated cyclic AMP production, levomedetomidine behaved as an inverse agonist by decreasing [Ca2+]i and increasing cyclic AMP production.
Other adrenoceptor subtypes have also been studied to observe inverse agonism in such 'physiological studies'. The α2D-AR, endogenously expressed in RIN5AH cells and heterologously expressed in PC-12 cells, revealed inverse agonistic effects of rauwolscine in [35S]-GTPγS binding assays (Tian et al., 1994). Accordingly, isoprenaline increased GTPγS-induced adenylate cyclase activity via the β-AR expressed in turkey erythrocytes, while both propranolol and pindolol showed a decrease, thus behaving as inverse agonists (Götze & Jakobs 1994).
A single cell preparation from cardiac tissue was used in the following two examples. Mewes et al. (1993) recorded L-type calcium currents (ICa) on both guinea-pig and human ventricular myocytes as a means to study the effects of the β-AR antagonists atenolol and propranolol. The myocytes were first superfused with 0.5 μM forskolin, to make the cells more sensitive to receptor-mediated changes of ICa. Application of both antagonists led to a decrease in forskolin-stimulated ICa in all myocyte preparations. Successive exposures to atenolol, a hydrophilic compound that could be readily washed away, resulted in similar changes in ICa, suggesting that these inhibitory effects were not due to a potential competition with endogenous agonist but to inverse agonistic activity of these antagonists.
Hanf et al. (1993) performed a similar study also using cardiomyocytes, but from two different species, frog and rat. They studied ICa regulated by muscarinic receptors. To increase basal ICa, isoprenaline was added during the experiments with frog ventricular cells (Figure 1). Addition of ACh resulted in a decrease of this basal ICa, whereas addition of atropine had the opposite effect, namely an increase of ICa. A similar effect of atropine was also noticed in the absence of Ach. This effect was dose-dependent and reversible. Results obtained with rat myocytes did not require the addition of isoprenaline since basal ICa was already high enough to observe the effects produced by the agonist ACh and the inverse agonist atropine.
Figure 1.
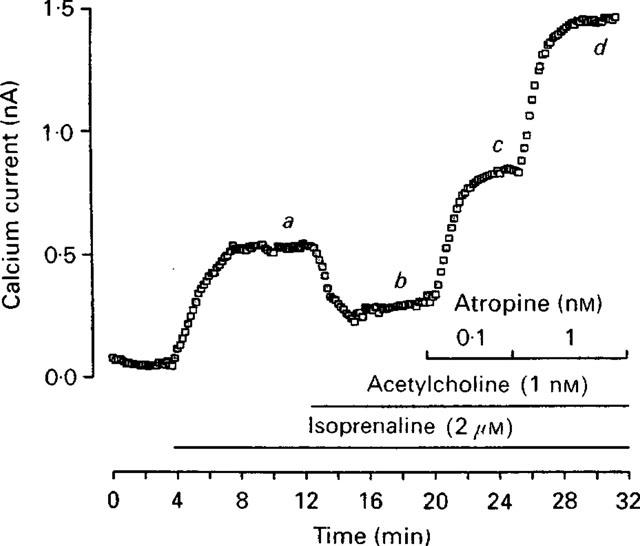
Atropine behaves as an inverse agonist on calcium currents (ICa) in frog ventricular cells (reproduced with permission from Hanf et al., 1993).
Organ bath preparations have also been used to detect inverse agonism (Noguera et al., 1996). Helically cut strips of rat thoracic aorta were prepared and the effects of α1-adrenoceptor antagonists on the regulation of the resting tone, induced by noradrenaline or Ca2+ exposure, were studied. Benoxathian and WB4101 not only inhibited the increase in resting tone induced by noradrenaline, but also blocked the response to noradrenaline in calcium-free medium. An explanation for these results was given within the concept of inverse agonism; it was suggested that both benoxathian and WB4101 acted as inverse agonists, decreasing the proportion of the receptor population in the active state (R*). Moreover, both compounds inhibited the increase in resting tone in the absence of agonist, providing a simple model for analysing inverse agonism in functional studies.
Other receptor types showing inverse agonism under 'physiological conditions' are the bradykinin BK2 receptor and the δ opioid receptor. Briefly, different bradykinin antagonists decreased basal IP production in rat myometrial cells endogenously expressing the BK2 receptor, while increasing concentrations of bradykinin increased basal IP production (Leeb-Lundberg et al., 1994). Furthermore, ICI-174,864 was shown to behave as an inverse agonist at the δ opioid receptor, endogenously expressed in NG108-15 cells, in both GTPase activity measurements and [35S]-GTPγS binding (Costa & Herz, 1989; Szekeres & Traynor, 1997).
It may be concluded from the above examples, that even in 'classical' organ bath or cell preparations inverse agonism may be readily demonstrated. This led us to examine older studies that show indirect evidence of some form of inverse agonism 'avant la lettre'.
Further indirect indications of inverse agonism
Within the concept of a (simplified) two-state receptor model (Figure 2), ligand efficacy may be redefined as the differential affinity of the ligand for the two conformational states (R, R*) with full agonists exhibiting higher affinity for the 'active' conformation (R*) and full inverse agonists for the 'inactive' conformation (R) (Monod et al., 1965; Leff, 1995). This differential affinity may be detected in radioligand binding studies with the use of appropriate 'modulators' of the receptor state. For example, in several receptor systems it is known that guanylyl nucleotides (e.g. GTP in the μM–mM range) uncouple the G protein from the receptor leading to a 'low' affinity state of the receptor for agonists (Chang & Snyder, 1980; De Lean et al., 1980; Stiles, 1988; Lohse et al.,1984). Thus, 'GTP-shifts' have been extensively used, in some GPCR fields, as a parameter to discriminate full from partial agonists due to the differential affinity of these ligands for the two receptor states (Ijzerman et al., 1996; Ehlert et al., 1985).
Figure 2.
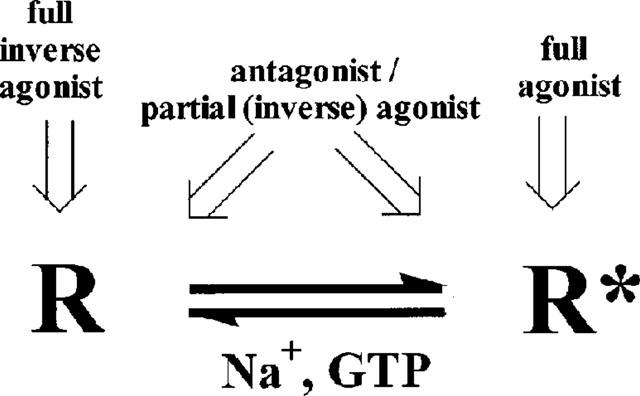
Simplified model representing the two conformational states of a receptor (inactive R, and active R*) and their differential affinities for ligands.
However, extrapolation of this concept to antagonists that are able to discriminate between the free and G protein-bound form of the receptor may reflect, and even correlate with, the extent of inverse agonistic properties (negative intrinsic activity) of these antagonists. Although a regulatory role of GTP on antagonist binding, which is inverse to the role of GTP on agonist binding, has been previously suggested (Burgisser et al., 1982; Ströher et al., 1989) we should like to point out its significance and consequence that seems to have escaped attention somewhat.
