

Abstract
Recruitment to activated tyrosine kinase growth factor receptors of Grb2 and p21ras leads to downstream activation of the kinases Raf, MAPK/Erk kinase (Mek) and, subsequently, extracellular signal-regulated kinase (Erk). Activated Erk phosphorylates specific serine residues within cytosolic phospholipase A2 (PLA2), promoting enzyme translocation to membranes and facilitating liberation of arachidonic acid (AA).
In the A549 human adenocarcinoma cell line dexamethasone inhibited epidermal growth factor (EGF)-stimulated cytosolic PLA2 (cPLA2) activation and AA release by blocking the recruitment of Grb2 to the activated EGF receptor (EGF-R) through a glucocorticoid receptor (GR)-dependent (RU486-sensitive), transcription-independent (actinomycin-insensitive), mechanism.
The dexamethasone-induced block of Grb2 recruitment was parallelled by changes in phosphorylation status and subcellular localization of lipocortin 1 (LC1) and an increase in the amount of the tyrosine phosphoprotein co-localized with EGF-R.
Like dexamethasone, peptides containing E-Q-E-Y-V from the N-terminal domain of LC1 also blocked ligand-induced association of Grb2, p21ras and Raf.
Our results point to an unsuspected rapid effect of glucocorticoids, mediated by occupation of GR but not by changes in gene transcription, which is brought about by competition between LC1 and Grb2 leading to a failure of recruitment off signalling factors to EGF-R
Keywords: Lipocortin 1, annexin 1, Grb2, mitogen-activated protein kinase, phospholipase A2, glucocorticoids, epidermal growth factor, epidermal growth factor receptors, A549 cells
Introduction
Recruitment to activated protein tyrosine kinase (PTK) growth factor receptors of the adapter protein Grb2 and the small GTPase p21ras initiates transduction of the mitogenic signalling mechanisms leading to downstream activation of the kinases Raf, MAPK/Erk kinase (Mek) and, subsequently, extracellular signal-regulated kinase (Erk) (Ahn et al., 1991; de Vries-Smits et al., 1992). Activated Erk phosphorylates specific serine (Ser) residues within cytosolic phospholipase A2 (cPLA2), promoting enzyme translocation to membranes and facilitating liberation of arachidonic acid (AA) (Lin et al., 1993).
Using the A549 human adenocarcinoma cell line we have shown that epidermal growth factor (EGF) can rapidly activate cPLA2 and AA release by a pertussis toxin-sensitive mechanism (Croxtall et al., 1995; 1996). More importantly, we have shown that the synthetic glucocorticoid, dexamethasone, inhibits the activation of AA release by EGF by a mechanism which apparently does not involve the suppression of cPLA2 expression. Rather, cPLA2 activity is instead inhibited, as shown by an increased proportion of the unphosphorylated (inactive) form of the enzyme revealed on an electrophoretic mobility shift assay following dexamethasone treatment (Croxtall et al., 1996). These rapid changes in the phosphorylation/dephosphorylation status of cPLA2 have led us to postulate that this mechanism permits a more responsive control over arachidonic acid release without a need for invoking gross changes in cPLA2 protein expression (Croxtall et al., 1996). The pathway of activation of cPLA2 by EGF is already well documented as is outlined above. However, it is clear that dexamethasone must somehow also regulate this process.
We have previously shown that the effects of dexamethasone on cPLA2 activity and AA release are mediated via the protein lipocortin 1 (LC1, annexin 1) and in particular, we have demonstrated the importance of the N-terminal domain sequence containing the pharmacophore glutamate-glutamine-glutamate-tyrosine-valine (EQEYV) (Croxtall et al., 1998). We describe here how rapid changes in the phosphorylation status of LC1 induced by dexamethasone may regulate signalling mechanisms leading to the activation of cPLA2.
Methods
Cell culture and immunprecipitation
A549 cells (European Collection of Cell Cultures, Salisbury, Wiltshire) were maintained in continuous log-phase growth in Dulbecco's modified Eagle's medium/F-12 (DMEM/F-12) containing phenol red and 10% foetal calf serum (FCS) in T-150 flasks (Greiner Labortechnik Ltd., Stonehouse, Gloucestershire, U.K.) at 37°C, 5% CO2. For EGF receptor (EGF-R) complex immunoprecipitation and Western blotting cells were deprived of FCS and phenol red for 24 h prior to treatment with dexamethasone (usually 1 μM, 5 min–3 h, together with a simultaneous addition of 10 μM RU486 where appropriate) and/or EGF (10 nM, 5–30 min) or LC1-derived peptide.
Following experimental treatment, the medium was aspirated and the cell monolayer was washed with phosphate-buffered saline (PBS) containing 1 mM EDTA to remove adherent surface-bound proteins. The monolayer was dispersed with 0.05% trypsin in PBS containing 10 mM EDTA; following centrifugation, cell pellets were snap-frozen in 3 ml PBS containing 10 mM EDTA, 1 mg ml−1 soybean trypsin inhibitor, 0.01% w v−1 leupeptin, 1 mM phenylmethylsulphonyl fluoride and 1 mM sodium orthovanadate. Once thawed, cell lysates were clarified by centrifugation at 13,000 × g for 20 min to prepare a cytosol fraction for immunoprecipitation analysis. The cell pellet was resuspended in 3 ml PBS containing 10 mM EDTA, 1% v v−1 Triton X-100 and protease inhibitors for 30 min at 4°C and clarified again; the supernatant was retained as a membrane fraction.
Protein concentrations were measured by Bradford assay and identical concentrations were used in each immunoprecipitation. For immunoprecipitation, 1 ml cell lysate was incubated with 5 μg precipitating monoclonal antibody for 16 h with continuous rocking. Then, 20 mg Protein A-Sepharose was added for a further 2 h. The Protein A-Sepharose-bound immunocomplexes were washed three times in PBS containing 10 mM EDTA and then incubated with 250 μl sample buffer for 5 min at 90°C prior to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE) analysis by Western blotting and detection by diaminobenzidine (DAB).
Arachidonic acid release
Sub-confluent cells were seeded into 12-place multiwell plates (Falcon Plastics, Cowley, Oxfordshire, U.K.) at three times 105 cells ml−1 well−1 in DMEM/F-12 containing 10% FCS and incubated overnight. [3H]-Arachidonic acid in ethanol was evaporated to dryness under N2 and resuspended in an appropriate volume of DMEM/F-12 (without phenol red) and, after vortex mixing, left at 37°C for 1 h. After washing the cells with PBS, 9.25 kBq [3H]-AA in 0.5 ml DMEM/F-12 (without phenol red) was added to each well and incubated overnight. The medium containing free [3H]-AA was then removed and the cells washed three times with 1 ml DMEM/F-12 containing 1 mg ml−1 bovine serum albumin. The cells thus labelled with [3H]-AA were then treated with 1 μM dexamethasone in the presence and absence of 10 μM RU486 (1 min–3 h, added simultaneously), actinomycin D (0.1 μM, 3 h) or vehicle control diluted into DMEM/F-12 (without phenol red). Then 10 nM EGF and 50 nM thapsigargin was added for 30 min. After incubation, 0.4 ml medium was removed from each well for scintillation counting.
Grb2-SH2-protein a binding to EGF-R
Ten μg ml−1 EGF-R (Sigma Chemical Co., Poole, Dorset, U.K.) was incubated with 1 μg ml−1 Grb2-SH2-Protein A and 10 μg ml−1 human IgG in 1 ml PBS containing 1 mM sodium orthovanadate and 1 mM NaF for 18 h at 4°C with continuous stirring. The complex was precipitated with the addition of 100 μg ml−1 Protein A-Sepharose and the precipitate washed three times in PBS prior to boiling with sample buffer. Immunoprecipitated EGF-R was detected by Western blotting with PY20 anti-phosphotyrosine monoclonal antibody.
