

Abstract
The effects of cilostamide, a cyclic nucleotide phosphodiesterase 3 (PDE3) selective inhibitor, on vascular intimal hyperplasia were evaluated using a single-balloon injury model and a double-injury model in which the rat common carotid artery was subjected to a second injury at a site injured 14 days previously.
In the double-injury model, the second balloon injury caused more severe intimal hyperplasia (intima/media (IM) ratio, 1.88±0.10) than in the single-injury model (1.09±0.08). Histopathological study revealed that vascular smooth muscle cells (VSMC) were the predominant cell-type in the affected neointimal area.
Oral administration of cilostamide for 2 weeks after the second injury suppressed intimal hyperplasia in the double-injury model (30 mg kg−1 bid, 83% inhibition in terms of the IM ratio, P<0.05; 100 mg kg−1 bid, 69% inhibition, P<0.05). Similar effects were also observed in the single-injury model with oral administration of cilostamide for 2 weeks (100 mg kg−1 bid, 36% inhibition, P<0.01).
Cilostamide inhibited DNA synthesis of cultured VSMC stimulated by foetal calf serum or different kinds of growth factors, but did not affect their migration stimulated by platelet-derived growth factor (PDGF)-BB. Cilostamide significantly increased the cyclic AMP concentration of VSMC dose-dependently.
These results indicate that cilostamide suppresses intimal hyperplasia both in the single- and double-injury models of rat, presumably by inhibiting proliferation rather than migration of VSMC. It is suggested that PDE3 inhibitors might find application in preventing intimal hyperplasia following angioplasty such as percutaneous transluminal coronary angioplasty (PTCA) or stent.
Keywords: Phosphodiesterase, cyclic AMP, cilostamide, intimal hyperplasia, vascular smooth muscle cells, cell proliferation, double-injury model
Introduction
PTCA has rapidly gained acceptance as the procedure of choice for cases of coronary artery disease. Despite improvements in equipment and techniques, however, acute reocclusion after successful angioplasty occurs in approximately 5–10% of patients, and late restenosis, which is one of the most serious problems after coronary angioplasty, occurs in 30–50% (Gurlek et al., 1995; Guerin et al., 1996). PTCA performed with an inflated balloon is associated with endothelial denudation; early accumulation of platelets and fibrin, splitting of the intima and media, stretching of the medial layer and over distension of the adventia (Baptista et al., 1995). Platelet adhesion and aggregation induced by endothelial injury after PTCA have been shown to result in the release of several mitogenes, including PDGF and epidermal growth factor (EGF), from platelets. They cause migration from the media to the intima (Moreno et al., 1996), and proliferation of VSMC, resulting in intimal hyperplasia, one of the critical processes for restenosis (Davies & Hagen, 1994). VSMC, activated endothelial cells and macrophages may also release several kinds of growth factors and extracellular matrixes. However, the molecular mechanisms of restenosis remain to be clarified.
For the investigation of the restenosis process after PTCA and examination of the influence of exogenous agents, models of balloon injury in rat or rabbit arteries have been widely used. Numerous agents such as heparin, angiotensin converting enzyme (ACE) inhibitors and Ca2+ antagonists have thus been shown to prevent intimal lesion formation. However, although these agents are effective in animal models, they have not proved to be beneficial for reducing the incidence of restenosis in human clinical trials (Muller et al., 1992; Jackson, 1994). One explanation for the discrepancy between the results might be differences between the human patient and animal model with respect to the condition of vessel walls. Animal models generally rely on injury to the media of normal arteries without intima, while PTCA in human is performed for atherosclerotic lesions with intima. To resolve this problem, a restenosis model with balloon injury to the femoral artery of hypercholesterolemic rabbits with pre-existing intima has been developed, and evaluated for the effect of hirudin and heparin (Sarembock et al., 1991; Gimple et al., 1991). In contrast to the rabbit, the rat does not respond to cholesterol feeding by forming atherosclerotic lesions with intima. Therefore, we previously designed a balloon double-injury model for rat common carotid arteries (Inoue et al., 1995). In this, a second balloon injury is applied to a rat carotid artery with intima preformed due to a first balloon injury, whereas a single balloon injury is applied to media of rat artery without intima in the traditional balloon injury model. So far, it has been demonstrated that heparin and anti-fibroblast growth factor (FGF)2 antibody, effective in the single injury model, were ineffective for control of intimal hyperplasia in the double injury model (Capron et al., 1997; Koyama & Reidy, 1997).
It is known that cyclic AMP analogues and agents which increase intracellular cyclic AMP concentration inhibit platelet aggregation and proliferation of various kinds of cells. Cyclic nucleotide phosphodiesterases (PDE) are the major enzymes involved in metabolic processing of cyclic AMP and cyclic GMP. In mammalian tissues, at least 10 PDE isozymes, PDE1 ∼PDE10, have been indentified (Beavo et al., 1994; Conti & Jin, 1999; Fujishige et al., 1999), demonstrating different tissue- and cell-distribution patterns. PDE3 is the only cyclic AMP-regulating isozyme expressed in both VSMC and platelets and its inhibition has been shown to block platelet aggregation and proliferation of VSMC in vitro (Hidaka et al., 1979; Souness et al., 1992; Pan et al., 1994; Sudo et al., 2000). Therefore, PDE3 inhibitors may have potential for alleviating restenosis after PTCA.
Among selective PDE3 inhibitors, cilostamide and its derivatives have been widely used as probes to assess physiological functions, as ligands for affinity chromatography to purify the enzyme and as clinical drugs for ischaemic diseases (Hidaka et al., 1979; Degerman et al., 1987; Kimura et al., 1985; Manganiello et al., 1995; Umekawa et al., 1984; Dawson et al., 1998; Sudo et al., 2000). In the present study, we evaluated the inhibitory activity of cilostamide against intimal hyperplasia in arteries using balloon single- and double-injury models. Parts of the present study were previously reported in abstract form (Inoue et al., 1994; 1995).
Methods
Animals
For all the in vivo experiments male 6 week-old Sprague-Dawley rats (Clea Japan, Inc) weighing 180–200 g were used. The animals were housed under controlled temperature (22±2°C) and lighting conditions (a 12-h light-dark cycle) with access to food and water ad libitum. Rats were accustomed to the laboratory conditions for 1 week before the beginning of experimentation.
Arterial balloon injury
Endothelial denudation and injury to the vascular wall was achieved in the left common carotid artery of rats. The rats were anaesthetized with an intraperitoneal administration of sodium pentobarbitone (40 mg kg−1 of body weight) and a 2F Fogarty arterial embolectomy balloon catheter was inserted through the right iliac artery and advanced to the left carotid artery via the aorta. The cervical skin was sectioned and the position of the balloon, which was placed under the muscle layer in the carotid artery, was checked visually. The balloon was then inflated by injecting 30 μl of saline using a 1-ml syringe attached to the end of the catheter, and was passed through the entire length of the artery five times to cause balloon injury. After removal of the catheter from the iliac artery, the wound was ligated and closed. For the double-injury group the catheter was inserted through the left iliac artery and advanced to the left carotid artery and injury caused in the same site as before. The contralateral artery (right carotid artery) remained untreated and served as the intra individual control.