Examples of guanylyl nucleotide enhancement of binding of several antagonists (as have been reported in literature) are shown in Table 3. Both affinity and Bmax values of A1 adenosine receptor antagonists [3H]-XAC and [3H]-DPCPX, on rat adipocyte and guinea-pig brain membranes, respectively (Ramkumar & Stiles, 1988; Ströher et al., 1989), were increased significantly in the presence of GTP, indicating a preference of these antagonists for the uncoupled receptor state. A similar increase in binding was observed with two muscarinic cholinergic antagonists, [3H]-QNB on frog heart membranes (Burgisser et al., 1982) and [N-methyl-3H]-scopolamine methyl chloride on rat heart membranes (Berrie et al., 1979).
Table 3.
Indirect indications of inverse agonism in vitro

Accordingly, differential increase in binding of antagonists has also been observed in pertussis-toxin-mediated uncoupling of receptor and G protein; Costa & Herz (1989) were once more the first to show a leftward shift (increase in binding) upon toxin treatment in the competition curve of the δ opioid receptor antagonist ICI-174,864. The absence of any shift of antagonist MR2266 indicated a potential correlation of the negative intrinsic activity of ICI-174,864 with its greater affinity for the uncoupled form of the receptor.
Along these lines, interaction of sodium ions with several GPCRs is presumed to result in a stabilization of the low affinity (R) conformation of receptors (Green, 1984; Nunnari et al., 1987). It is believed that this effect of Na+ ions is linked to an aspartate residue in transmembrane helix II, conserved in virtually all GPCRs (Horstman, 1990). This effect of Na+ ions has been shown to be correlated with intrinsic activity of ligands (Tsai & Lefkowitz, 1978), the largest Na+-induced reduction in binding being observed for full agonists. However, although Na+ ions in almost all cases inhibit strongly agonist binding, their effect on 'antagonist' binding has been variable, ranging from no effect to an increase in binding (Chang & Synder, 1980; Pert & Snyder, 1974; Nunnari et al., 1987; Green, 1984). Although no explanation was given for this variability of effect among the different 'antagonists', it is now tempting to speculate that the inverse agonist behaviour of some may be responsible for the degree of 'Na+-shift' they exhibit.
Hence, upon retrospective consideration, extrapolation of this concept may shed a new light on the observed effects of Na+ ions. The increase in binding of some antagonists in the presence of Na+ ions, such as [3H]-naloxone to the μ opioid receptor (Pert & Snyder, 1974), ICI-174,864 to the δ opioid receptor (Appelmans et al., 1986), [3H]-yohimbine to the α2-AR (Nunnari et al., 1987) or [125I]-epidepride to the D2 receptor (Neve et al., 1990), may be indicative of the inverse agonistic properties of these ligands. The fact that naloxone has been proposed to act as an inverse opiate agonist in a guinea-pig ileum preparation (Cruz et al., 1996) is further support for this correlation.
Finally, one may note that apart from differential binding of ligands, the effect of Na+ on the R⇆R* equilibrium is also apparent in the reduction of basal levels of [35S]-GTPγS binding (Szekeres & Traynor, 1997) or GTPase activity (Gierschik et al., 1989; Costa et al., 1990) in in vitro assays. This reduction is not only similar to the effect of inverse agonists themselves on these assays (decrease of basal receptor activity), but confirms the presence of constitutively active receptors in these assay systems. Thus, although the presence of Na+ ions in radioligand binding assays may help to identify inverse agonists, their absence improves the ability of the [35S]-GTPγS assay to expose inverse agonism.
Tonically active systems: a connection with constitutive receptor activity?
In physiology there are many examples of receptor systems tonically regulating a certain effect. This 'tone' has traditionally been attributed to the presence of the endogenous agonist of the receptor system involved that interacts at a continuous level with its receptors. Interestingly, under the new light shed by the concepts of the two-state receptor in equilibrium, it is tempting to set forth constitutive activity of the receptor system as the potential mechanism of receptor-mediated tone regulation, instead of -or in addition to- the role of endogenous agonists.
As an example, the cannabinoid receptor system tonically regulates thermal nociceptive thresholds in mice. SR141716A, characterized as an inverse agonist at CB1 receptors (Landsman et al., 1998; MacLennan et al., 1998; Rinaldi-Carmona et al., 1998), produces hyperalgesia in mice (in contrast to agonists in this system that produce analgesia) by inhibiting this tone (Richardson et al., 1997). The same compound alone increased voltage-dependent Ca2+ currents in neurons microinjected with cloned CB1 receptor RNA, reversing the tonic CB1 receptor activity (Pan et al., 1998). Again, for an 'antagonist' to elicit such an effect some receptors must be tonically active. Since care was taken to ensure the absence of endogenous agonists in this experimental set-up, the 'tone' could only be attributed to constitutive receptor activity and its reversal only to 'antagonists' with inverse agonist properties.
Another related example is the marked relaxation by oxytocin receptor antagonists F314 and F792 of spontaneous contractility of human myometrium in vitro (Kinsler et al., 1996). It is interesting to postulate whether or not spontaneous uterine contractions during preterm labour are due to constitutive oxytocin receptor activity and whether, therefore, inverse agonists should be preferred over 'neutral' antagonists in treating such a condition. It should be mentioned in this respect that the levels of Gs vary dramatically in pregnancy, and hence the coupling with the oxytocin receptor. During gestation Gs α-subunit levels are substantially increased in human myometrium, whereas a significant downregulation of Gs is observed during parturition (Europe-Finner et al., 1994).
Naturally occurring inverse agonists
The potential existence of naturally occurring or endogenous inverse agonists would be another line of evidence in favour of the physiological relevance of inverse agonism. Although not without dispute, agouti protein is considered an endogenous inverse agonist. This protein consists of 131 amino acids and is encoded by the agouti gene. Lu et al. (1994) were the first to report that agouti protein competitively antagonized melanocyte stimulating hormone (α-MSH) binding to its receptor, currently referred to as the melanocortin MC1 receptor, with high affinity in the subnanomolar range. Siegrist et al. (1997) followed this observation and investigated the interactions between α-MSH, agouti protein, cyclic AMP elevating agents and phorbol ester on mouse B16 melanoma cells that endogenously express the MC1 receptor. Cell proliferation, measured in one of the assays, was inhibited by α-MSH. Also agouti protein dose-dependently inhibited B16 cell growth, thus acting as an agonist. However, low concentrations of α-MSH counteracted the growth inhibition induced by agouti protein. Furthermore, melanin production and MC1 receptor regulation were studied. Agouti was shown not only to decrease basal levels of melanin but also α-MSH-induced melanin production. In contrast to the cell proliferation assay, agouti protein had effects opposite to α-MSH, acting as an inverse agonist. However, the effects of α-MSH and agouti protein on receptor regulation were again comparable, i.e. both ligands downregulated the MC1 receptor over a similar time course and to a similar extent. The authors provided two possible explanations for these differential effects. Either agouti protein could be interacting at a second site downstream in the MSH signalling pathway, implying a second putative agouti receptor, or agouti protein may be regarded as an inverse agonist on the MC1 receptor. If this were the case agouti protein would interfere with a certain level of constitutive activation of the MC1 receptor. However, constitutive activity of this receptor was not clearly shown in either study, preventing an explicit demonstration of inverse agonism. Therefore, the conclusion that agouti protein indeed is an inverse agonist on the MC1 receptor cannot be drawn decisively at present.