Materials
EGF, EGF-R, thapsigargin, geldanamycin, actinomycin D, Grb2-SH2-Protein A, Protein A sepharose, all anti-phosphoprotein monoclonal antibodies and all other general purpose, cell culture or blotting reagents were from Sigma. PP2 was from Calbiochem-Novabiochem (Beeston, Nottinghamshire, U.K.). RU486 was a gift from Roussel-Uclaf (Romainville, France). [5,6,8,9,11,12,14,15-3H]-(N)-arachidonic acid was from NEN Du Pont (Zaventem, Belgium). Specific polyclonal anti-LC1 for Western blotting was raised in-house using a denatured form of LC1 as immunogen in sheep. Monoclonal anti-LC1 (mA 1B) for immunoprecipitation was a gift from Dr Jeff Browning, Biogen, Boston, MA, U.S.A. Immunoprecipitation of activated EGF-R and Mek1, Mek2, Erk1 and Erk2 and Western blotting for Grb2, p21ras and Raf was performed using monoclonal antibodies from Transduction Laboratories/Becton Dickinson (Cowley, Oxfordshire, U.K.). Immunoprecipitation of Jun N-terminal kinase 1 (Jnk1), glucocorticoid receptor (GR), heat shock protein 90 (Hsp90) and Src was performed with monoclonal antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Peptide fragments of LC1 were kindly prepared by Dr Stefan Peters (Boehringer Ingelheim KG, Ingelheim-am-Rhein, Germany).
Data analysis and statistics
All arachidonic acid release experiments were performed in triplicate and each experiment presented is a typical example of at least three such experiments. Results are expressed as the mean±1 s.d. and presented as per cent changes. However, all statistical calculations were performed on the raw numerical data. Student's t-test (unpaired) with the Bonferonni correction to allow for multiple testing within each group was used to determine statistical significance with P<0.001 defined as significant.
Western blots were scanned using a ScanMaker (Microtek, CA, U.S.A.) and the image composite transferred into PowerPoint (Microsoft, WA, U.S.A.) running on an Apple Macintosh computer. Densitometric analysis was performed with NIH Image 1.54 software and relative band intensities reported as per cent changes within each blot. The values given are semi-quantitative and are only meant to give some numerical guide to the ratio of band intensities. The blots presented are typical examples of at least three such experiments. Although overall band intensities varied between experiments, the ratio of band intensities remained the same.
Results
Inhibition of recruitment of signalling intermediates to EGF-R
We measured ligand-induced activation of the EGF-R in A549 cells by monitoring the phosphorylation of its tyrosine (Tyr)-containing domains following immunoprecipitation of cell extracts with a specific anti-EGF-R monoclonal antibody and subsequently SDS–PAGE and Western blotting using a specific anti-phosphotyrosine monoclonal antibody (PY20).
Thirty-minute treatment of cells with 10 nM EGF increased phosphorylation of the EGF-R complex by approximately 7 fold and activated EGF-R bound increased amounts of immunoreactive Grb2, p21ras and Raf (Figure 1). However, this association of signalling intermediates was robustly diminished following pre-treatment of cells (0.5–3 h) with 1 μM dexamethasone whereas the Tyr phosphorylation of EGF-R remained unaffected. These results imply that dexamethasone inhibits the association of Grb2, p21ras and Raf without influencing the binding of EGF, EGF-R phosphorylation or subsequent homodimerization.
Figure 1.
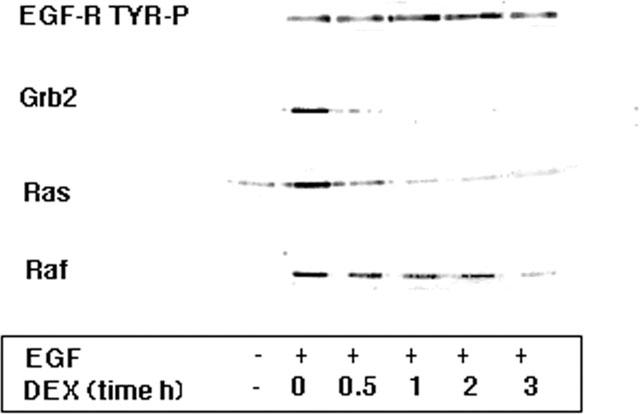
Effects of glucocorticoids on the association of signalling intermediates with activated EGF-R. A549 cells activated with 10 nM EGF for 30 min were pre-treated with 1 μM dexamethasone for the times indicated. Ligand-induced activation of the EGF-R immunoprecipitate was increased over 7 fold as assessed by Western blotting with an anti-phosphotyrosine antibody PY20 and this activation was unaffected by treatment for up to 3 h with dexamethasone. Western blotting of the EGF-R immunoprecipitate with specific antibodies to Grb2, p21ras and Raf showed that the co-association of these intermediates was increased 7–10 fold following ligand activation. However, this association was diminished following dexamethasone (DEX, 1 μM) treatment. After 3 h, association of Grb2 was undetectable, p21ras was reduced to 12% and Raf reduced to 26% of control (EGF alone) values.
The nature and time-dependence of the dexamethasone effect were examined further with the aid of the GR antagonist RU486. Blockade of signalling activation (but not EGF-R phosphorylation) with 1 μM dexamethasone, as assessed by a reduction in the recruitment of Grb2 and other intermediates, was detectable within 5–10 min of treatment (the shortest practicable time points for preparing cell extracts; Figure 2a). Increasing the incubation time to 1 h or 3 h (Figure 2b) produced qualitatively the same response. In the presence of 10 μM RU486 the ability of the glucocorticoid to inhibit association of Grb2, p21ras and Raf with activated EGF-R at all time points was substantially reversed. Furthermore, the dose-dependence of this inhibitory effect of dexamethasone was essentially the same at both 5 min and 3 h, with an optimal effect at 1 μM and an EC50 of approximately 50 nM (data not shown). These results imply that this very rapid effect of dexamethasone is dependent upon GR occupation and that it is not caused by a genome-dependent transcriptional event or non-specific (e.g. physicochemical) phenomenon. These data also suggest that glucocorticoids should exhibit a very rapid blocking action on AA release itself. Figure 3a shows that this is indeed the case: dexamethasone (1 μM) produced an RU486-reversible blockade of AA release within 1 min of administration (the shortest practicable time point for estimating AA release) that was only marginally less than that seen after 10 min incubation. However, the inhibition by dexamethasone increased from 40 to 70% with longer contact times. Actinomycin D (0.1 μM) was found to be ineffective at preventing acute (<10 min) effects of dexamethasone but, in contrast, totally blocked the effect at 3 h (Figure 3b), confirming previous observations in this cell line. This finding suggests that some factor, which is initially present, is being consumed, thus maintaining the dexamethasone blockade.
Figure 2.
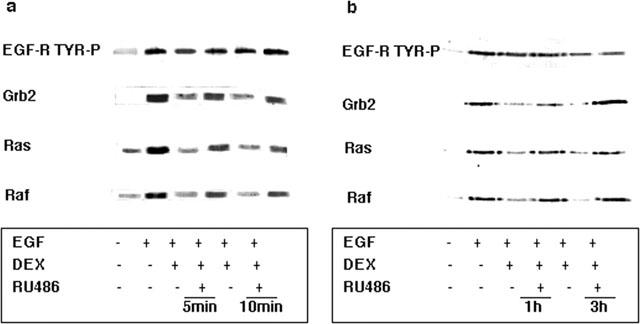
Inhibition by dexamethasone of the co-association of Grb2, p21ras and Raf with activated EGF-R occurs within minutes and is GR-mediated. (a) A549 cells activated with 10 nM EGF for 5 min were pre-treated with 1 μM dexamethasone for the times indicated in the presence and absence of 10 μM RU486. Western blotting of the EGF-R immunoprecipitate showed that inhibition of the co-association of Grb2 by dexamethasone was reversed by RU486 to 71% (5 min) and 60% (10 min) of control values (EGF alone). Similarly, p21ras association was reversed to 67% (5 min) and 50% (10 min), and Raf association was restored to 100% (5 min) and 68% (10 min) of control values. (b) A549 cells activated with 10 nM EGF for 30 min were pretreated with 1 μM dexamethasone (DEX) for the times indicated in the presence and absence of 10 μM RU486. Western blotting of the EGF-R immunoprecipitate showed that inhibition of the co-association of Grb2 by dexamethasone was restored by RU486 to 86% (1 h) and 100% (3 h) of control values (EGF alone). Similarly, p21ras association was restored to 87% (1 h) and 80% (3 h), and Raf association was restored to 100% (1 and 3 h) of control values. Activation of the EGF-R immunoprecipitate itself was unaffected by steroid treatment as shown by Western blotting with PY20.