Histopathological study
For histological evaluation, carotid arteries were fixed by perfusion at 120 mmHg with 10% formalin in neutral phosphate buffered solution via a large cannula placed in the left ventricle. Paraffin sections of the arteries were prepared and stained with Elastica-van Gieson (E.V.). The arteries were stained immunohistochemically with HHF-35 to identify VSMC, and antibody to proliferating cell nuclear antigen (PCNA), which is essential for DNA synthesis, to identify the cells that had been stimulated to enter the cell cycle (Groves et al., 1995). Counterstaining was with methylgreen.
In an additional group of experiments, we quantified the time course of changes in area, cell number and population of PCNA positive cells, separately within the media and intima. Subgroups of animals were sacrificed 0, 1, 2, 3, 5, 7, 10, 14, and 28 days after the first balloon injury in the ‘single-injury group' and 1, 2, 3, 5, 7 and 14 days after the second injury in the ‘double-injury group'. Cross sections from three separate arterial segments of each animal were stained with anti-PCNA. The stained nuclei were counted by an investigator blind to the nature of the specimen and were averaged for each animal. The PCNA index was defined as the number of PCNA positive cells divided by the sum of all cells evaluated, expressed as a percentage. Neointimal and medial areas were measured with an NIH image analyzer. The mean intimal and medial areas for each artery were determined from three sections obtained from the isolated portion of the artery.
In the single-injury experiment, the rats were allocated to six groups. Cilostamide was dissolved in 1% hydroxy propyl methyl cellulose (HPMC) and administered orally at doses of 0, 3, 10, 30 or 100 mg kg−1. On the 14th day after the operation, the rats were anaesthetized with ether, injected with heparin (150 u rat −1) sacrificed and their left and right carotid arteries were removed for histopathological study and analysis of DNA contents.
In the double-injury experiment, the rats were allocated to seven groups and subjected to balloon double-injury. Cilostamide was dissolved in HPMC, and administered orally in doses of 0, 3, 10, 30 or 100 mg kg−1, 1 h before the second injury and thereafter twice a day for 2 weeks. On the 28th day after the first operation, rats were anaesthetized with ether, injected with heparin (150 u rat−1), sacrificed and their left and right carotid arteries were removed for histopathological study and analysis of DNA contents.
Analysis of DNA in carotid arteries
DNA contents of 1 cm length of carotid arteries were analysed with a fluorescent method (Kissane & Robins, 1958) as the marker of intimal hyperplasia. The tissues were immersed in 1.0 ml of 0.5 N NaOH and incubated at 37°C overnight until no residues were evident, and neutralized with 0.1 ml of 5 N HCl. Aliquots of 0.2 ml of 3,5-diamino benzoic acid dihydrochloride (250 mg ml−1 H2O) were added, followed by incubation at 60°C for 45 min, cooling to room temperature and mixing with 2 ml of 1.0 N HCl. Fluorescence (peak excitation wavelength, 415 nm; peak emission wavelength, 515 nm) was measured with a fluorimeter (MTP-100F CORONA). A solution of calf thymus DNA in ammonium hydroxide were used as the DNA standard.
VSMC culture
Rat carotid VSMC were prepared by an enzymatic method. After 1 h of digestion at 37°C in Dulbecco's modified Eagle's medium (DMEM), pH 7.4, containing 1 mg ml−1 collagenase S-1, 25 mM HEPES and 10% FCS, carotid arteries were cleaned of adventitia and immersed again in the same media at 24°C for 16 h. The cells were centrifuged for 5 min at 1000 r.p.m, the supernatant was discarded and the pellets were resuspended in a DMEM containing 10% FCS and seeded in T-25 flasks. The medium was renewed twice a week. Cells were used after 3–5 passages.
VSMC DNA synthesis
Measurements of DNA synthesis were carried out according to the method of Morisaki et al. (1988). VSMC were plated into 24-well plates at 2×104 cells well−1 in 1 ml of DMEM containing 10% FBS for 3 days and then made quiescent (G0 stage) by placement in serum free medium for 2 days. Then the medium was changed to 1 ml of DMEM containing cilostamide in absolute dimethyl sulphoxide (DMSO) at a final concentration of 0.05% and 1 μCi 3H-thymidine (925GBq/mmol). Cells were incubated for 24 h, supplemented with transferrin (5 μg ml−1), insulin (3 μg ml−1) and sodium selenite (5 ng ml−1), under the following different conditions: (1) Serum free medium; (2) 1% FCS; (3) EGF (10 ng ml−1, 1.6 nM); (4) FGF (1 ng ml−1, 0.06 nM); (5) PDGF-AA (10 ng ml−1, 0.36 nM); (6) PDGF-AB (10 ng ml−1, 0.37 nM); (7) PDGF-BB (10 ng ml−1, 0.41 nM); (8) transforming growth factor (TGF)-β1 (1 ng ml−1, 0.04 nM). In the last case, cells were cultured for 48 h in serum free medium containing 1 ng ml−1 TGF-β1 and cilostamide, and were pulsed for 24 h by adding [3H]-thymidine. The cells were washed with 1 ml phosphate-buffered saline (PBS, pH 7.4, containing 0.1 mM CaCl2 and 1 mM MgCl2) , and after the addition of 1 ml of ice cold 5% trichloroacetic acid (TCA) to each well, the plates were kept cool for another 30 min. Then the TCA was aspirated and the cells were washed with PBS (pH 7.4) twice, 0.5 ml of 0.5 N NaOH was added to each well and the plates were incubated for 16 h at 37°C before being neutralized with 5 N HCl. Radioactivity was assayed in a liquid scintillation counter and counts were normalized for DNA content as determined by a fluorescent method (Kissane & Robins, 1958).
Lactate dehydrogenase (LDH) activity
Quiescent VSMC were incubated for 24 h at 37°C in 1% FCS with cilostamide. Nifedipine was used as a positive control. After the incubation, the medium was collected and stored at −70°C in a deep freezer until assayed for LDH activity. For this purpose 5 μl aliquots of cell-free supernatant was measured spectrophotometrically at 340 nm using a commercial kit (LDH OPTIMIZED).
VSMC migration
The procedure for assessing VSMC migration has been described previously (Ohlstein et al., 1993). Carvedilol was used as a positive control. Briefly, VSMC were suspended (6.67×105 cells ml−1) in DMEM supplemented with 10% FCS. Migration assays were performed in modified Boyden chambers using a chemotaxicell® cell culture chambers with a polycarbonate membrane (pore size, 8 μm). PDGF-BB (10 ng m−1) was dissolved in DMEM and placed in the lower compartment. Cilostamide and carvedilol were placed in the upper compartment. Aliquots of VSMC (2×105 cells) were then loaded in the upper compartment and incubated for 6 h at 37°C in a CO2 incubator. Nonmigrating cells on the upper surface were scraped away gently. Filters were fixed in 10% formalin and stained with crystal violet. The number of VSMC per x 100 high-power field (HPF) that had migrated to the lower surface of the filters was determined microscopically. Five HPFs were counted per filter. Experiments were performed either in duplicate or in triplicate.
Cyclic AMP measurement
The effects of cilostamide on cyclic AMP content in rat carotid VSMC were assessed by enzyme immunoassay (EIA). Quiescent VSMC were incubated for 3 h at 37°C in 1 ml of DMEM containing 1% FCS with cilostamide. The incubation was terminated by adding 2 ml of ice-cold ethanol, extracts were transferred to fresh tubes, and the cells were washed with 65% ice-cold ethanol. The ethanol was evaporated off and the extracts were dried and stored at −20°C until assay. The cyclic AMP content in VSMC was determined using a cyclic AMP EIA kit.