Exendin-(9-39) is a truncated form of exendin-4, a peptide isolated from the venom of the lizard Heloderma suspectum. It acts as an inverse agonist on the murine glucagon-like peptide-1 (GLP-1) receptor (Serre et al., 1998). Exendin-(9-39) dose-dependently decreased basal cyclic AMP levels in murine β-cells, which endogenously express the GLP-1 receptor. Neither endogenous GLP-1 nor preproglucagon mRNA could be demonstrated in these cells. Therefore exendin-(9-39) did not act as a 'neutral' antagonist preventing endogenously produced GLP-1 from binding to the GLP-1 receptor. Moreover the inverse agonistic effect of exendin-(9-39) was also observed one step downstream the signalling pathway. In mouse β-cells GLP-1 receptor activation results in the stimulation of glucose-induced secretion of insulin. Exedin-(9-39) inhibited this insulin secretion, corroborating the concept that inverse agonism has strong physiological relevance. Inhibition of glucose-induced insulin secretion by exendin-(9-39) acting via human GLP-1 receptors has not been shown yet. In conclusion, exendin-(9-39) is an inverse agonist from an animal species, although acting on a receptor of another species. This finding could stimulate the search for truly 'endogenous' peptides as inverse agonists.
The opposite 'combination' has also been described. Interferon-γ-inducible protein 10 (IP-10) is an endogenous ligand, acting as an inverse agonist on a viral receptor. Human IP-10 was shown to inhibit the basal activiy of a GPCR encoded within the genome of Kaposi's sarcoma-associated herpes virus/human herpes virus 8 (Geras-Raaka et al., 1998). This GPCR shows homology with chemokine C-X-C receptors and is also referred to as ORF-74 (Rosenkilde et al., 1999). C-X-C receptors are the natural target of IP-10 where activation of these receptors results in IP production. However, on ORF-74, which is constitutively active, IP-10 acts as an inverse agonist, decreasing basal IP levels. Human monokine induced by interferon-γ (Mig) is also a C-X-C receptor agonist, but lacks the ability to decrease basal signalling of ORF-74.
Rosenkilde et al. (1999), when analysing various other C-X-C receptor ligands, such as Growth-Related Oncogenes, Stromal cell-Derived Factor 1α (SDF-1α) and viral Macrophage Inflammatory Protein-II (vMIP-II), showed that besides IP-10, SDF-1α and vMIP-II behaved as inverse agonists on ORF-74. However vMIP-II, like ORF-74 itself, is also encoded by the human herpes virus 8 and acts as an antagonist on multiple human chemokine receptors. Thus, endogenous ligands (agouti or IP-10) have been identified as inverse agonists although not always acting on receptors present in the same organism (e.g. IP-10 for ORF-74 or exendin-(9-39) for GLP-1).
Implications for pharmacotherapy and drug design
Since the emergence of the concept of inverse agonism, (re)classification of existing and new compounds in an agonist-antagonist-inverse agonist continuum has become necessary.
Although it is hard to avoid reference to 'neutral antagonists' vs to inverse agonists, we are now becoming more aware that such a class of compounds does not really exist, or if so it is extremely limited. That is because an accurate definition of a neutral antagonist would be reserved for a ligand with exactly the same affinity for the active and inactive receptor conformation, something probably very rare. On the contrary, it is easier to speculate on 'partial inverse agonists' with various degrees of efficacy and variable affinity for these two receptor states. Thus, maybe we should realise that there is only one class of ligands after all, i.e. agonists, whether they are full, partial, partial inverse or full inverse. However, for convenience we will continue hereby to use the term 'antagonist' and /or 'neutral antagonist'.
It is now obvious that both mechanism of action and side-effect profile may largely vary for the two classes, antagonists and inverse agonists, especially in pathological conditions. Therefore it is imperative in drug therapy to clearly identify therapeutic agents that may act as either antagonists or inverse agonists. In some cases desired effects may be obtained by treatment with an antagonist, while in other situations an inverse agonist might be more effective. We will now discuss some observations that may have clinical relevance or strong implications for lead finding in drug discovery.
Effects of long-term treatment with inverse agonists
It has been shown for various receptor types that long-term treatment with inverse agonists is associated with upregulation of the receptor involved. The increase in receptor density may be associated with drug tolerance and withdrawal effects. For example, the marketed histamine 'antagonists' cimetidine, ranitidine and famotidine, which have been shown to be inverse agonists, upregulated rat histamine H2 receptor numbers (Smit et al., 1996). Theoretically, upregulation of the receptor involved could account for withdrawal effects and deterioration of the disease upon cessation of the drug. In such a case, treatment with a neutral antagonist rather than with an inverse agonist should be considered a more rational choice of therapy. However, upregulation by cimetidine was less pronounced in studies with human H2 receptors (150% over control cells versus 180% at rat H2 receptors) (Alewijnse et al., 1998). Moreover, a correlation between the level of upregulation of the receptor and withdrawal effects has not been found yet. To our knowledge, clinical relevance of histamine H2 receptor upregulation has not been reported so far.
Upregulation of β-adrenoceptors upon antagonist treatment has been reported on many occasions. In one of the earlier studies the receptor density on membranes prepared from lymphocytes of healthy human subjects was determined after 8 days of drug treatment (Molinoff & Aarons, 1983). Propranolol increased β-AR density 140% over basal, while the agonists ephedrine and terbutaline decreased receptor density to approximately 50%. Pindolol, a so-called β-blocker which can express intrinsic sympathomimetic activity (hence, a partial agonist), also reduced receptor levels, but to a lesser extent. Abrupt discontinuation of propranolol treatment from patients with ischaemic heart disease is known to result in withdrawal effects such as increasingly severe and frequent anginal attacks, arrythmias and myocardial infarction. These side effects might be correlated with the increase in receptor number. Since pindolol decreased the β-AR density, compared to an increase of receptors by propranolol, it was suggested that discontinuation of long-term pindolol administration would not lead to outspoken withdrawal symptoms. However, the effect of drug treatment on receptor density appeared different in other studies with different experimental settings. For example, Hughes et al. (1988) reported downregulation of β-AR by propranolol in cultured lymphoma and muscle cells. Extrapolation of observations from such studies to the clinic is difficult and should be done with great care.
Upregulation of opiate receptors has also been reported. Quantitative autoradiography after chronic naloxone infusion, showed increased amounts of κ opioid receptors in specific regions in rat brain (Morris et al., 1988). Furthermore, naloxone has been used to try and elucidate the mechanism of opiate tolerance/dependence and withdrawal (Cruz et al., 1996). Its effects were assayed in situ in both morphine-treated and control guinea-pig ilea. In this study, naloxone counteracted morphine-induced neurodepression, but it also caused an (undesired) abstinence response. This withdrawal phenomenon, especially seen after bolus dosing, was interpreted as an abrupt change from the active (R*) to the inactive (R) receptor conformation. In this hypothesis tolerance to morphine is a reflection of the persistence of the active state of the receptor.