Figure 3.
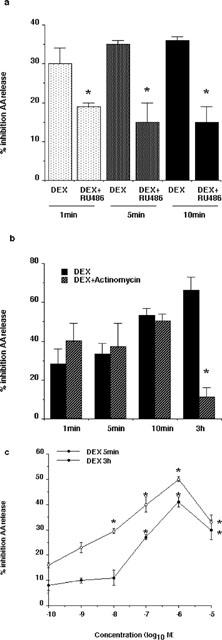
Dexamethasone inhibition of EGF-stimulated AA release occurred within minutes through a GR-dependent, transcription-independent mechanism. (a) A549 cells activated with 10 nM EGF and 50 nM thapsigargin for 30 min were pre-treated with 1 μM dexamethasone (DEX) for the indicated times in the presence and absence of 10 μM RU486. Data are presented as per cent inhibition by dexamethasone compared to EGF/thapsigargin treatment alone. RU486 significantly (*P<0.001, n=3) reversed the action of dexamethasone at 1, 5 and 10 min of treatment. (b) A549 cells activated with 10 nM EGF and 50 nM thapsigargin for 30 min were pre-treated with 1 μM dexamethasone for the indicated times in the presence and absence of actinomycin D (0.1 μM, 3 h). The inhibitory effect of dexamethasone was unaffected by actinomycin D at 1, 5 and 10 min but was significantly (*P<0.001, n=3) reversed at 3 h. (c) A549 cells activated with 10 nM EGF and 50 nM thapsigargin were pre-treated with the indicated concentrations of dexamethasone for either 5 min or 3 h. In both cases an optimal inhibition of arachidonic acid release was seen at 1 μM and an EC50 of approximately 50 nM was observed (*P<0.001, n=3).
The concentration dependence of dexamethasone's inhibition of arachidonic acid release is shown in Figure 3c. Again, although the inhibitory effect was increased with longer contact times, the optimal inhibitory concentration was 1 μM at 5 min and 3 h with an EC50 of approximately 50 nM at both time points. The transcription-independence of the acute dexamethasone inhibition of AA release was further confirmed when pre-incubation of A549 cells with the NF-κ B inhibitor, ammonium pyrrolidinedithiocarbamate, for 3 h failed to reverse the inhibitory effect (data not shown).
Inhibition of MAP kinase pathways
The inhibitory action of dexamethasone on the recruitment of key signalling intermediates such as p21ras and Raf implies that activation of downstream components of Raf-linked pathways should also be prevented. We examined this possibility by immunoprecipitating several downstream kinases with specific monoclonal antibodies and evaluating their phosphorylation status by Western blotting with specific anti-phosphoserine (PS), anti-phosphothreonine (PT) or anti-phosphotyrosine (PY20) antibodies. EGF treatment of A549 cells resulted in phosphorylation of both Mek1 and Mek2 on Ser residues but only phosphorylation of Mek1 was inhibited by dexamethasone (Figure 4). This is consistent with data from other groups who have observed that Mek1, but not Mek2, forms specific complexes with Raf (Jelinek et al., 1994). The effects of 1 μM dexamethasone on Mek1 phosphorylation were reversed by a 10 fold excess of RU486, again confirming the GR-dependence of this effect.
Figure 4.
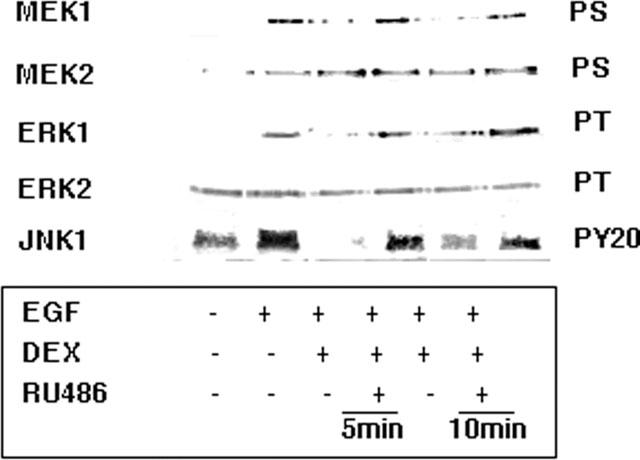
Selective inhibition by dexamethasone of MAPK pathways activated by EGF by a GR-dependent mechanism. A549 cells activated with 10 nM EGF for 30 min were pre-treated with 1 μM dexamethasone (DEX) for the times indicated in the presence and absence of 10 μM RU486. Immunoprecipitates of the Mek1 & 2, Erk1 & 2 and Jnk1 kinases were examined for activation status by Western blotting with specific antibodies to phosphoserine (PS), phosphothreonine (PT) or phosphotyrosine (PY20) motifs. Phosphorylation of Ser residues of Mek1 and Mek2 was increased by EGF (10–4 fold, respectively) but only phosphorylation of Mek1 was inhibited by dexamethasone treatment (at 5 min reduced to 17% of EGF control value). Similarly, Erk1 was phosphorylated on Thr residues (7.5 fold) also in a dexamethasone-sensitive manner (at 5 min reduced to 16% of EGF alone value) whereas Erk2 appeared to be constitutively activated. Jnk1 was activated on Tyr residues (2 fold) following EGF and reduced below basal levels by both 5 and 10 min of dexamethasone treatment. Dexamethasone inhibition of Mek1 activation was restored to 77% of the EGF control value at 1 h, ErK1 activation was restored to 100% of the EGF control value at 1 h and Jnk1 activation was restored to 73% of the EGF alone value at 10 min, by RU486.
The activation of the Mek substrates Erk1 and Erk2 were examined in a similar manner. Phosphorylation of Erk1 on threonine (Thr) residues was increased following EGF treatment in a dexamethasone-sensitive manner whereas Erk2 appeared to be constitutively phosphorylated and was unaffected by the presence of the hormone (Figure 4). Recently, other groups have implicated Jnk as an important mediator in A549 cell proliferation (Bost et al., 1997) and suggested that there is cross-talk between this pathway and activation of nuclear hormone receptors (Caelles et al., 1997). We confirmed that the Tyr phosphorylation of Jnk1 is elevated almost 2 fold following EGF treatment and that this is sensitive to dexamethasone treatment (Figure 4). Taken together, these results imply that the dexamethasone inhibition of Raf recruitment leads to a specific inhibition of Mek1 and, thereby, of Erk1 activation, blocking the activation and translocation of cPLA2 and, perhaps, other key kinase pathways.
Activation of LC1 phosphorylation
Dexamethasone inhibition of cPLA2 activation and AA release in A549 and other cells is associated with the appearance in the membrane of increased amounts of the annexin LC1 (Croxtall & Flower, 1992). This protein has, within its N-terminal, a domain containing a high homology to the preferred EGF tyrosine kinase substrate sequence (Croxtall et al., 1998). In A549 cells, specific anti-LC1 neutralizing monoclonal antibodies readily reverse (Croxtall & Flower, 1992) – while the addition of human recombinant LC1 or the N-terminal peptide LC113–25 mimics – the effect of dexamethasone (Croxtall et al., 1998). To provide an explanation for the effects of LC1 and its peptides, as well as the changes in location of LC1 in the cell which are observed during dexamethasone treatment, we examined the phosphorylation status of LC1 and its subcellular distribution and monitored its appearance in the immunoprecipitated EGF-R complex.