Intracellular Ca2+ measurement
Measurement of intracellular Ca2+ concentration ([Ca2+]i) was carried out with Fura-2 according to the method of Escobales et al. (1996). Rat carotid VSMC (5×106 cells ml−1) were loaded with Fura-2 by exposure to Fura-2/AM (5 μM), for 20 min at 37°C. Loaded cells were washed free of the extracellular dye. Cells were then incubated with 100 μM cilostamide dissolved in dimethyl formamide at a final concentration of 0.05% for 15 min at room temperature then transferred to a cuvette and placed in a temperature controlled chamber (37°C). Angiotensin II and arginine vasopressin (AVP) were added at final concentrations of 100 nM. The dye was alternately excited at 340 and 380 nm and fluorescent was measured at 510 nm in a Hitachi F-2000 spectrofluorophotometer and [Ca2+]i was calculated.
Statistical analysis
All results are expressed as mean±s.e.mean values for n observations. The statistical significance of differences between multiple groups was evaluated using one-way analysis of variance followed by Dunnett's test (two-tailed). The statistical analysis system (SAS) was adopted for these statistical analyses. In all cases, P<0.05 was considered significant.
Drugs and materials
Cilostamide was synthesized at the Tokushima Research Laboratories of Otsuka Pharmaceutical Co. Ltd. (Tokushima, Japan). A 2F Fogarty arterial embolectomy balloon catheter was purchased from Baxter Healthcare (CA, U.S.A.), muscle actin antibody (HHF-35) and antibody to PCNA from DAKO A/S (Glostrup, Denmark), 3,5-Diamino benzoic acid dihydrochloride from Aldrich Chemical Ltd. (Steinheim, Germany), HPMC from Shinetsu Chemical Industry (Tokyo, Japan), calf thymus DNA, transferrin, sodium selenite, insulin, LDH OPTIMIZED, angiotensin II and AVP from Sigma Chemical Company (St. Louis, MO, U.S.A.), DMEM from Nissui Pharmaceutical Co Ltd (Tokyo, Japan), collagenase S-1 from Nitta gelatin Co. (Osaka Japan), FCS from ICN Biomedical (Australia), cultured flasks and plates from Corning Glass Works (NY, U.S.A.), [3H]-thymidine and a cyclic AMP EIA kit from Amersham International (U.K.), EGF from Upstate Biotechnology Inc. (NY, U.S.A.), bovine brain basic FGF from R & D (Minesota, U.S.A.), PDGF-AA, -AB and -BB, and TGF-β1 from Gibco BRL (U.K.), and Chemotaxicell® from Kurabo (Osaka, Japan). Fura-2/AM was from Dojindo (Kumamoto, Japan). Carvedilol was extracted from Artist® tablets purchased from Daiichi Pharmaceutical Co. LTD (Tokyo Japan).
Results
Histopathological study
A continuous series of histological changes over time were observed by light microscopy in the single- and double-balloon injury models (Figure 1). In the single-balloon injury model, medial areas within the injured carotid arteries were slightly and rapidly increased up to 3 days after the injury and thereafter remained stationary (Figure 2a, day 0, 0.060±0.003 mm2; day 14, 0.081±0.002 mm2). Neointima appeared at 5 days after the first injury and the intimal area increased up to 14 days (0.088±0.010 mm2), with decrease in the lumen area, thereafter becoming stationary (Figure 2b,c). This demonstrated that the decrease in lumen area resulted from the increase of intima, not media. In the double-injury model, the second injury applied at 14 days after the first balloon injury caused the appearance of additional neointima and resulted in more intimal hyperplasia than after the first injury, the intimal area reaching 0.160±0.004 mm2 at 14 days after the second injury. The medial area after the second injury was not significantly changed. IM ratios both in the single- and double-injury model showed similar patterns in each intimal area, and were 1.09±0.08 at 14 days after the first injury and 1.88±0.10 at 14 days after the second injury, respectively (Figure 2d).
Figure 1.
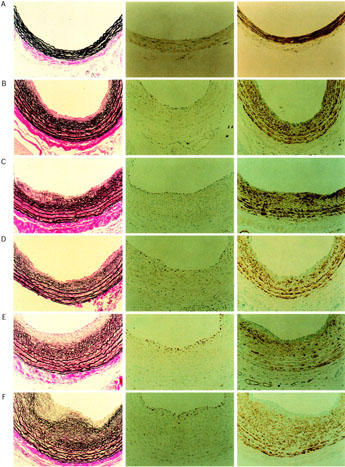
Microphotographs of cross sections from rat left carotid arteries after balloon injury, stained with Elastica van Gieson (left column), and immunohistochemically for PCNA (middle column) and HHF-35 (right column). Day 0 (A) is the day of balloon injury, Day 14 (B) and Day 28 (C) are days after the first injury. Day 14+1 (D), Day 14+3 (E), and Day14+14 (F) are days after the first injury + days after the second injury.
Figure 2.
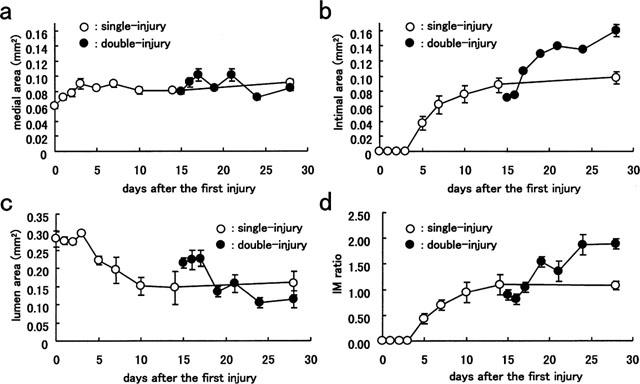
Time course of change in intimal hyperplasia in rat left carotid arteries after balloon injury in single- and double-injury models. (a) Medial area; (b) intimal area; (c) lumen area; (d) intima/media ratio. Each value is the mean±s.e.mean of data for 4–6 rats.
PCNA-positive cells were identified all over the media mostly at 2 and 3 days after the injury in the single-injury model (data not shown), whereas they were identified in the superficial areas of neointima (adjacent to the lumen) in the double-injury model (Figure 1). Areas with PCNA-positive cells were weakly stained with HHF-35 and lacked organized elastic fibres. In both single- and double-injury models the vessels were markedly stained with HHF-35, indicating VSMC to be the predominant cell-type. Macroscopic thrombus formation in response to single or double injury was not observed.
Time course of cell number and PCNA expression
In the single-injury model, the cell number in the media began to increase 2 days after balloon injury and reached a maximum after 5 days (Table 1). Intima cells appeared 5 days after the balloon injury and the number of intimal cells increased thereafter. In the double-injury model, the second injury resulted in further increase of cell number in the intima, but not in the media. Cell proliferation was not observed within the media of uninjured segments of artery (Table 1, day 0, PCNA labelling index: 0%). The DNA content in the carotid artery increased with the formation of intima after the first balloon injury, with a maximum value observed after 7 days, and then increased further after the second injury with neointima formation (Figure 3). Expression of PCNA was maximally induced in the media underlying an intact internal elastic lamina (PCNA labelling index: 19.1±4.9%) at 2 days after the first balloon injury and diminished thereafter. At 5 days after the first injury, the PCNA labelling index in the intima reached a maximum (20.0±4.3%). In the double-injury model, the second balloon injury induced PCNA in the neointima, but essentially not in the media, reaching a maximum level after 2 days (22.6±4.2%). The maximum PCNA labelling indices were not significantly different from those in the single-injury model.