Long-term treatment with inverse agonists might have effects on other receptors as well. The G protein involved or receptors sharing the same signalling pathway as the target receptor could be affected. Bouaboula et al. (1999) described cross talk between different receptors via Gi proteins. SR144,528 not only acted as an inverse agonist on the cannabinoid CB2 receptor (see above), it also prevented MAPK activation in response to insulin or lysophosphatidic acid by inhibiting Gi activity. Furthermore, Western blot analysis revealed Gi protein upregulation upon sustained treatment with SR144,528, which was reversed by washing and further exposure of the cells to the agonist CP-55940. To account for these observations, the existence of an inactive CB2/Gi protein complex, in which the Gi protein is physically trapped and thus unavailable for other receptors, was suggested.
MacEwan & Milligan (1996), however, did not observe upregulation of Gs protein upon long-term treatment with inverse agonists for the β2-AR, expressed in NG108-15 cells. Levels of other G protein subtypes (Gq and G11) were also unaffected by sustained treatment with inverse agonists. Therefore, the regulatory properties of SR144528 on G protein levels might not be a common feature of all inverse agonists.
Recently, Berg et al. (1999) reported on the long-term treatment of the serotonin 5-HT2c receptor. In CHO cells, expressing the 5-HT2c receptor at a relatively low expression level (∼250 fmol mg−1 protein), prolonged incubation with the inverse agonist SB206553 led to an increased responsiveness of the receptor to agonists (homologous sensitization). This was measured as an increase in maximal IP production induced by the agonist DOI. Interestingly, IP production was also enhanced by ATP, mediated via endogenous purinergic P2 receptors (heterologous sensitization). These findings are quite comparable to the cross talk between receptors discussed by Bouaboula et al. (1999). In conclusion, long-term effects may be a sensitive measure of ligand properties.
Inverse agonists and somatic receptor mutations
Somatic receptor mutations leading to constitutively active receptors are a causal factor in certain diseases (Table 4). Inverse agonists may be beneficial here, since they would decrease the high basal activity induced by the mutation, while antagonists would have no effect. These mutations have been reviewed by Spiegel (1996), hence, we will discuss one example only.
Table 4.
Diseases associated with activating mutations (Spiegel, 1996)

The mutations H223R and T410P in the human receptor for parathyroid hormone (PTH) and parathyroid hormone-related peptide (PTHrP) have been found in patients with Jansen's metaphyseal chondrodysplasia. Both mutant receptors, expressed in COS-7 cells, caused an increase in basal cyclic AMP production, but to a different extent. Furthermore, they had apparent binding affinities for the natural ligands (PTH and PTHrP) that were approximately two times higher than those of the wild type receptor (Schipani et al., 1996). Gardella et al. (1996) used these two mutant receptors to screen for peptide ligands as inverse agonists.
An interesting and somewhat related phenomenon is the RNA editing of receptors. This is a post-transcriptional modification of RNA transcripts due to the enzymatic conversion of adenosine to inosine. Niswender et al. (1999) demonstrated that as a result, the human 5HT2C receptor exists in several isoforms in human brain. These isoforms all displayed less constitutive activity than the non-edited receptor upon expression in NIH3T3 fibroblasts and lower agonist potencies. This may imply that RNA editing is a physiological mechanism for fine-tuning synaptic signalling.
Inverse agonists and 'auto-receptor antibodies'
Inverse agonists may also play an important role in the treatment of autoimmune diseases. Mijares et al. (1996) analysed an antibody against a peptide corresponding to the second extracellular loop of the human β2-AR. Monoclonal antibodies for this peptide were obtained from rabbits and their effects on guinea-pig cardiomyocytes were studied. A whole-cell patch clamp technique was used to assess the influx of Ca2+ ions by L-type calcium channels (ICa). Zinterol, a specific β2-selective partial agonist, increased ICa 14% compared to the 100% increase in ICa induced by isoprenaline, a non-selective full agonist. Addition of the purified antipeptide antibodies to the cardiomyocytes increased ICa to a similar extent as zinterol. This effect appeared to be mediated by β-adrenoceptors and was β2-selective. Addition of the neutral antagonist alprenolol did not decrease antibody-stimulated ICa, and on the contrary a significant increase was observed. ICI-118,551, an inverse agonist, blocked the effect of the antibodies completely, decreasing ICa. The results were interpreted in the light of the two-state receptor model based on the hypothesis that the antibodies recognized an epitope which is only presented at the active conformation of the receptor (R*). ICI-118,551 would shift the equilibrium towards the inactive conformation of the receptor (R), covering up the epitope recognized by the antibody. Consequently, the antibody is no longer able to bind and activate the receptor and therefore, an antibody-mediated change in ICa is not observed. Alprenolol, on the other hand, does not change the existing equilibrium between R and R* in this concept. Since some of the receptors are present in the R* conformation, the antibodies can still recognize the epitope on the receptor and elicit an increase of ICa.
It has been reported that autoantibodies against the second extracellular loop of certain GPCRs are involved in the pathology of human autoimmune diseases. Examples are listed in Table 5. These autoantibodies are believed to behave in a similar way compared to the anti-peptide antibodies described above. If the autoantibodies do activate the receptor, drug therapy with inverse agonists could be more effective than treatment with neutral antagonists. Moreover, conformation 'sensitive' antibodies can be used as a research tool to distinguish inverse agonists from neutral antagonists.
Table 5.
Activating antibodies in autoimmune disease

Inverse agonism as screening tool in drug discovery
Nowadays molecular genetic approaches have provided numerous orphan GPCRs with unknown function and/or ligand. Therefore robust functional assays need to be developed, preferably in high throughput screening formats. The concept of inverse agonism might provide new opportunities in such screening strategies. A functional assay in which a certain level of spontaneous activity is established could not only identify activating compounds (agonists) but also compounds with opposite effect (inverse agonists). In a classical screening assay, i.e. without constitutive activity, the latter ligands would not be detected at all. Screening strategies implementing inverse agonism may thus be more successful, significantly expanding 'hit' chances. Depending on the role of the orphan receptor, one of the classes can be further explored to discover and/or design new therapeutics. Furthermore, if inverse agonists are identified in such an assay, the medicinal chemist may use them as lead structures for further optimization and the discovery of e.g. more or less 'neutral' antagonists.
Epilogue
The physiological relevance of inverse agonism is not only becoming clearer, but is slowly taking a place and establishing a role in drug therapy. Further, it dictates a new field in drug research and especially in drug design, since it is obvious that new structure-activity-relationships for inverse agonists versus those for neutral antagonists must be sought for and established.
Acknowledgments
The authors acknowledge support from the EU BIOMED2 programme 'Inverse agonism. Implications for drug design' (#BMH4-CT97-2152). We thank our colleagues in this programme Tommaso Costa, Susanna Cotecchia, Rob Leurs, Martin Lohse, Graeme Milligan, Nigel Shankley and Sir James Black. Their comments, discussions and critical reading of this manuscript were essential. A. Kourounakis also wishes to gratefully acknowledge financial support from the EU Biotechnology programme 'Novel concepts in the interaction between adenosine receptors and ligands: inverse agonists and allosteric enhancers' (#BIO4-CT97-5138).