Following treatment with 1 μM dexamethasone (±10 μM RU486) for 5 min, 10 min or 3 h, A549 cells were immediately lysed and LC1 immunoprecipitated with a specific anti-LC1 monoclonal antibody mA 1B. This enabled us to evaluate both the mass and phosphorylation status of cellular LC1 using SDS–PAGE and Western blotting with PS, PT or PY20 antibodies or a sheep polyclonal anti-LC1 antiserum. Phosphorylation of LC1 was increased on Ser, Thr and Tyr residues by approximately 4 fold after only 5–10 min of treatment with dexamethasone and this occurred against a background of no apparent change in total LC1 expression (Figure 5). Subsequently, after 3 h of treatment, total LC1 expression was increased approximately 2 fold by dexamethasone, in agreement with our previous observations (Croxtall & Flower, 1992). Both the early effects on phosphorylation and the late effects on expression were optimal at 1 μM dexamethasone (with an EC50 of approximately 50 nM, data not shown) and were reversed by the GR antagonist RU486.
Figure 5.
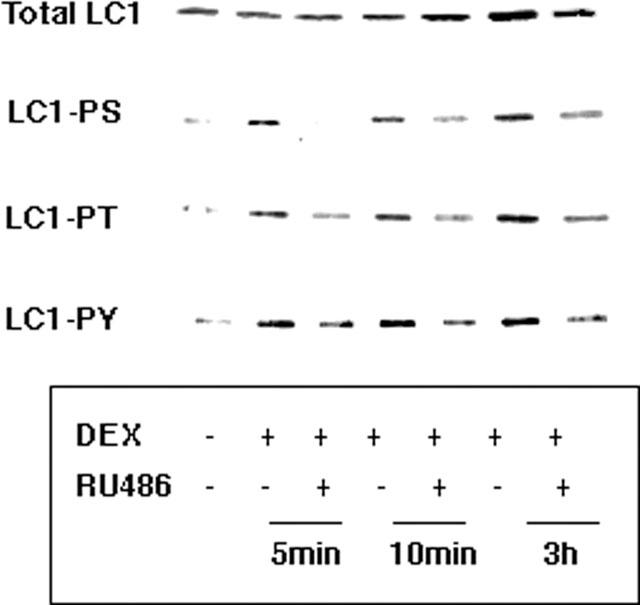
Rapid GR-dependent effect of dexamethasone on the phosphorylation of LC1. A549 cells were treated with 1 μM dexamethasone for the indicated times in the presence and absence of 10 μM RU486. LC1 immunoprecipitates from total cell lysates were evaluated for phosphorylation status by Western blotting with anti-phosphoserine (PS), anti-phosphothreonine (PT) or anti-phosphotyrosine (PY20) antibodies. Total LC1 expression was evaluated by blotting with anti-LC1. The phosphorylation of Ser, Thr and Tyr residues was increased approximately 4 fold at all times. The effects of dexamethasone on LC1 phosphorylation status were reversed by RU486 at all time points. The expression of LC1 was not significantly changed after 5 and 10 min of dexamethasone (DEX) treatment but was increased approximately 2 fold after 3 h. This effect was also reversed by RU486.
Next, we determined the phosphorylation status of the major subcellular pools of LC1 described by our laboratory and by others. A preliminary EDTA wash of intact A549 cells to remove calcium-dependent external membrane-bound proteins, a cytosolic fraction (prepared from freeze–thawed cells in the presence of EDTA) containing intracellular LC1 and the Triton X-100 extractable membrane protein pool were prepared. Again, LC1 was immunoprecipitated from these respective pools and phosphorylation status evaluated. Treatment of the cells with dexamethasone increases the proportion of Ser and Thr- phosphorylated LC1 in the membrane pool (Figure 6). This effect of dexamethasone occurs within 5 min (the shortest time point at which this can be measured), is reversed by RU486, and no further increase in effect is seen with incubations as long as 3 h.
Figure 6.
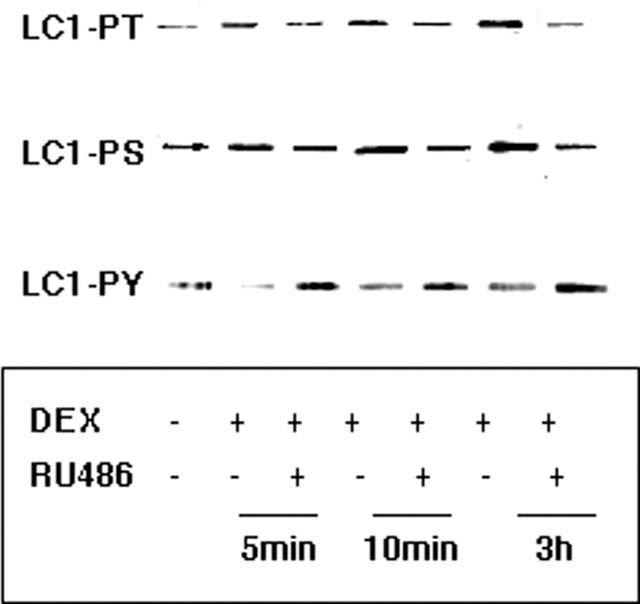
Influence of dexamethasone on the phosphorylation status of subcellular pools of LC1 within minutes by a GR-dependent mechanism. A549 cells were treated with 1 μM dexamethasone (DEX) for the indicated times in the presence and absence of 10 μM RU486. LC1 immunoprecipitates from the cell membrane were evaluated for phosphorylation status by Western blotting with anti-phosphoserine (PS), anti-phosphothreonine (PT) or anti-phosphotyrosine (PY20) antibodies. In the membrane the phosphorylation of Thr and Ser residues was enhanced (∼2 fold each at all time points) by dexamethasone at all time points examined. The effects of dexamethasone on LC1 phosphorylation status were reversed by RU486 at all time points.
LC1 is a well characterized substrate for protein kinase C, which phosphorylates the residues Thr24, Ser27 and Ser28 equally in vitro (Summers & Creutz, 1985; Schlaepfer & Haigler, 1988), and for PTK. LC1 is a low (50 nM) km substrate for the EGF-R in A431 cells (Pepinsky & Sinclair, 1986) and in fibroblasts and is also a substrate for the insulin receptor kinase in rat liver (Karasik et al., 1988), and probably other kinases also. Both EGF-R and insulin receptor kinases phosphorylate LC1 Tyr21 (Tyr26 in LC2) exclusively (Karasik et al., 1988; Schlaepfer & Haigler, 1987). We therefore have investigated the role of dexamethasone pre-treatment combined with EGF on the phosphorylation of LC1, its subsequent subcellular distribution and appearance in the EGF-R complex.
Dexamethasone treatment increased the Ser phosphorylation of LC1 within the membrane compartment (Figure 7) and this was accompanied by a reciprocal decrease in the appearance of the phosphoprotein in the cytosol (not shown). Changes in Thr phosphorylation were less well defined although, once again, dexamethasone resulted in an overall increase of the phosphorprotein in the membrane pool. This increase in membrane phospho-LC1 is mirrored by an increase in EGF-R-bound LC1, but a combination of EGF and dexamethasone led to the appearance of substantial amounts of EDTA extractable LC1 on the cell surface which apparently arose from the membrane compartment. Subsequent analysis (not shown) revealed that this cell surface pool was heavily phosphorylated on Thr, Ser and Tyr residues. Taken together these results imply that dexamethasone, by changing LC1 phosphorylation status, enables the recruitment of LC1 to the membrane EGF-R and subsequently to the external ‘EDTA soluble' pool where it is eventually proteolytically clipped (as evidenced by the appearance of a doublet) and may be released (presumably in an inactive form) into the medium.