Table 1.
Numbers of cells and cell proliferative activity within the media and intima after injury
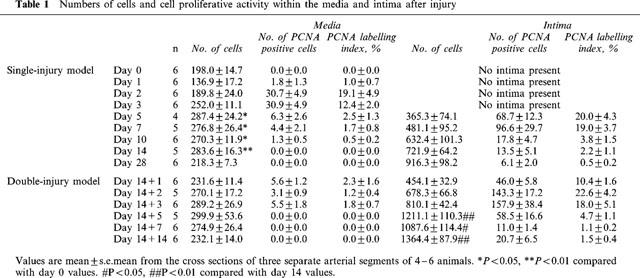
Figure 3.
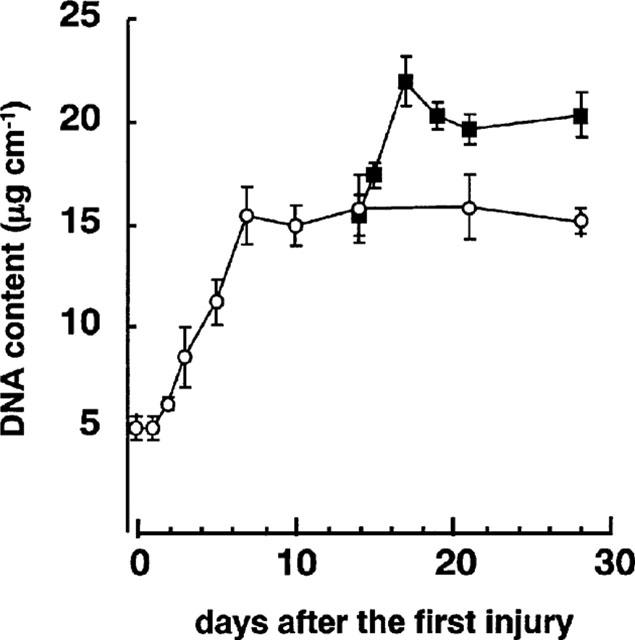
Time course of change in DNA content in rat left carotid arteries after balloon injury in single- and double-injury models. Each value is the mean±s.e.mean of data for five rats on days 0–14, six rats on day 21, and 10 rats on day 28 in the single-injury group. In the double-injury group, means are for five rats except for days 17 and 28 (mean of four rats each).
Effects of cilostamide on intimal hyperplasia in the single- and double-injury models
In the single-injury model, cilostamide orally administered for 14 days after the injury significantly suppressed the increase of the intimal area at 100 mg kg−1 bid (42% inhibition, P<0.01, Table 2), but had no significant effect on medial area. It also decreased the IM ratio at 100 mg kg−1 bid (36% inhibition, P<0.01) and suppressed the decrease of lumen area (45% inhibition, P<0.01). Cilostamide did not affect in normal right carotid arteries (data not shown). In the double-injury model, cilostamide was orally administered 1 h before and for 14 days after the second injury. It almost completely suppressed the additional increase of neointima by the second balloon injury at 30 and 100 mg kg−1 bid (30 mg kg−1, 79% inhibition, P<0.05; 100 mg kg−1, 91% inhibition, P<0.05, Table 3). The increase of IM ratio by the second injury was also significantly reduced (30 mg kg−1, 83% inhibition, P<0.01; 100 mg kg−1, 69% inhibition, P<0.05). Cilostamide almost completely eliminated the decrease of lumen area by the second injury (100 mg kg−1, 100% inhibition, P<0.01). The effective dose was one-third of that in the single injury model.
Table 2.
Effects of cilostamide on intimal hyperplasia in the rat balloon single-injury model
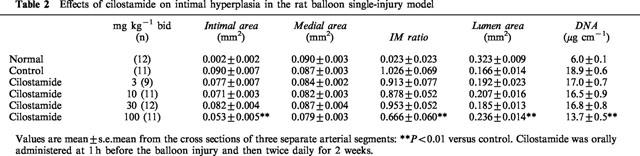
Table 3.
Effects of cilostamide on intimal hyperplasia in the rat balloon double-injury model

Effects of cilostamide on cultured VSMC
Cilostamide inhibited the DNA synthesis of cultured VSMC induced by FCS, EGF, FGF, PDGF-AA, PDGF-AB, PDGF-BB or TGF-β1 in a dose-dependent manner (Figure 4). The similar potency observed with several kinds of growth factors indicated that this was independent of the type of stimulation. Cilostamide also exhibited inhibitory effects on DNA synthesis in the absence of FCS or growth factors, suggesting an action on proliferation of VSMC by autocrine secretion of growth factors such as smooth muscle cell-derived growth factor (SDGF) (Morisaki et al., 1989). Under these experimental conditions, cilostamide did not affect the DNA content (data not shown). Examination of the effects of cilostamide on PDGF-BB (10 ng ml−1) stimulation of VSMC migration with the Boyden chamber method did not demonstrate any effect up to 100 μM (Figure 5). In contrast, carvedilol (100 μM) completely inhibited the migration. Cilostamide (1–100 μM) significantly increased the cyclic AMP concentration in quiescent VSMC cultured with 1% FCS for 3 h (Figure 6). The effect was dose-dependent and the highest concentration of cilostamide tested (100 μM) caused an approximately 2 fold increase in cyclic AMP accumulation. The effect of cilostamide on Ca2+-elevation stimulated by AVP and angiotensin II in rat carotid VSMC. Cilostamide did not affect their Ca2+-elevation up to 100 μM (Figure 7 and data not shown). Cytotoxicity of cilostamide was evaluated by assessment of LDH release from VSMC into the culture medium. No effects were observed up to 100 μM (Figure 8), which almost completely inhibited DNA synthesis in VSMC (Figure 4). In contrast, 30 μM of nifedipine significantly increased LDH release.
Figure 4.
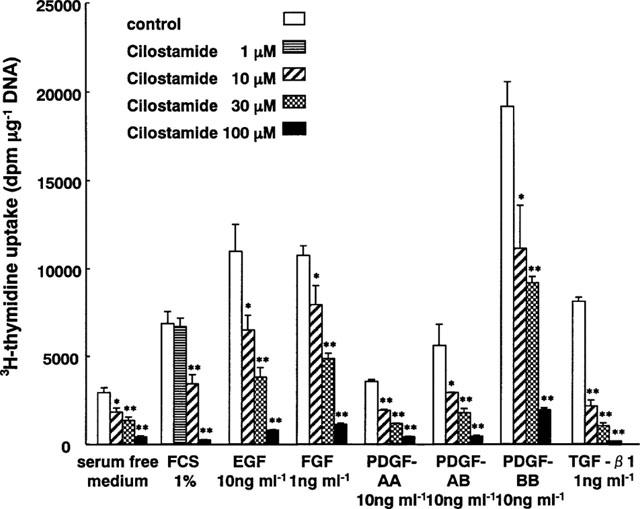
Inhibitory effects of cilostamide on DNA synthesis in VSMC. Quiescent cells were stimulated with FCS, EGF, FGF, PDGF-AA, PDGF-AB, PDGF-BB and TGF-β1 in the presence of various amounts of cilostamide. *P<0.05 and **P<0.01 compared with the control level.
Figure 5.