Abbreviations
- Ado
adenosine
- AM630
6-iodopravadoline
- AR
adrenoceptor
- BK
bradykinin
- CAM
constitutively active mutant
- CB
cannabinoid
- CHO
Chinese hamster ovary
- COS
African green monkey kidney fibroblast-like cell line
- CP-55940
(−)-3-[2-hydroxy-4-(1,1-dimethylhyptyl)phenyl]-4(3-hydroxypropyl)cyclohexan-1-ol
- CT
calcitonin
- 5-CT
5-carboxamidotryptamine
- D
dopamine
- DMCM
methyl 6,7-dimethoxy-4-ethyl-β-carboline-e-carboxylate
- DOI
(±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane
- DPCPX
1,3-DiPropyl-8-cyclopentyladenosine
- F314
[Mpa, D-Tyr(Ethyl),Thr, Orn]-oxytocin
- FMLP
N-formyl-L-methionyl-L-leucyl-L-phenylalanine
- FP
formyl peptide
- FSH
follicle-stimulating hormone
- GLP-1
glucagon-like peptide-1
- GPCR
G protein-coupled receptor
- GTP
guanosine-5′-triPhosphate
- GTPase
guanosine-5′-triphosphatase
- [35S]-GTPγS
-[35S] guanosine 5′-0-(thiotriphosphate)
- H
histamine
- HEK293
human embryonic kidney
- H.E.L. 92.1.17
human erythroleukemia
- HL-60
human leukemia
- ICa
L-type calcium currents
- ICI-118,551
(±)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol
- ICI-174,864
N-N-diallyl-Tyr-Aib-Aib-Phe-Thr
- IP
inositol phosphates
- IP-10
interferon-γ-inducible protein 10
- LH
luteinizing hormone
- M
muscarinic
- MAPK
mitogen-activated protein kinase
- MC
melanocortin
- Mig
monokine induced by interferon-γ
- α-MSH
melanocyte stimulating hormone
- NG108-15
neuroblastoma X glioma hybrid
- O
opioid
- P
purinergic
- PTH
parathyroid hormone
- PTHrP
parathyroid hormone-related peptide
- QNB
R(−)-quinuclidinylbenzilate
- RIN5AH
rat insulinoma-derived
- Sf9
Spodoptera frugiperda 9
- SB206553
N-3-pyridinyl-3,5-dihydro-5-methylbenzo[1,2-b:4,5-b′]dipyrrole-1(2H)-carboxamide hydrochloride
- SDF-1α
stromal cell-derived factor 1α
- SR141,716A
N-(piperidino-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide
- SR144,528
N-([1s]-endo-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl)-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide
- vMIP-II
viral macrophage inflammatory protein-II
- WAY100,635
N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)-cyclohexane-carboxamide
- WIN55,212-2
R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-napthalenyl)methanone mesylate
- WB4101
2-(2,6-dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane
- XAC
xanthin amino congener
References
- ALEWIJNSE A.E., SMIT M.J., HOFFMANN M., VERZIJL D., TIMMERMAN H., LEURS R. Constitutive activity and structural instability of the wild type human H2 receptor. J. Neurochem. 1998;71:799–807. doi: 10.1046/j.1471-4159.1998.71020799.x. [DOI] [PubMed] [Google Scholar]
- APPELMANS N., CARROLL J.A., RANCE M.J., SIMON E.J., TRAYNOR J.R. Sodium ions increase the binding of the antagonist peptide ICI 174864 to the delta opiate receptor. Neuropeptides. 1986;7:139–143. doi: 10.1016/0143-4179(86)90089-2. [DOI] [PubMed] [Google Scholar]
- BERG K.A., STOUT B.D., CROPPER J.D., MAAYANI S., CLARKE W.P. Novel actions of inverse agonists on 5-HT2C receptor systems. Mol. Pharmacol. 1999;55:863–872. [PubMed] [Google Scholar]
- BERRIE C.P., BIRDSALL N.J., BURGEN A.S., HULME E.C. Guanine nucleotides modulate muscarinic receptor binding in the heart. Biochem. Biophys. Res. Commun. 1979;87:1000–1005. doi: 10.1016/s0006-291x(79)80006-6. [DOI] [PubMed] [Google Scholar]
- BOND R.A., LEFF P., JOHNSON T.D., MILANO C.A., ROCKMAN H.A., MCMINN T.R., APPARSUNDARAM S., HYEK M.F., KENAKIN T.P., ALLEN L.F., LEFKOWITZ R.J. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the beta(2)-adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- BOUABOULA M., DESNOYER N., CARAYON P., COMBES T., CASELLAS P. Gi protein modulation induced by a selective inverse agonist for the peripheral cannabinoid receptor CB2: implication for intracellular signalization cross-regulation. Mol. Pharmacol. 1999;55:473–480. [PubMed] [Google Scholar]
- BOUABOULA M., PERRACHON S., MILLIGAN L., CANAT X., RINALDI-CARMONA M., PORTIER M., BARTH F., CALANDRA B., PECCEU F., LUPKER J., MAFFRAND J.P., LE FUR G., CASELLAS P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- BRAESTRUP C., SCHMIECHEN R., NEEF G., NIELSEN M., PETERSEN E.N. Interaction of convulsive ligands with benzodiazepine receptors. Science. 1982;216:1241–1243. doi: 10.1126/science.6281892. [DOI] [PubMed] [Google Scholar]
- BURGISSER E., DE LEAN A., LEFKOWITZ R.J. Reciprocal modulation of agonist and antagonist binding to muscarinic cholinergic receptor by guanine nucleotide. Proc. Natl. Acad. Sci. U.S.A. 1982;79:1732–1736. doi: 10.1073/pnas.79.6.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURSTEIN E.S., SPALDING T.A., BRANN M.R. Pharmacology of muscarinic receptor subtypes constitutively activated by G proteins. Mol. Pharmacol. 1997;51:312–319. doi: 10.1124/mol.51.2.312. [DOI] [PubMed] [Google Scholar]
- CHANG R.S.L., SYNDER S.H. Histamine H1-receptor binding sites in guinea pig brain membranes: regulation of agonist interactions by guanine nucleotides and cations. J. Neurochem. 1980;34:916–922. doi: 10.1111/j.1471-4159.1980.tb09666.x. [DOI] [PubMed] [Google Scholar]
- CHIALE P.A., ROSENBAUM M.B., ELIZARI M.V., HJALMARSON A., MAGNUSSON Y., WALLUKAT G., HOEBEKE J. High prevalence of antibodies against beta 1- and beta 2-adrenoceptors in patients with primary electrical cardiac abnormalities. J. Am. Coll. Cardiol. 1995;26:864–869. doi: 10.1016/0735-1097(95)00262-2. [DOI] [PubMed] [Google Scholar]
- CHIDIAC P., HEBERT T.E., VALIQUETTE M., DENNIS M., BOUVIER M. Inverse agonist activity of beta-adrenergic antagonists. Mol. Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- COHEN D.P., THAW C.N., VARMA A., GERSHENGORN M.C., NUSSENZVEIG D.R. Human calcitonin receptors exhibit agonist-independent (constitutive) signaling activity. Endocrinology. 1997;138:1400–1405. doi: 10.1210/endo.138.4.5046. [DOI] [PubMed] [Google Scholar]