Figure 7.
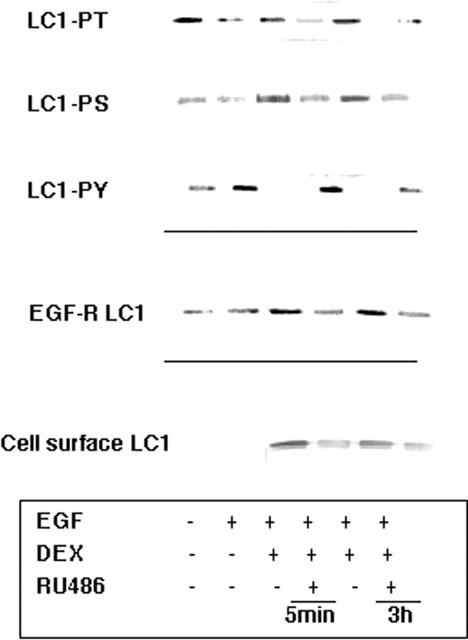
Influence of EGF and dexamethasone on the phosphorylation status of subcellular pools of LC1. A549 cells activated with 10 nM EGF for 30 min were pre-treated with 1 μM dexamethasone for the times indicated in the presence and absence of 10 μM RU486. LC1 immunoprecipitates from the cell membrane were evaluated for phosphorylation status by Western blotting with phosphoserine (PS), phosphothreonine (PT) or phosphotyrosine (PY20) antibodies. In the membrane phosphorylation status of LC1 at Ser and Thr residues was diminished (49 and 38% respectively of control) by EGF treatment and Tyr phosphorylation was enhanced (2.4 fold). The effects of dexamethasone on the phosphorylation status of LC1 in the membrane were reversed in the presence of RU486 at both 1 and 3 h. Immunoprecipitates of activated EGF-R contained elevated levels (2 fold) of LC1 following dexamethasone treatment. Subsequent externalization of LC1 as a ‘clipped' doublet band was seen in the cell surface wash of A549 cells following a combination of dexamethasone and EGF treatment.
The appearance of the increased amounts of phospho-LC1 in the membrane, in the EGF-R complex and in the EDTA soluble cell surface pool coincides with the onset of the dexamethasone blockade of signalling factor recruitment. One potential explanation for this effect of LC1 might be that it is phosphorylated by EGF-R in competition with the autocatalytic phosphorylation of Tyr1036, thereby reducing the density of high affinity SH2 binding sites on the activated complex. This option would seem unlikely given that no obvious changes in EGF-R phosphorylation were observed in the presence of dexamethasone. Alternatively, the Tyr21 phospho-LC1 may act as an alternative ‘decoy' ligand for Grb2 binding again blocking recruitment to the signalling complex.
Competition with Grb2-SH2 binding domains
The inhibitory effect of human recombinant (h) LC1 on cell proliferation and AA release in A549 cells may be mimicked by the addition of peptides taken from the N-terminal region of the molecule, particularly those which contain the sequence LC113–25, containing the Tyr21 phosphorylation site (Croxtall et al., 1993; 1998) which has a high homology with the preferred substrate site of EGF-R. Figure 8 illustrates that the addition of the peptide LC113–25 or LC118–25 to cultured A549 cells, in concentrations (54 and 89 μM respectively) which block cell AA release and cell proliferation, potently inhibited the recruitment of signalling intermediates but were without discernible effect on EGF-R phosphorylation. A scrambled LC113–25 peptide control was without effect, as was the peptide LC11–12, which we have previously shown to be inactive in this system (Croxtall et al., 1993; 1995; 1996).
Figure 8.
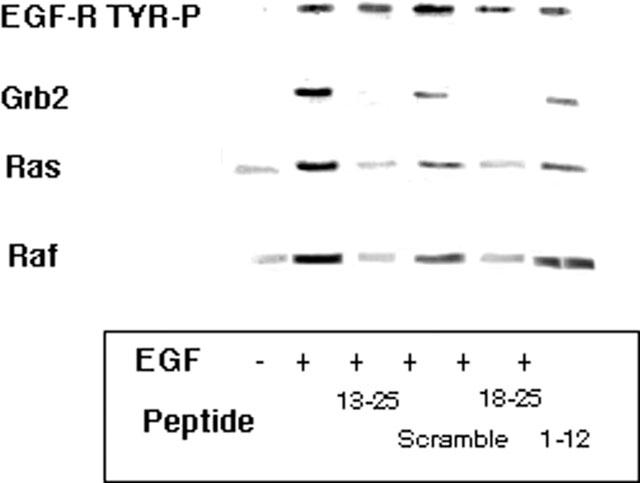
Inhibition of the association of Grb2, p21ras and Raf to activated EGF-R by N-terminal peptide fragments of LC1. A549 cells activated with 10 nM EGF for 30 min were pretreated for 3 h with 100 μg ml−1 of the LC1 peptide sequences indicated. Western blotting of immunoprecipitated EGF-R with specific antibodies to these signalling intermediates showed that the co-association of Grb2 was inhibited by peptide LC113–25 by 95% and by LC118–25 by 100% of EGF alone values. Co-association of p21ras was reduced by LC113–25 by 80% and by LC118–25 by 75% of EGF alone values. Co-association of Raf was reduced by LC113–25 by 80% and by LC118–25 by 70% of EGF alone values. A scrambled sequence of LC113–25 and peptide LC11–12 had no significant effect on the co-association of any of these intermediates.
The ability of these peptides directly to compete with Grb2 for binding to EGF-R was investigated further in a cell-free assay using a Grb2 SH2-domain-Protein A fusion protein. This construct readily binds to purified EGF-R as revealed by precipitation of the complex with human IgG and Protein A-Sepharose (Figure 9). However, the inclusion of LC113–25 significantly reduced the co-precipitation of EGF-R, whereas a scrambled peptide control was without effect. Furthermore, LC121–33 also reduced the association of EGF-R with the Grb2 construct, whereas the Tyr21-substituted variant LC121–33(Phe) was without effect. Taken together, these results show that in intact cells dexamethasone treatment increases the proportion of LC1 associated with the EGF-R complex. This is accompanied by a reduction in the association of Grb2 and other downstream signalling intermediates. More directly, using a cell free assay, we have shown that peptide fragments containing the active pharmacophore of LC1 are able to interrupt the association of EGF-R for Grb2. Once again the availability of Tyr21 appears to be crucial.
Figure 9.

Competition of peptide fragments from the N-terminus of LC1 with a Grb2-SH2 fusion protein for binding to purified EGF-R. Ten μg ml−1 EGF-R was incubated with 1 μg ml−1 Grb2-SH2-Protein A and 10 μg ml−1 human IgG in 1 ml PBS and the complex precipitated with 100 μg ml−1 Protein A-Sepharose. The inclusion of peptides LC113–25 or LC121–33 at 100 μg ml−1 reduced the precipitation of EGF-R by Grb2-SH2-Protein A by 53 and 48%, respectively. A scrambled control was without effect, as was LC121–33(Phe) and LC11–12.