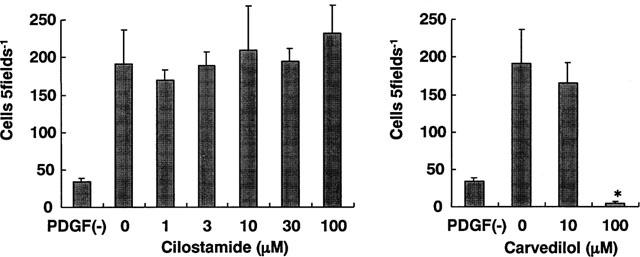
Effects of cilostamide on VSMC migration stimulated by PDGF-BB. Migration assays were performed in Chemotaxicell cell culture chambers. PDGF-BB (10 ng ml−1) was placed in the lower compartment together with the indicated concentration of cilostamide or carvedilol. VSMC (2×105 cells) were loaded in the upper compartment and incubated for 6 h. The number of VSMC per x 100 high-power field (HPF) that had migrated to the lower surface of the filters was determined. Five HPFs were counted per filter. Experiments were performed either in duplicate or in triplicate.
Figure 6.
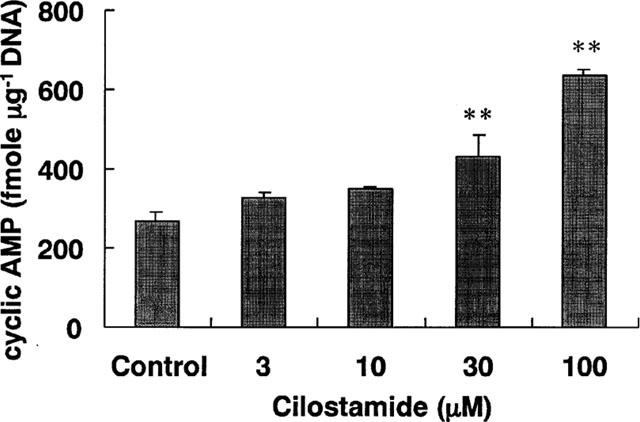
Effects of cilostamide on cyclic AMP concentration in VSMC. VSMC were cultured with cilostamide for 3 h. Data are means from three experiments and the vertical lines represent s.e.mean values. **P<0.01 compared with the control.
Figure 7.
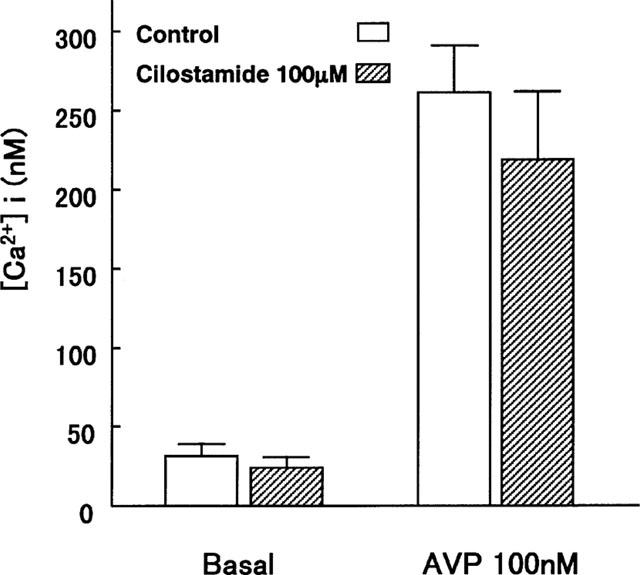
Effects of cilostamide on [Ca2+]i in VSMC. VSMC were pre-treated with 100 μM cilostamide for 15 min and stimulated with AVP. Data are means from five experiments and the vertical lines represent s.e.mean values.
Figure 8.
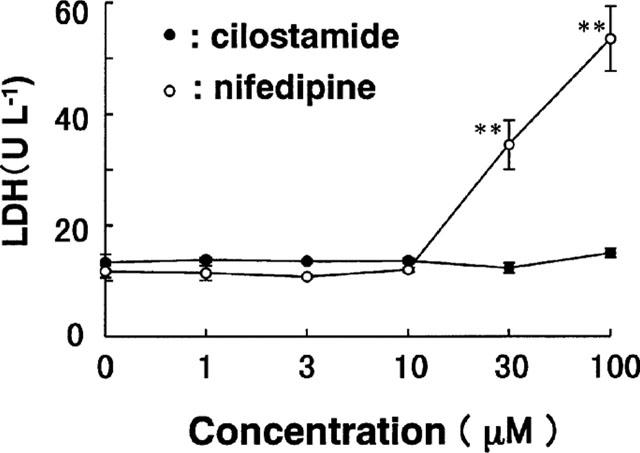
Effects of cilostamide on LDH release from VSMC. Quiescent VSMC were incubated for 24 h in the presence of the indicated concentrations of cilostamide and nifedipine. Data are means from three experiments and the vertical lines are s.e.mean values. **P<0.01 compared with the control level.
Discussion
Single- and double-injury models
The balloon single-injury model has been the most frequently employed in vivo model in restenosis research. Rats are commonly used due to the reproducible prominent formation of intimal hyperplasia within 1 or 2 weeks after balloon injury (Clowes & Schwartz, 1985). Since normal healthy arteries are injured by the balloon catheter in the model, the lack of preexisting plaque and intima in the injured artery is a definitive difference from human restenosis (Jackson, 1994). Rat carotid arteries only rarely demonstrate intimal cells and the rat also does not respond to cholesterol feeding, which causes atherosclerotic lesions with intima in the rabbit and pig. In the present evaluation of cilostamide, we therefore applied a balloon double-injury model using rat carotid arteries (Inoue et al., 1995).
In order to study the property of the double-injury model, we evaluated the time course of change in area and cell number within the media and intima after both the first and second injuries. The first injury resulted in an increase of area and cell number both in the media and intima (Figure 2, Table 1), as previously reported by Schwartz et al. (1995). The area and cell number in intima was prominently increased by the second injury from 3–5 days after the injury, while those in the media remained unchanged. The PCNA labeling index was very elevated in the intima between 1 and 3 days after the second injury, that is the period before increases of VSMC proliferation and intimal area, while only a few PCNA positive cells were identified in the media. These results suggest that proliferation of VSMC plays a critical role in the increase of the intimal area in this double-injury model. Evaluation of the PCNA labelling index indicated that the total proliferation activities of VSMC after the first and second injury were comparable. The big difference between the two cases was the response of medial cells, this being essentially lacking to the second injury. These results show that only intimal cells in the injured and pre-formed region proliferate after the second injury in the double-injury model, while both medial and intimal cells are involved in the single-injury model. The patterns of increase in the DNA content within injured arteries showed similar trends to the total cell number and total area of intima and media, suggesting that the DNA content might be a good index for evaluating intimal hyperplasia, not only with the single injury but also in the double-injury rat model.