- COSTA T., HERZ A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7321–7325. doi: 10.1073/pnas.86.19.7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSTA T., LANG J., GLESS C., HERZ A. Spontaneous association between opioid receptors and GTP-binding regulatory proteins in native membranes: specific regulation by antagonists and sodium ions. Mol. Pharmacol. 1990;37:383–394. [PubMed] [Google Scholar]
- COTECCHIA S., EXUM S., CARON M.G., LEFKOWITZ R.J. Regions of the alpha 1-adrenergic receptor involved in coupling to phosphatidylinositol hydrolysis and enhanced sensitivity of biological function. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2896–2900. doi: 10.1073/pnas.87.8.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRUZ S.L., VILLARREAL J.E., VOLKOW N.D. Further evidence that naloxone acts as an inverse opiate agonist: implications for drug dependence and withdrawal. Life Sci. 1996;58:L381–L389. doi: 10.1016/0024-3205(96)00250-0. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., STADEL J.M., LEFKOWITZ R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J. Biol. Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- DEL CASTILLO J., KATZ B. Interaction at end-plate receptors between different choline derivatives. Proc. Roy. Soc. Lond. B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- EHLERT F.J. The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol. Pharmacol. 1985;28:410–421. [PubMed] [Google Scholar]
- ELIES R., FERRARI I., WALLUKAT G., LEBESGUE D., CHIALE P., ELIZARI M., ROSENBAUM M., HOEBEKE J., LEVIN M.J. Structural and functional analysis of the B cell epitopes recognized by anti-receptor autoantibodies in patients with Chagas' disease. J. Immunol. 1996;157:4203–4211. [PubMed] [Google Scholar]
- ENG H., MAGNUSSON Y., MATELL G., LEFVERT A.K., SAPONJA R., HOEBEKE J. Beta 2-adrenergic receptor antibodies in myasthenia gravis. J. Autoimmun. 1992;5:213–227. doi: 10.1016/0896-8411(92)90201-z. [DOI] [PubMed] [Google Scholar]
- EUROPE-FINNER G.N., PHANEUF S., TOLKOVSKY A.M., WATSON S.P., LOPEZ BERNAL A. Down-regulation of G alpha s in human myometrium in term and preterm labor: a mechanism for parturition. J. Clin. Endocrinol. Metab. 1994;79:1835–1839. doi: 10.1210/jcem.79.6.7989491. [DOI] [PubMed] [Google Scholar]
- FU M.L., HOEBEKE J., MATSUI S., MATOBA M., MAGNUSSON Y., HEDNER T., HERLITZ H., HJALMARSON A. Autoantibodies against cardiac G-protein-coupled receptors define different populations with cardiomyopathies but not with hypertension. Clin. Immunol. Immunopathol. 1994;72:15–20. doi: 10.1006/clin.1994.1101. [DOI] [PubMed] [Google Scholar]
- GARDELLA T.J., LUCK M.D., JENSEN G.S., SCHIPANI E., POTTS J.T., JR, JËPPNER H. Inverse agonism of amino-terminally truncated parathyroid hormone (PTH) and PTH-related peptide (PTHrP) analogs revealed with constitutively active mutant PTH/PTHrP receptors. Endocrinology. 1996;137:3936–3941. doi: 10.1210/endo.137.9.8756569. [DOI] [PubMed] [Google Scholar]
- GERAS-RAAKA E., VARMA A., HO H., CLARK LEWIS I., GERSHENGORN M.C. Human interferon-gamma-inducible protein 10 (IP-10) inhibits constitutive signaling of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J. Exp. Med. 1998;188:405–408. doi: 10.1084/jem.188.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIERSCHIK P., SIDIROPOULOS D., STEISSLINGER M., JAKOBS K.H. Na+ regulation of formyl peptide receptor-mediated signal transduction in HL 60 cells. Evidence that the cation prevents activation of the G-protein by unoccupied receptors. Eur. J. Pharmacol. 1989;172:481–492. doi: 10.1016/0922-4106(89)90031-x. [DOI] [PubMed] [Google Scholar]
- GÖTZE K., JAKOBS K.H. Unoccupied beta-adrenoceptor-induced adenylyl cyclase stimulation in turkey erythrocyte membranes. Eur. J. Pharmacol. 1994;268:151–158. doi: 10.1016/0922-4106(94)90184-8. [DOI] [PubMed] [Google Scholar]
- GREEN R.D. Reciprocal modulation of agonist and antagonist binding to inhibitory adenosine receptors by 5′-guanylylimidodiphosphate and monovalent cations. J. Neurosci. 1984;4:2472–2476. doi: 10.1523/JNEUROSCI.04-10-02472.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIFFON N., PILON C., SAUTEL F., SCHWARTZ J.C., SOKOLOFF P. Antipsychotics with inverse agonist activity at the dopamine D3 receptor. J. Neural. Transm. 1996;103:1163–1175. doi: 10.1007/BF01271201. [DOI] [PubMed] [Google Scholar]
- GUILLET J.G., LENGAGNE R., MAGNUSSON Y., TATE K., STROSBERG A.D., HOEBEKE J. Induction of a pharmacologically active clonotypic B cell response directed to an immunogenic region of the human beta 2-adrenergic receptor. Clin. Exp. Immunol. 1992;89:461–467. doi: 10.1111/j.1365-2249.1992.tb06981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANF R., LI Y., SZABO G., FISCHMEISTER R. Agonist-independent effects of muscarinic antagonists on Ca2+ and K+ currents in frog and rat cardiac cells. J. Physiol. Lond. 1993;461:743–765. doi: 10.1113/jphysiol.1993.sp019539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILF G., JAKOBS K.H. Agonist-independent inhibition of G protein activation by muscarinic acetylcholine receptor antagonists in cardiac membranes. Eur. J. Pharmacol. 1992;225:245–252. doi: 10.1016/0922-4106(92)90026-r. [DOI] [PubMed] [Google Scholar]
- HORSTMAN D.A., BRANDON S., WILSON A.L., GUYER C.A., CRAGOE E.J., JR, LIMBIRD L.E. An aspartate conserved among G-protein receptors confers allosteric regulation of alpha 2-adrenergic receptors by sodium. J. Biol. Chem. 1990;265:21590–21595. [PubMed] [Google Scholar]
- HUGHES R.J., MAHAN L.C., INSEL P.A. Certain beta-blockers can decrease beta-adrenergic receptor number: II. Down-regulation of receptor number by alprenolol and propranolol in cultured lymphoma and muscle cells. Circ. Res. 1988;63:279–285. doi: 10.1161/01.res.63.2.279. [DOI] [PubMed] [Google Scholar]
- IJZERMAN A.P., VAN DER WENDEN E.M., ROELEN H.C.P.F., MATHÔT R.A.A., VON FRIJTAG DRABBE KÜNZEL J.K.Partial agonists for adenosine receptors Perspectives in Receptor Research 1996Amsterdam: Elsevier; 181–192.ed. Giardinà, D., Piergentili, A. & Pigini, M. pp [Google Scholar]