Activation of the glucocorticoid receptor complex
The mechanism by which dexamethasone phosphorylates LC1 is clearly very important in mediating the rapid inhibitory effects of the steroid. We have therefore attempted to evaluate the activation of the GR and proteins associated in the receptor complex following binding of dexamethasone. Immunoprecipitation from A549 cells of hsp90 co-precipitated both GR and Src (Figure 10a). However, following treatment with 1 μM dexamethasone for 5 min the precipitation of both of these components was robustly diminished. Geldanamycin is a benzoquinone ansamycin that selectively binds hsp90 and disrupts GR function (Whitesell & Cook, 1996) by preventing its nuclear translocation (Galigniana et al., 1998). Figure 10a shows that in A549 cells pre-treated with 10 μM geldanamycin, the rapid release of GR by dexamethasone from the hsp90 complex was inhibited but that the release of Src was unaffected. However, pre-treating A549 cells with 10 μM PP1, an inhibitor of the src family of kinases (Hanke et al., 1996), inhibited both the dexamethasone-induced release of GR and Src from the hsp90 complex. These results imply that the release of GR and Src from the hsp90 complex are separate events which may play distinct roles in glucocorticoid-mediated signalling and genomic effects. This is confirmed in Figures 10b,c, where the effects of these inhibitors on the rapid induction of phosphorylation of LC1 by dexamethasone were investigated. Pre-treatment of A549 cells with 10 μM PP1 significantly reduced the phosphorylation of LC1 on PT residues following 5 min 1 μM dexamethasone treatment, but 10 μM geldanamycin was without effect. Similarly, the rapid inhibition of AA release by dexamethasone was also inhibited by PP1 but unaffected by geldanamycin.
Figure 10.
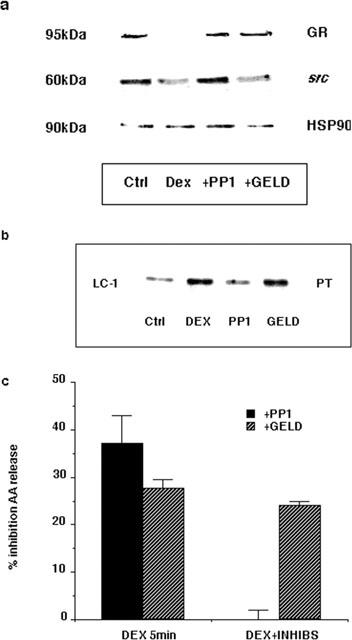
Effect of src and hsp90 inhibitors on GR assembly and rapid signalling effects. (a) A549 cells were pre-treated for 1 h with either 10 μM PP1 or 10 μM geldanamycin (GELD) prior to treatment with 1 μM dexamethasone for 5 min. Cell lysates were immunoprecipitated with anti-hsp90 polyclonal antibody then Western blotted with anti-hsp90, anti-GR and anti-Src antibodies. (b) Cell lysates were also immunoprecipitated with anti-LC1 monoclonal antibody mA 1B and the phosphorylation status determined by Western blotting with anti-PT antibodies. (c) A549 cells activated with 10 nM EGF and 50 nM thapsigargin for 30 min were pre-treated with either 10 μM PP1 or 10 μM geldanamycin for 1 h prior to treatment with 1 μM dexamethasone (DEX) for 5 min. Data are presented as a per cent inhibition by dexamethasone compared to EGF and thapsigargin alone (n=3).
Discussion
The observation that inhibitory effects of dexamethasone are seen at very early time points in this system was unexpected in the light of previous work from our and other labs which indicated that many aspects of glucocorticoid action were both time- and transcription-dependent. However, it has been appreciated for many years that some clinical effects of glucocorticoids occur too rapidly to be explained by genome-dependent processes: e.g. the rapid (within minutes) response to glucocorticoids of patients with acute adrenal insufficiency (Merry et al., 1994). Similarly, there is considerable evidence for a rapid feedback effect of glucocorticoids on the anterior pituitary gland that is clearly distinguished from their longer term actions (Buckingham, 1996). In the light of this, there is now an increasing awareness of glucocorticoid actions which, although mediated through GR-binding events, are nevertheless not dependent upon subsequent binding of the receptor to glucocorticoid responsive elements (GRE). A recent study, for example, showed that, in contrast to the lethal nature of a GR knock-out, mice bearing a mutation of the GR which disabled dimerization, and hence GRE binding, survived (Reichardt et al., 1998). The inference was that other transcriptional activators such as AP1 and NFκB, which are probably bound by the GR monomer (Heck et al., 1994), play a key role in mediating the action of glucocorticoids. Such effects clearly would also be transcription-dependent and may not explain the very rapid effects reported here. However, other recent studies confirm the importance of the GR per se in transduction of the glucocorticoid effect on phosphatase activity which are transcription-independent in some cells (Tumlin et al., 1997). Moreover, direct regulation of signalling mechanisms also appears to be a feature of the action of other steroid receptors, such as that for oestradiol (Migliaccio et al., 1996), and other nuclear hormone receptors (Bost et al., 1997). The key question is how steroid receptors could mediate these rapid signalling effects.
The unactivated (unligated) GR is retained in the cytoplasm as a multi-protein complex consisting of heat shock proteins (hsp) (Bamberger et al., 1996) and several kinases of the MAPK signalling system, including Src (Pratt, 1998). Following glucocorticoid binding the GR is released from this complex, thus revealing domains of the receptor that are able to bind specific GRE within DNA (Dalman et al., 1991). This mechanism accounts for the genome-mediated effects of glucocorticoids. However, the role of the remaining signalling components of the multi-protein complex is less clearly defined. We show here that both GR and Src are rapidly released from the hsp90 complex in A549 cells following treatment by dexamethasone. In cells pre-treated with geldanamycin, which disrupts GR function by preventing its nuclear translocation (Whitesell & Cook, 1996; Galigniana et al., 1998), the rapid release of GR by dexamethasone from the hsp90 complex is inhibited but the release of Src is unaffected. In these cell extracts the rapid phosphorylation of LC1 and inhibition of AA release by dexamethasone is also unaffected by geldanamycin treatment, indicating that nuclear translocation of the receptor is not required for this process. However, in cells pre-treated with the src inhibitor, PP1, not only is the dexamethasone-induced release of both GR and Src from the hsp90 complex inhibited but so is the rapid phosphorylation of LC1 and inhibition of AA release. Taken together, these results show that, following GR binding of dexamethasone, there is a rapid release of both GR and Src from the hsp90 complex. However, the release of GR and Src appear to be distinct events, since the nuclear translocation of GR is not required for the subsequent activation of LC1 and rapid inhibition of AA release. Rather, it appears that it is the rapid release of Src that fulfils this role.
The findings reported here provide novel information about the mechanisms utilized by dexamethasone – and therefore, presumably, other GCs – to control intracellular signalling mechanisms which culminate in, for example, the release of AA for the synthesis of inflammatory mediators. The inhibitory effect of dexamethasone in A549 cells, at these time points, appears to be specific for the Raf-linked Mek1 pathway whereas other EGF-activated pathways such as Mek2 are not affected, perhaps indicating that the inhibitory actions of LC1 are highly specific. Interestingly, several groups have described the presence of 36–38 kDa Tyr-phosphorylated proteins that participate in the EGF-R control of p21ras activity (Buday et al., 1994) or other signal transduction systems (Fukazawa et al., 1995). These proteins are probably members of the lnk family (Huang et al., 1995): although they do not seem to be related to the annexins it is noteworthy that they also contain the sequence E-Y-V-X-T found in the biologically active peptides derived from LC1 (Croxtall et al., 1998). There have been several other reports of regulatory factors operating at the level of the early stages of cell signalling to modify the activity of the PTK pathway. For example, the activity of p21ras is modulated by regulatory proteins such as GEFs and GAPs and may be inhibited by glucocorticoid-induced proteins such as Dexras1 (Kemppanien & Behrend, 1998) or the negative regulatory 66 kDa isoform of Shc (Okada et al., 1997). In the A549 system, glucocorticoids apparently block EGF signalling at the level of Grb2 recruitment rather than at the level of p21ras and this is apparently accomplished without influencing the tyrosine kinase activity of the receptor itself. Our work strongly implicates the annexin LC1 in this process and points to the importance of the N-terminal sequence containing the pharmacophore E-Q-E-Y-V in this action.
A similar conclusion about the blocking action of dexamethasone on signal transduction has been reached in other systems. Karasik et al. (1988) observed that rat liver insulin receptor kinase preferentially phosphorylates LC1 in vivo following dexamethasone treatment and speculated that competition between the abundant supply of LC1 relative to other signalling intermediates, coupled with the low km of LC1 compared with other known substrates, inhibited the phosphorylation of downstream signalling components. They proposed that this accounted for the anti-insulin effect of glucocorticoids. This was also apparently mediated through the N-terminal domain of LC1, as a peptide containing residues 16–30 mimicked the effect (Melki et al., 1994).