Balloon double-injury to carotid arteries has been previously applied in rabbits (Jorgensen et al., 1988; Niimi et al., 1994; Gerdes et al., 1996), the second injury leading to more severe intimal hyperplasia than after the first injury, in accordance with our results with rats, and a high rate of earlier thrombotic occlusion. In contrast to rats and rabbits, it seems that the second injury to pig coronary artery does not lead to additional intimal hyperplasia (Bonan et al., 1996). Recently, heparin and anti-FGF2 antibody were both not effective at depressing intimal hyperplasia in the rat double-injury model (Capron et al., 1997; Koyama & Reidy, 1997), although these could be demonstrated in the rat single-injury model. In the rabbit, short-term treatment of recombinant hirudin (i.v. bolus immediately before injury, followed by i.v. infusion for 2 h) reduced neointimal formation in both the single- and double-injury model (Gerdes et al., 1996). In the single-injury model of rats, however, no inhibition of neointimal growth was observed after short-term treatment, although long term treatment of hirudin (3 or 14 days infusion) was effective. Gerdes et al. (1996) proposed that differences in responsiveness to hirudin treatment between rats and rabbits might be explained by differential activity of the coagulation system or by variable thrombogenicity of the injured vessel wall surface. The change in intimal size in the double-injury model of rat might be attributed to an increase in matrix synthesis (Koyama & Reidy, 1997). Although this parameter was not examined in the present study, Strauss et al. (1994) quantified matrix synthesis in the double-injury model of rabbit arteries and noted an increase in extracellular matrix. In another study, the second injury increased the whole mass (wet weight) of intima-media, its DNA, elastin content, and the I/M ratio; but the collagen content remained unaffected (Capron et al., 1997). Usage of the double-injury model of rat has several limitations. Double-injury to rat carotid artery is a highly simplified model for the study of restenosis after angioplasty in human. The injured vessel wall lacks several features of atherosclerotic plaques such as extracellular and intracellular lipid deposition, infiltration by macrophages and T-cells, and neovascularization. With the exception of proliferation and migration of VSMC and extracellular matrix synthesis, many aspects likely to be involved in the arterial reaction to angioplasty, such as plaque rupture, mural thrombus formation, elastic recoil and remodelling, are absent. However, the present study indicated that intimal hyperplasia in the double-injury model of rat was strongly affected by the proliferation of VSMC.
Inhibitory effect of cilostamide on VSMC hyperplasia
For the purpose of developing drugs that prevent restenosis after PTCA, we have focused on PDE3 inhibitors. The present study indicated that cilostamide suppressed intimal hyperplasia not only by single-balloon but also by double-injury. In order to elucidate the mechanism of anti-intimal hyperplasia, we examined the effects of cilostamide on the proliferation and migration of cultured rat carotid VSMC, because both of these parameters are highly relevant to intimal hyperplasia (Ross, 1993; Schwartz et al., 1995). Cilostamide inhibited DNA synthesis of cultured VSMC in vitro, but not their migration, suggesting that anti-intimal hyperplasia effects of cilostamide may be due to anti-VSMC proliferation rather than anti-migration effects. The present LDH release study (Figure 8) demonstrated that the inhibitory effect of cilostamide on VSMC proliferation in vitro is not due to cytotoxicity. In addition, 100 mg kg−1 bid of cilostamide did not cause any toxic effects in vivo (data not shown).
Whereas intimal hyperplasia in the single-injury model results from proliferation of VSMC in media followed by the migration to the intima and proliferation there, additional intimal hyperplasia in the double-injury model seems to result from proliferation of VSMC in the intima. The fact that cilostamide suppressed intimal hyperplasia in the double-injury model more potently than in the single injury model, thus provides further evidence of VSMC proliferation and not migration as the most important process.
It has been shown that cilostamide strongly inhibits platelet aggregation (Hidaka et al., 1979; Sudo et al., 2000) and this might contribute to suppression of intimal hyperplasia in vivo, because growth factors, like PDGF, released from activated platelets stimulate proliferation of VSMC. However, in the rat model of single-injury, platelets and PDGF were found not to play such a role, rather regulating migration of VSMC from the media into the intima (Fingerle et al., 1989; Ferns et al., 1991). In the present study, we employed 3 mg kg−1 bid, which exhibits no effect on platelet aggregation ex vivo in rat, to 100 mg kg−1 bid, which potently inhibits the aggregation. While administration of 10 mg kg−1 of cilostamide inhibits ex vivo aggregation (data not shown), this dose did not suppress intimal hyperplasia in either single- or double-injury models. Therefore, it is unclear to what extent anti-platelet actions of cilostamide contribute to its anti-intimal hyperplasia effects.
It has been reported that dipyridamole (PDE5 inhibitor), AH-P719 (PDE3 inhibitor) and SCH51866 (PDE1 and 5 inhibitor) inhibit VSMC proliferation in intimal hyperplasia models in the rabbit and rat (Wojenski et al., 1988; Singh et al., 1994; Vemulapalli et al., 1996). In addition, it was recently shown that other clinically available PDE3 inhibitors, aminophylline, amrinone and cilostazol may suppress formation of intima with local administration but not intravenous administration in the single-injury model (Indolfi et al., 1997; Ishizaka et al., 1999). Agents which increase the intracellular cyclic AMP concentration are known to inhibit cell growth (Drees et al., 1993). In this study with rat carotid artery VSMC, the fact that the PDE3 inhibitor cilostamide increased the cyclic AMP level (Figure 6) thus suggests that its inhibitory effects on VSMC proliferation were due to this elevation. Many agents, like dipyridamole, ACE inhibitors and Ca2+ antagonists, have not exhibited protective effects against restenosis after PTCA in clinical trials (Schwartz et al., 1988; Muller et al., 1992; Jackson, 1994). This also suggests that intimal hyperplasia in rat and rabbit models could only partly reflect the pathogenesis of human restenosis.
Cross-talk between the cyclic AMP-dependent protein kinase (PKA) pathway and the mitogen-activated protein kinase (MAPK) cascade has been proposed (Wu et al., 1993; Cook & McCormick, 1993; Graves et al., 1993; Marx, 1993). Increase of the intracellular cyclic AMP level activates PKA and this in turn phosphorylates Raf-1 and inhibits its kinase activity. This hinders the MAPK cascade and results in attenuation of cell proliferation. In mesangial cells, it has been reported that activation of PKA by cilostamide decreases MAPK activity and cell growth (Matousovic et al., 1995). In cultured pig aortic VSMC, four PDE isozymes, PDE1, 3, 4 and 5, have been identified (Souness et al., 1992; Xiong et al., 1995). PDE3 and 4 hydrolyze cyclic AMP and regulate the intracellular concentration in VSMC. It has been reported that a PDE3 inhibitor (SKF94836) and a PDE4 inhibitor (Ro20-1724) reduce DNA synthesis and proliferation of pig aortic VSMC through elevation of intracellular cyclic AMP, whereas zaprinast, an inhibitor of PDE5, involved in hydrolysis of cyclic GMP, was without effect (Souness, 1992). Pan et al. (1994) reported that cilostamide suppresses DNA synthesis in the VSMC cell line, A10, with an EC50 of 5.3 μM, while the inhibitory potentials of the PDE4 inhibitors, rolipram and Ro20-1724, were very weak (EC50=111 and 100 μM, respectively). These data suggest that PDE3 is the major PDE isozyme regulating DNA synthesis and proliferation of VSMC.
In conclusion, the present study showed that a second balloon injury to rat carotid arteries further stimulates proliferation of neointimal VSMC. Cilostamide inhibited this neointimal formation with significant anti-VSMC proliferation activity without cytotoxicity or effects on migration. The results suggest that cilostamide or similar PDE3 inhibitors might be promising agents in preventing intimal hyperplasia after angioplasties such as PTCA or stent in humans. In this context, it should be noted that there are several recent clinical reports that cilostazol, a derivative of cilostamide, reduced restenosis after directional coronary atherectomy and PTCA by inhibiting neointimal proliferation (Tsuchikane et al., 1998; 1999).