- JAKUBÍK J., BAčÁAKOVÁ L., EL-FAKAHANY E.E., TUčEK S. Constitutive activity of the M1-M4 subtypes of muscarinic receptors in transfected CHO cells and of muscarinic receptors in the heart cells revealed by negative antagonists. FEBS Lett. 1995;377:275–279. doi: 10.1016/0014-5793(95)01360-1. [DOI] [PubMed] [Google Scholar]
- JANSSON C.C., KUKKONEN J.P., NÄSMAN J., HUIFANG G., WURSTER S., VIRTANEN R., SAVOLA J.M., COCKCROFT V., ÅKERMAN K.E. Protean agonism at alpha2A-adrenoceptors. Mol. Pharmacol. 1998;53:963–968. [PubMed] [Google Scholar]
- KINSLER V.A., THORNTON S., ASHFORD M.L., MELIN P., SMITH S.K. The effect of the oxytocin antagonists F314 and F792 on the in vitro contractility of human myometrium. Br. J. Obstet. Gynaecol. 1996;103:373–375. doi: 10.1111/j.1471-0528.1996.tb09745.x. [DOI] [PubMed] [Google Scholar]
- LANDSMAN R.S., MAKRIYANNIS A., DENG H., CONSROE P., ROESKE W.R., YAMAMURA H.I. AM630 is an inverse agonist at the human cannabinoid CB1 receptor. Life Sci. 1998;62:L109–L113. doi: 10.1016/s0024-3205(97)01187-9. [DOI] [PubMed] [Google Scholar]
- LEEB-LUNDBERG L.M.F., MATHIS S.A., HERZIG M.C.S. Antagonists of bradykinin that stabilize a G-protein-uncoupled state of the B2 receptor act as inverse agonists in rat myometrial cells. J. Biol. Chem. 1994;269:25970–25973. [PubMed] [Google Scholar]
- LEFF P. The two-state model of receptor activation. Trends Pharmacol. Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- LEURS R., SMIT M.J., ALEWIJNSE A.E., TIMMERMAN H. Agonist-independent regulation of constitutively active G-protein-coupled receptors. Trends Biochem. Sci. 1998;23:418–422. doi: 10.1016/s0968-0004(98)01287-0. [DOI] [PubMed] [Google Scholar]
- LOHSE M.J., LENSCHOW V., SCHWABE U. Two affinity states of Ri adenosine receptors in brain membranes. Analysis of guanine nucleotide and temperature effects on radioligand binding. Mol. Pharmacol. 1984;26:1–9. [PubMed] [Google Scholar]
- LU D., WILLARD D., PATEL I.R., KADWELL S., OVERTON L., KOST T., LUTHER M., CHEN W., WOYCHIK R.P., WILKISON W.O.Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor Nature 1994371799–802.et al [DOI] [PubMed] [Google Scholar]
- MACEWAN D.J., MILLIGAN G. Inverse agonist-induced up-regulation of the human beta2-adrenoceptor in transfected neuroblastoma X glioma hybrid cells. Mol. Pharmacol. 1996;50:1479–1486. [PubMed] [Google Scholar]
- MACLENNAN S.J., REYNEN P.H., KWAN J., BONHAUS D.W. Evidence for inverse agonism of SR141716A at human recombinant cannabinoid CB1 and CB2 receptors. Br. J. Pharmacol. 1998;124:619–622. doi: 10.1038/sj.bjp.0701915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGNUSSON Y., MARULLO S., HOYER S., WAAGSTEIN F., ANDERSSON B., VAHLNE A., GUILLET J.G., STROSBERG A.D., HJALMARSON A., HOEBEKE J. Mapping of a functional autoimmune epitope on the beta 1-adrenergic receptor in patients with idiopathic dilated cardiomyopathy. J. Clin. Invest. 1990;86:1658–1663. doi: 10.1172/JCI114888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEWES T., DUTZ S., RAVENS U., JAKOBS K.H. Activation of calcium currents in cardiac myocytes by empty beta-adrenoceptors. Circulation. 1993;88:2916–2922. doi: 10.1161/01.cir.88.6.2916. [DOI] [PubMed] [Google Scholar]
- MIJARES A., LEBESGUE D., ARGIBAY J., HOEBEKE J. Anti-peptide antibodies sensitive to the 'active' state of the beta2-adrenergic receptor. FEBS Lett. 1996;399:188–191. doi: 10.1016/s0014-5793(96)01321-x. [DOI] [PubMed] [Google Scholar]
- MILLIGAN G., MACEWAN D.J., MERCOURIS M., MULLANEY I. Inverse agonism at adrenergic and opioid receptors: studies with wild type and constitutively active mutant receptors. Receptors Channels. 1997;5:209–213. [PubMed] [Google Scholar]
- MOLINOFF P.B., AARONS R.D. Effects of drugs on beta-adrenergic receptors on human lymphocytes. J. Cardiovasc. Pharmacol. 1983;5:S63–S67. doi: 10.1097/00005344-198300051-00010. [DOI] [PubMed] [Google Scholar]
- MONOD J., WYMAN J., CHANGEAUX J-P. On the nature of allosteric transitions: a plausible model. J. Biol. Chem. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- MORRIS B.J., MILLAN M.J., HERZ A. Antagonist-induced opioid receptor up-regulation. II. Regionally specific modulation of mu, delta and kappa binding sites in rat brain revealed by quantitative autoradiography. J. Pharmacol. Exp. Ther. 1988;247:729–736. [PubMed] [Google Scholar]
- NAGARAJA S., IYER S., LIU X., EICHBERG J., BOND R.A. Treatment with inverse agonists enhances baseline atrial contractility in transgenic mice with beta2-adrenoceptor activation. Br. J. Pharmacol. 1999;127:1099–1104. doi: 10.1038/sj.bjp.0702645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEVE K.A., HENNINGSEN R.A., KINZIE J.M., DE PAULIS T., SCHMIDT D.E., KESSLER R.M., JANOWSKY A. Sodium-dependent isomerization of dopamine D2 receptors characterized using [125I]epidepride, a high-affinity substituted benzamide ligand. J. Pharmacol. Exp. Ther. 1990;252:1108–1116. [PubMed] [Google Scholar]
- NEWMAN-TANCREDI A., CONTE C., CHAPUT C., SPEDDING M., MILLAN M.J. Inhibition of the constitutive activity of human 5-HT1A receptors by the inverse agonist, spiperone but not the neutral antagonist, WAY 100,635. Br. J. Pharmacol. 1997;120:737–739. doi: 10.1038/sj.bjp.0701025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NISWENDER C.M., COPELAND S.C., HERRICK-DAVIS K., EMESON R.B., SANDERS-BUSH E. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J. Biol. Chem. 1999;274:9472–9478. doi: 10.1074/jbc.274.14.9472. [DOI] [PubMed] [Google Scholar]
- NOGUERA M.A., IVORRA M.D., D'OCON P. Functional evidence of inverse agonism in vascular smooth muscle. Br. J. Pharmacol. 1996;119:158–164. doi: 10.1111/j.1476-5381.1996.tb15689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUNNARI J.M., REPASKE M.G., BRANDON S., CRAGOE E.J., JR, LIMBIRD L.E. Regulation of porcine brain alpha 2-adrenergic receptors by Na+, H+ and inhibitors of Na+/H+ exchange. J. Biol. Chem. 1987;262:12387–12392. [PubMed] [Google Scholar]