Skouteris & Schroder (1996) investigated the signal transduction events following the stimulation of A549 cells with hepatocyte growth factor (HGF). In line with the events described in this paper, LC1 was rapidly phosphorylated on Tyr21 within 10 min of addition of HGF and the protein translocated to the membrane fraction for up to 6 h following stimulation. Furthermore, as was also the case with the EGF-R in our studies, LC1 was found to associate with the HGF receptor complex. However, in contrast to our findings, incubation with LC1 antisense agents reduced the ability of HGF to stimulate cell proliferation, as did the addition of an anti-LC1 antibody. This led these authors to conclude that LC1 provided an important link in the signal transduction chain activated by the HGF receptor and speculated that it may function as a signal amplifier.
A consistent, if puzzling, observation made by ourselves and others is that externally applied LC1 can produce the same inhibitory effect on AA release as glucocorticoid treatment of cells and that externally applied anti-LC1 monoclonal antibodies can reverse the effect of dexamethasone. We show here that externally applied LC1 N-terminal peptide fragments interrupt signalling in the same fashion as dexamethasone treatment but without the necessity for internal changes in LC1 phosphorylation. Presumably, this pool can interact directly with a membrane bound target from within as well as without the cell. Indeed, the existence of such proteinaceous LC1 binding sites in the membrane has been repeatedly observed (Goulding et al., 1996). We have previously reported that the methoxy-Tyr21 peptide, which cannot be phosphorylated, was inactive as an inhibitor of AA release in these cells (Croxtall et al., 1998). The notion that the peptide needs to be phosphorylated supports the concept that it may act as a decoy substrate.
If the intracellular levels of LC1 are important in controlling signal transduction, and thus the activity of cPLA2, then deletion of LC1 using antisense or other techniques should potentiate cPLA2 activity in cells. Vishwanath et al. (1992) observed that phospholipase A2 levels were inversely related to intracellular LC1 levels in adrenalectomized animals, thus providing presumptive evidence that this is indeed the case. Transient transfection of A549 cells with antisense LC1 cDNA oligonucleotides blocked the de novo synthesis of LC1, increased the amount of prostaglandins and AA released and lead to an increase in cell growth (Croxtall & Flower, 1994). (Solito et al. (1998), using U937 cells stably transfected with LC1 sense and antisense, also observed an increase in intracellular PLA2 activity in cells bearing the antisense construct.
Inhibition of PTK signalling pathways as a means of controlling tumour cell growth is currently under intensive investigation. Blocking the interaction between Tyr-phosphorylated PTK receptors and Grb2 is considered by some to be an attractive strategy for inhibiting p21ras-dependent pathways involved in mitogenesis (Furet et al., 1998; Smithgall, 1995). Extending this concept to other members of the annexin family, we may perhaps anticipate that LC2, as a substrate for pp60v-src oncogene kinases (Radke & Martin, 1979) as well as the insulin receptor kinase (Karasik et al., 1988), will have an analogous effect on other src-dependent signalling pathways within the cell.
Acknowledgments
We gratefully acknowledge Dr Stefan Peters and Dr Gerd Schnorrenberg of Boehringer Ingelheim who prepared the N-terminal peptides of LC1. We are also grateful to Dr Peter Van Hal of AZR-Dijkzigt, Rotterdam, for his critical comments during the preparation of this manuscript. This work was funded by The Wellcome Trust, of which R.J. Flower is a Principal Research Fellow; we wish to record our gratitude for their support.
Abbreviations
- AA
arachidonic acid
- DAB
diaminobenzidine
- EGF
epidermal growth factor
- Erk
extracellular signal-regulated kinase
- FCS
foetal calf serum
- GR
glucocorticoid receptor
- GRE
glucocorticoid response elements
- Hsp
heat shock protein
- Jnk
Jun N-terminal kinase
- LC1
lipocortin 1
- MAPK
mitogen-activated protein kinase
- Mek
MAPK/Erk kinase
- PBS
phosphate-buffered saline
- PP1
4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PTK
protein tyrosine kinase
- SDS–PAGE
sodium dodecyl sulphate polyacrylamide gel electrophoresis
- Ser
serine
- Thr
threonine
- Tyr
tyrosine
References
- AHN N.G., SEGER R., BRATLIEN R.L., DILTZ C.D., TONKS N.K., KREBS E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. J. Biol. Chem. 1991;266:4220–4227. [PubMed] [Google Scholar]
- BAMBERGER C.M., SCHULTE H.M., CHROUSOS G.P. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocrin. Rev. 1996;17:245–261. doi: 10.1210/edrv-17-3-245. [DOI] [PubMed] [Google Scholar]
- BOST F., MCKAY R., DEAN N., MERCOLA D. The JUN kinase/stress-activated protein kinase pathway is required for epidermal growth factor stimulation of growth of human A549 lung carcinoma cells. J. Biol. Chem. 1997;272:33422–33429. doi: 10.1074/jbc.272.52.33422. [DOI] [PubMed] [Google Scholar]
- BUCKINGHAM J.C. Stress and the neuroendocrine-immune axis: the pivotal role of glucocorticoids and lipocortin 1. Br. J. Pharmacol. 1996;118:1–19. doi: 10.1111/j.1476-5381.1996.tb15360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUDAY L., EGAN S.E., VICIANA P.R., CANTRELL D.A., DOWNWARD J. A complex of Grb2 adaptor protein, Sos exchange factor, and a 36-kDa membrane-bound tyrosine phosphoprotein is implicated in Ras activation in T cells. J. Biol. Chem. 1994;269:9019–9023. [PubMed] [Google Scholar]
- CAELLES C., GONZALEZ-SANCHO J., MUNOZ A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Inhibitory effect of peptides derived from the N-terminus of lipocortin 1 on arachidonic acid release and proliferation in the A549 cell line: identification of E-Q-E-Y-V as a crucial component. Br. J. Pharmacol. 1998;123:975–983. doi: 10.1038/sj.bjp.0701679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., NEWMAN S., FLOWER R.J. Lipocortin 1 and the control of cPLA2 activity in A549 cells. Biochem. Pharmacol. 1996;52:351–356. doi: 10.1016/0006-2952(95)02442-5. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., TOKUMOTO H., FLOWER R.J. Lipocortin 1 and the control of arachidonic acid release in cell signalling. Biochem. Pharmacol. 1995;50:465–474. doi: 10.1016/0006-2952(95)00156-t. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., FLOWER R.J. Lipocortin 1 mediates dexamethasone-induced growth arrest of the A549 lung adenocarcinoma cell line. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3571–3575. doi: 10.1073/pnas.89.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., FLOWER R.J. Antisense oligonucleotides to human lipocortin-1 inhibit glucocorticoid-induced inhibition of A549 cell growth and eicosanoid release. Biochem. Pharmacol. 1994;48:1729–1734. doi: 10.1016/0006-2952(94)90458-8. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., WAHEED S., CHOUDHURY Q., ANAND R., FLOWER R.J. N-terminal peptide fragments of lipocortin-1 inhibit A549 cell growth and block EGF-induced stimulation of proliferation. Int. J. Cancer. 1993;54:153–158. doi: 10.1002/ijc.2910540124. [DOI] [PubMed] [Google Scholar]