Acknowledgments
We thank M. Nagamoto, Y. Tanaka, T. Utsunomiya, K. Hayashi, Y. Nagamura, H. Ito and K. Maeda (Otsuka Pharmaceutical Co. Ltd.) for their technical assistance and advice and Dr Malcolm Moore for critical reading of the manuscript.
Abbreviations
- ACE
angiotensin converting enzyme
- AVP
arginine vasopressin
- DMSO
dimethyl sulphoxide
- EGF
epidermal growth factor
- EIA
enzyme immunoassay
- FCS
foetal calf serum
- FGF
fibroblast growth factor
- IM ratio
intima/media ratio
- LDH
lactate dehydrogenase
- MAPK
mitogen-activated protein kinase
- PBS
phosphate buffered saline
- PCNA
proliferating cell nuclear antigen
- PDE
cyclic nucleotide phosphodiesterase
- PDGF
platelet-derived growth factor
- PKA
cyclic AMP-dependent protein kinase
- PTCA
percutaneous transluminal coronary angioplasty
- SDGF
smooth muscle cell-derived growth factor
- VSMC
vascular smooth muscle cells
References
- BAPTISTA J., UMANS V.A., DI MARIO C., ESCANED J., DE FEYTER P., SERRUYS P.W. Mechanism of luminal enlargement and quantification of vessel wall trauma following balloon coronary angioplasty and directional atherectomy. Eur. Heart. J. 1995;16:1603–1612. doi: 10.1093/oxfordjournals.eurheartj.a060784. [DOI] [PubMed] [Google Scholar]
- BEAVO J.A., CONTI M., HEASLIP R.J. Multiple cyclic nucleotide phosphodiesterases. Mol. Pharmacol. 1994;46:399–405. [PubMed] [Google Scholar]
- BONAN R., PAIEMENT P., LEUNG T.K. Swine model of coronary restenosis: effect of a second injury. Cathet. Cardiovasc. Diagn. 1996;38:44–49. doi: 10.1002/(SICI)1097-0304(199605)38:1<44::AID-CCD10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- CAPRON L., JARNET J., HEUDES D., MONROSE D.J., BRUNEVAL P. Repeated balloon injury of rat aorta: A model of neointima with attenuated inhibition by heparin. Arterioscler. Thromb. Vasc. Biol. 1997;17:1649–1656. doi: 10.1161/01.atv.17.9.1649. [DOI] [PubMed] [Google Scholar]
- CLOWES A.W., SCHWARTZ S.M. Significance of quiescent smooth muscle migration in the injured rat carotid artery. Circ. Res. 1985;56:139–145. doi: 10.1161/01.res.56.1.139. [DOI] [PubMed] [Google Scholar]
- CONTI M., JIN S.L.C. The molecular biology of cyclic nucleotide phosphodiesterases. Prog. Nucleic. Acid Res. Mol. Biol. 1999;63:1–38. doi: 10.1016/s0079-6603(08)60718-7. [DOI] [PubMed] [Google Scholar]
- COOK S.J., MCCORMICK F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–1072. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- DAVIES H.G., HAGEN P.O. Pathobiology of intimal hyperplasia. Br. J. Surgery. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- DAWSON D.L., CUTLER B.S., MEISSNER M.H., STRANDNESS D.E. Cilostazol has beneficial effects in treatment of intermittent claudication. Circulation. 1998;98:678–686. doi: 10.1161/01.cir.98.7.678. [DOI] [PubMed] [Google Scholar]
- DEGERMAN E., BELFRAGE P., NEWMAN A.H., RICE K.C., MANGANIELLO V.C. Purification of the putative hormone-sensitive cyclic AMP phosphodiesterase from rat adipose tissue using a derivative of cilostamide as a novel affinity ligand. J. Biol. Chem. 1987;262:5797–5807. [PubMed] [Google Scholar]
- DREES M., ZIMMERMANN R., EISENBRAND G. 3′, 5′-cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993;53:3058–3061. [PubMed] [Google Scholar]
- ESCOBALES N., CASTRO M., ALTIERI P.I., SANABRIA P. Simvastatin releases Ca2+ from a thapsigargin-sensitive pool and inhibits InsP3-dependent Ca2+ mobilization in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1996;27:383–391. doi: 10.1097/00005344-199603000-00011. [DOI] [PubMed] [Google Scholar]
- FERNS G.A.A., RAINES E.W., SPRUGEL K.H., MOTANI A.S., REIDY M.A., ROSS R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- FINGERLE J., JOHNSON R., CLOWES A.W., MAJESKY M.W., REIDY M.A. Role of platelets in smooth muscle cell proliferation and migration after vascular injury in rat carotid artery. Proc. Natl. Acad. Sci. 1989;86:8412–8416. doi: 10.1073/pnas.86.21.8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUJISHIGE K., KOTERA J., MICHIBATA H., YUASA K., TAKEBAYASHI Si., OKUMURA K., OMORI K. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A) J. Biol. Chem. 1999;274:18438–18445. doi: 10.1074/jbc.274.26.18438. [DOI] [PubMed] [Google Scholar]
- GERDES C., FABER-STEINFELD V., YALKINOUGLU O., WOHLFEIL S. Comparison of the effects of the thrombin inhibitor r-hirudin in four animal models of neointima formation after arterial injury. Arterioscler. Thromb. Vasc. Biol. 1996;16:1306–1311. doi: 10.1161/01.atv.16.10.1306. [DOI] [PubMed] [Google Scholar]
- GIMPLE L.W., GERTZ S.D., HABER H.L., RAGOSTA M., POWERS E.R., ROBERTS W.C., SAREMBOCK I.J. Effect of chronic subcutaneous or intramural administration of heparin on femoral artery restenosis after balloon angioplasty in hypercholesterolemic rabbits. Circulation. 1991;86:1536–1546. doi: 10.1161/01.cir.86.5.1536. [DOI] [PubMed] [Google Scholar]
- GRAVES L.M., BORNFELDT K.E., RAINES E.W., POTTS B.C., MACDONALD S.G., ROSS R., KREBS E.G. Protein kinase A antagonizes platelet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A.. 1993;90:10300–10304. doi: 10.1073/pnas.90.21.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GROVES P.H., BANNING A.P., PENNY W.J., LEWIS M.J., CHEADLE H.A., NEWBY A.C. Kinetics of smooth muscle cell proliferation and intimal thickening in a pig carotid model of balloon injury. Atherosclerosis. 1995;117:83–96. doi: 10.1016/0021-9150(95)05562-b. [DOI] [PubMed] [Google Scholar]
- GUERIN Y., SPAULDING C., DESNOS M., FUNCK F., RAHAL S., PY A., BESSE B., TSOCANAKIS O., GUERIN F., GUEROT C. Rotational atherectomy with adjunctive balloon angioplasty versus conventional percutaneous transluminal coronary angioplasty in type B2 lesions; results of a randomized study. Am. Heart J. 1996;131:879–883. doi: 10.1016/s0002-8703(96)90168-4. [DOI] [PubMed] [Google Scholar]
- GURLEK A., DAGALP Z., ORAL D., OMURLU K., EROL C., AKYOL T., TUTAR E. Restenosis after transluminal coronary angioplasty: a risk factor analysis. J. Cardiovasc. Risk. 1995;2:51–55. [PubMed] [Google Scholar]