- PAN X., IKEDA S.R., LEWIS D.L. SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2+ currents by reversal of tonic CB1 cannabinoid receptor activity. Mol. Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- PERT C.B., SNYDER S.H. Opiate receptor binding of agonists and antagonists affected differentially by sodium. Mol. Pharmacol. 1974;10:868–870. [Google Scholar]
- POZVEK G., HILTON J.M., QUIZA M., HOUSSAMI S., SEXTON P.M. Structure/function relationships of calcitonin analogues as agonists, antagonists, or inverse agonists in a constitutively activated receptor cell system. Mol. Pharmacol. 1997;51:658–665. doi: 10.1124/mol.51.4.658. [DOI] [PubMed] [Google Scholar]
- RAMKUMAR V., STILES G.L. Reciprocal modulation of agonist and antagonist binding to A1 adenosine receptors by guanine nucleotides is mediated via a pertussis toxin-sensitive G protein. J. Pharmacol. Exp. Ther. 1988;246:1194–1200. [PubMed] [Google Scholar]
- RICHARDSON J.D., AANONSEN L., HARGREAVES K.M. SR 141716A, a cannabinoid receptor antagonist, produces hyperalgesia in untreated mice. Eur. J. Pharmacol. 1997;319:R3–R4. doi: 10.1016/s0014-2999(96)00952-1. [DOI] [PubMed] [Google Scholar]
- RINALDI-CARMONA M., LE DUIGOU A., OUSTRIC D., BARTH F., BOUABOULA M., CARAYON P., CASELLAS P., LE FUR G. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J. Pharmacol. Exp. Ther. 1998;287:1038–1047. [PubMed] [Google Scholar]
- ROSENKILDE M.M., KLEDAL T.N., BRÄUNER-OSBORNE H., SCHWARTZ T.W. Agonists and inverse agonists for the herpesvirus 8-encoded constitutively active seven-transmembrane oncogene product, ORF-74. J. Biol. Chem. 1999;274:956–961. doi: 10.1074/jbc.274.2.956. [DOI] [PubMed] [Google Scholar]
- SAMAMA P., BOND R.A., ROCKMAN H.A., MILANO C.A., LEFKOWITZ R.J. Ligand-induced overexpression of a constitutively active beta2-adrenergic receptor: pharmacological creation of a phenotype in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 1997;94:137–141. doi: 10.1073/pnas.94.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMAMA P., COTECCHIA S., COSTA T., LEFKOWITZ R.J. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- SCHIPANI E., LANGMAN C.B., PARFITT A.M., JENSEN G.S., KIKUCHI S., KOOH S.W., COLE W.G., JÜPPNER H. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen's metaphyseal chondrodysplasia [see comments] N. Engl. J. Med. 1996;335:708–714. doi: 10.1056/NEJM199609053351004. [DOI] [PubMed] [Google Scholar]
- SERRE V., DOLCI W., SCHAERER E., SCROCCHI L., DRUCKER D., EFRAT S., THORENS B. Exendin-(9-39) is an inverse agonist of the murine glucagon-like peptide-1 receptor: implications for basal intracellular cyclic adenosine 3′,5′-monophosphate levels and beta-cell glucose competence. Endocrinology. 1998;139:4448–4454. doi: 10.1210/endo.139.11.6295. [DOI] [PubMed] [Google Scholar]
- SIEGRIST W., DROZDZ R., COTTI R., WILLARD D.H., WILKISON W.O., EBERLE A.N. Interactions of alpha-melanotropin and agouti on B16 melanoma cells: evidence for inverse agonism of agouti. J. Recept. Signal Transduct. Res. 1997;17:75–98. doi: 10.3109/10799899709036595. [DOI] [PubMed] [Google Scholar]
- SMIT M.J., LEURS R., ALEWIJNSE A.E., BLAUW J., VAN NIEUW AMERONGEN G.P., VAN DE VREDE Y., ROOVERS E., TIMMERMAN H. Inverse agonism of histamine H2 antagonist accounts for upregulation of spontaneously active histamine H2 receptors. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6802–6807. doi: 10.1073/pnas.93.13.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPIEGEL A.M. Defects in G protein-coupled signal transduction in human disease. Annu. Rev. Physiol. 1996;58:143–170. doi: 10.1146/annurev.ph.58.030196.001043. [DOI] [PubMed] [Google Scholar]
- STADEL J.M., WILSON S., BERGSMA D.J. Orphan G protein-coupled receptors: a neglected opportunity for pioneer drug discovery. Trends Pharmacol. Sci. 1997;18:430–437. doi: 10.1016/s0165-6147(97)01117-6. [DOI] [PubMed] [Google Scholar]
- STILES G.L. A1 adenosine receptor-G protein coupling in bovine brain membranes: effects of guanine nucleotides, salt, and solubilization. J. Neurochem. 1988;51:1592–1598. doi: 10.1111/j.1471-4159.1988.tb01129.x. [DOI] [PubMed] [Google Scholar]
- STRÖHER M., NANOFF C., SCHÜTZ W. Differences in the GTP-regulation of membrane-bound and solubilized A1-adenosine receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989;340:87–92. doi: 10.1007/BF00169212. [DOI] [PubMed] [Google Scholar]
- SZEKERES P.G., TRAYNOR J.R. Delta opioid modulation of the binding of guanosine-5′-O-(3-[S-35]thio)triphosphate to NG108-15 cell membranes: Characterization of agonist and inverse agonist effects. J. Pharmacol. Exp. Ther. 1997;283:1276–1284. [PubMed] [Google Scholar]
- TIAN W.-N., DUZIC E., LANIER S.M., DETH R.C. Determinants of alpha 2-adrenergic receptor activation of G proteins: evidence for a precoupled receptor/G protein state. Mol. Pharmacol. 1994;45:524–531. [PubMed] [Google Scholar]
- TIBERI M., CARON M.G. High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J. Biol. Chem. 1994;269:27925–27931. [PubMed] [Google Scholar]
- TSAI B.S., LEFKOWITZ R.J. Agonist-specific effects of monovalent and divalent cations on adenylate cyclase-coupled alpha adrenergic receptors in rabbit platelets. Mol. Pharmacol. 1978;14:540–548. [PubMed] [Google Scholar]
- WENZEL-SEIFERT K., HURT C.M., SEIFERT R. High constitutive activity of the human formyl peptide receptor. J. Biol. Chem. 1998;273:24181–24289. doi: 10.1074/jbc.273.37.24181. [DOI] [PubMed] [Google Scholar]
- WREGGETT K.A., DE LEAN A. The ternary complex model. Its properties and application to ligand interactions with the D2-dopamine receptor of the anterior pituitary gland. Mol. Pharmacol. 1984;26:214–227. [PubMed] [Google Scholar]