- DALMAN F.C., SCHERRER L.C., TAYLOR L.P., AKIL H., PRATT W.B. Localization of the 90 kDa heat shock protein binding site within the hormone binding domain of the glucocorticoid receptor by peptide competition. J. Biol. Chem. 1991;266:3482–3490. [PubMed] [Google Scholar]
- DE VRIES-SMITS A.M.M., BURGERING B.M.T., LEEVERS S.J., MARSHALL C.J., BOS J.L. Involvement of p21ras in activation of extracellular signal-related kinase 2. Nature. 1992;357:602–604. doi: 10.1038/357602a0. [DOI] [PubMed] [Google Scholar]
- FUKAZAWA T., REEDQUIST K.A., PANCHAMORTHY G., SOLTOFF S., TRUB T., DRUKER B., CANTLEY L., SHOELSON S.E., BAND H. T cell activation-dependent association between the p85 subunit of the phosphatidylinositol 3-kinase and Grb2/phospholipase Cγ1-binding phosphotyrosyl protein pp36/38. J. Biol. Chem. 1995;270:20177–20182. doi: 10.1074/jbc.270.34.20177. [DOI] [PubMed] [Google Scholar]
- FURET P., GAY B., CARAVATTI G., GARCIA-ECHEVERRIA C., RAHUEL J., SCHOEPFER J., FRETZ H. Structure-based design and synthesis of high affinity tripeptide ligands of the Grb2-SH2 domain. J. Med. Chem. 1998;41:3442–3449. doi: 10.1021/jm980159a. [DOI] [PubMed] [Google Scholar]
- GALIGNIANA M.D., SCRUGGS J.L., HERRINGTON J., WELSH M.J., CARTER-SU C., HOUSLEY P.R., PRATT W.B. Heat shock protein90-dependent (geldanamycin-inhibited) movement of the glucocorticoid receptor through the cytoplasm to the nucleus requires intact cytoskeleton. Mol. Endocrinol. 1998;12:1903–1913. doi: 10.1210/mend.12.12.0204. [DOI] [PubMed] [Google Scholar]
- GOULDING N.J., PAN L., K., WARDWELL K., GUYRE V.C., GUYRE P.M. Evidence for specific annexin I-binding proteins on human monocytes. Biochem. J. 1996;316:593–597. doi: 10.1042/bj3160593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANKE J.H., GARDNER J.P., DOW R.L., CHANGELIAN P.S., BRISSETTE W.H., WERINGER E.J., POLLOK B.A., CONNELLY P.A. Discovery of a novel, potent, and src-family-selective tyrosine kinase inhibitor: study of Lck- and Fyn-dependent T cell activation. J. Biol. Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- HECK S., KULLMANN M., GAST A., PONTA H., RAHMSDORF H.J., HERRLICH P., CATO A.C.B. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG X., LI Y., TANAKA K., GREGORY MOORE K., HAYASHI J.I. Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase Cγ1, Grb2, and phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11618–11622. doi: 10.1073/pnas.92.25.11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JELINEK T., CATLING A.D., RUETER C.M.W., MOODIE S.A., WOLFMAN A., WEBER M.J. RAS and RAF-1 form a signalling complex with MEK-1 but not MEK-2. Mol. Cell. Biol. 1994;14:8212–8218. doi: 10.1128/mcb.14.12.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARASIK A., PEPINSKY R.B., SHOELSON S.E., KAHN C.R. Lipocortins 1 and 2 as substrates for the insulin receptor kinase in rat liver. J. Biol. Chem. 1988;263:11862–11867. [PubMed] [Google Scholar]
- KEMPPANIEN R.J., BEHREND E.N. Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J. Biol. Chem. 1998;273:3129–3131. doi: 10.1074/jbc.273.6.3129. [DOI] [PubMed] [Google Scholar]
- LIN L.L., WARTMANN M., LIN A.Y., KNOPF J.L., SETH A., DAVIS R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- MELKI V., HULLIN F., MAZARGUIL H., FAUVEL J., RAGAB-THOMAS J.M.F., CHAP H. Annexin I as a potential inhibitor of insulin receptor tyrosine kinase. Biochem. Biophys. Res. Commun. 1994;203:813–819. doi: 10.1006/bbrc.1994.2255. [DOI] [PubMed] [Google Scholar]
- MERRY W.H., CAPLAN R.H., WICKUS G.G., REYNERTSON R.H., KISKEN W.A., COGBILL T.H., LANDERCASPER J. Post operative acute adrenal failure caused by transient corticotropin deficiency. Surgery. 1994;116:1095–1100. [PubMed] [Google Scholar]
- MIGLIACCIO A., DI DOMENICO M., CASTORIA G., DE FALCO A., BONTEMPO P., NOLA E., AURRICHIO F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- OKADA S., KAO A.W., CERESA B.P., BLAIKIE P., MARGOLIS B., PESSIN J.E. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem. 1997;272:28042–28049. doi: 10.1074/jbc.272.44.28042. [DOI] [PubMed] [Google Scholar]
- PEPINSKY R.B., SINCLAIR L.K. Epidermal growth factor-dependent phosphorylation of lipocortin. Nature. 1986;321:81–85. doi: 10.1038/321081a0. [DOI] [PubMed] [Google Scholar]
- PRATT W.B. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc. Soc. Exp. Biol. Med. 1998;217:420–434. doi: 10.3181/00379727-217-44252. [DOI] [PubMed] [Google Scholar]
- RADKE K., MARTIN G.S. Transformation by Rous sarcoma virus: effect of src gene expression on the synthesis and phosphorylation of cellular polypeptides. Proc. Natl. Acad. Sci. U.S.A. 1979;76:5212–5216. doi: 10.1073/pnas.76.10.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REICHARDT H.M., KAESTNER K.H., TUCKERMANN J., KRETZ O., GASS P., SCHMID W., HERRLICH P., ANGEL P., SCHUTZ G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- SCHLAEPFER D.D., HAIGLER H.T. Characterization of Ca2+-dependent phospholipid binding and phosphorylation of lipocortin 1. J. Biol. Chem. 1987;262:6931–6937. [PubMed] [Google Scholar]
- SCHLAEPFER D.D., HAIGLER H.T. In vitro protein kinase C phosphorylation sites of placental lipocortin. Biochemistry. 1988;27:4253–4258. doi: 10.1021/bi00412a008. [DOI] [PubMed] [Google Scholar]
- SKOUTERIS G.G., SCHRODER C.H. The hepatocyte growth factor receptor kinase-mediated phosphorylation of lipocortin-1 transduces the proliferating signal of the hepatocyte growth factor. J. Biol. Chem. 1996;271:27266–27273. doi: 10.1074/jbc.271.44.27266. [DOI] [PubMed] [Google Scholar]
- SMITHGALL T.E. SH2 and SH3 domains: potential targets for anti-cancer drug design. J. Pharmacol. Toxicol. Methods. 1995;34:125–132. doi: 10.1016/1056-8719(95)00082-7. [DOI] [PubMed] [Google Scholar]
- SOLITO E., RAGUENES-NICOL C., DE COUPADE C., BISAGNE-FAURE A., RUSSO-MARIE F. U937 cells deprived of endogenous annexin 1 demonstrate an increased PLA2 activity. Br. J. Pharmacol. 1998;124:1675–1683. doi: 10.1038/sj.bjp.0701991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUMMERS T.A., CREUTZ C.E. Phosphorylation of a chromaffin granule-binding protein by protein kinase C. J. Biol. Chem. 1985;260:2437–2443. [PubMed] [Google Scholar]
- TUMLIN J.A., LEA J.P., SWANSON C.E., SMITH C.L., EDGE S.S., SOMEREN J.S. Aldosterone and dexamethasone stimulate calcineurin activity through a transcription-independent mechanism involving steroid receptor-associated heat shock proteins. J. Clin. Invest. 1997;99:1217–1223. doi: 10.1172/JCI119278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VISHWANATH B.S., FREY F.J., BRADBURY M.J., DALLMAN M.F., FREY B.M. Adrenalectomy decreases lipocortin-I messenger ribonucleic acid and tissue protein content in rats. Endocrinology. 1992;130:585–591. doi: 10.1210/endo.130.2.1531128. [DOI] [PubMed] [Google Scholar]
- WHITESELL L., COOK P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol. Endocrinol. 1996;10:705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]