- HIDAKA H., HAYASHI H., KOHRI H., KIMURA Y., HOSOKAWA T., IGAWA T., SAITOH Y. Selective inhibitor of platelet cyclic adenosine monophosphate phosphodiesterase, cilostamide, inhibits platelet aggregation. J. Pharmacol. Exp. Ther. 1979;211:26–30. [PubMed] [Google Scholar]
- INDOLFI C., AVVEDIMENTO E.V., LORENZO E.D., ESPOSITO G., RAPACCIUOLO A., GIULIANO P., GRIECO D., CAVUTO L., STINGONE A.M., CIULLO I., CONDORELLI G., CHIARIELLO M. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nature Med. 1997;3:775–779. doi: 10.1038/nm0797-775. [DOI] [PubMed] [Google Scholar]
- INOUE Y., KIMURA Y., TOCHIZAWA S., TOGA K., TACHIBANA K., YOSHIDA Y., HIDAKA H.A new model of rat arterial restenosis. -Two stage injury model- Thromb. Haemostasis. 1995731335(Abstract form) [Google Scholar]
- INOUE Y., TOGA K., TACHIBANA K., KIMURA Y., HIDAKA H.Prevention of intimal proliferation by cilostamide Atherosclerosis 1994109253(Abstract form) [Google Scholar]
- ISHIZAKA N., TAGUCHI J., KIMURA Y., IKARI Y., AIZAWA T., TOGO M., MIKI K., KUROKAWA K., OHNO M. Effects of a single local administration of cilostazol on neointimal formation in balloon-injured rat carotid artery. Atherosclerosis. 1999;142:41–46. doi: 10.1016/s0021-9150(98)00147-6. [DOI] [PubMed] [Google Scholar]
- JACKSON C.L. Animal models of restenosis. Trends Cardiovasc. Med. 1994;4:122–130. doi: 10.1016/1050-1738(94)90064-7. [DOI] [PubMed] [Google Scholar]
- JORGENSEN L., GROTHE A.G., GROVES H.M., KINLOUGH-RATHBONE R.L., RICHARDSON M., MUSTARD J.F. Sequence of cellular responses in rabbit aortas following one and two injuries with a balloon catheter. Br. J. Exp. Path. 1988;69:473–486. [PMC free article] [PubMed] [Google Scholar]
- KIMURA Y., TANI T,. , KANBE T., WATANABE K. Effect of cilostazol on platelet aggregation and experimental thrombosis. Arzneim.-Forsch./Drug Res. 1985;35:1144–1149. [PubMed] [Google Scholar]
- KISSANE J.M., ROBINS E. The fluorometric measurement of deoxyribonucleic acid in animal tissue with special reference to the central nervous system. J. Biol. Chem. 1958;233:184–188. [PubMed] [Google Scholar]
- KOYAMA H., REIDY M.A. Reinjury of arterial lesions induces intimal smooth muscle cell replication that is not controlled by fibroblast growth factor 2. Circ. Res. 1997;80:408–417. [PubMed] [Google Scholar]
- MANGANIELLO V.C., TAIRA M., DEGERMAN E., BELFRAGE P. Type III cGMP-inhibited cyclic nucleotide phosphodiesterases (PDE3 gene family) Cell. Signal. 1995;7:445–455. doi: 10.1016/0898-6568(95)00017-j. [DOI] [PubMed] [Google Scholar]
- MARX J. Two major signal pathways linked. Science. 1993;262:988–990. doi: 10.1126/science.8257559. [DOI] [PubMed] [Google Scholar]
- MATOUSOVIC K., GRANDE J.P., CHINI C.C.S., CHINI E.N., DOUSA T.P. Inhibitors of cyclic nucleotide phosphodiesterase isozymes type-III and type-IV suppresses mitogenesis of rat mesangial cells. J. Clin. Invest. 1995;96:401–410. doi: 10.1172/JCI118049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORENO P.R., BERNARDI V.H, LOPEZ-CUELLAR J., NEWELL J.B., MCMELLON C., GOLD H.K., PALACIOS I.F., FUSTER V., FALLON J.T. Macrophage infiltration predicts restenosis after coronary intervention in patients with unstable angina. Circulation. 1996;94:3098–3102. doi: 10.1161/01.cir.94.12.3098. [DOI] [PubMed] [Google Scholar]
- MORISAKI N., KANZAKI T., MOTOYAMA N., SAITO Y., YOSHIDA S. Cell cycle-dependent inhibition of DNA synthesis by prostaglandin I2 in cultured rabbit aortic smooth muscle cells. Atherosclerosis. 1988;71:165–171. doi: 10.1016/0021-9150(88)90140-2. [DOI] [PubMed] [Google Scholar]
- MORISAKI N., KOYAMA N., MORI S., KANZAKI T., KOSHIKAWA T., SAITO Y., YOSHIDA S. Effects of smooth muscle cell derived growth factor (SDGF) in combination with other growth factors on smooth muscle cells. Atherosclerosis. 1989;78:61–67. doi: 10.1016/0021-9150(89)90159-7. [DOI] [PubMed] [Google Scholar]
- MULLER D.W.M., ELLIS S.G., TOPOL E.J. Experimental models of coronary artery restenosis. J. Am. Coll. Cardiol. 1992;19:418–432. doi: 10.1016/0735-1097(92)90500-m. [DOI] [PubMed] [Google Scholar]
- NIIMI Y., AZUMA H., HIRAKAWA K. Repeated endothelial removal augments intimal thickening and attenuates EDRF release. Am. J. Physiol. 1994;266:H1348–H1356. doi: 10.1152/ajpheart.1994.266.4.H1348. [DOI] [PubMed] [Google Scholar]
- OHLSTEIN E.H., DOUGLAS S.A., SUNG C.P., YUE T.L., LOUDEN C., ARLETH A., POSTE G., RUFFOLO R.R., FEUERSTEIN G.Z. Carvedilol, a cardiovascular drug, prevents vascular smooth muscle cell proliferation, migration and neointimal formation following vascular injury. Proc. Natl. Acad. Sci. 1993;90:6189–6193. doi: 10.1073/pnas.90.13.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAN X., ARAUZ E., KRZANOWSKI J.J., FITZPATRICK D.F., POLSON J.B. Synergistic interactions between selective pharmacological inhibitors of phosphodiesterase isozyme families PDEIII and PDEIV to attenuate proliferation of rat vascular smooth muscle cells. Biochem. Pharmacol. 1994;48:827–835. doi: 10.1016/0006-2952(94)90062-0. [DOI] [PubMed] [Google Scholar]
- ROSS R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- SAREMBOCK I.J., GERTZ S.D., GIMPLE L.W., OWEN R.M., POWERS E.R., ROBERTS W.C. Effectiveness of recombinant desulphatohirudin in reducing restenosis after balloon angioplasty of atherosclerotic femoral arteries in rabbits. Circulation. 1991;84:232–243. doi: 10.1161/01.cir.84.1.232. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ L., BOURASSA M.G., LESPERANCE J., ALDRIDGE H.E., KAZIM F., SALVATORI V.A., HENDERSON M., BONAN R., DAVID P.R. Aspirin and dipyridamole in the prevention of restenosis after percutaneous transluminal coronary angioplasty. New Engl. J. Med. 1988;318:1714–1719. doi: 10.1056/NEJM198806303182603. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ S.M., DEBLOIS D., O'BRIEN E.R.M. The intima. Soil for atherosclerosis and restenosis. Circ. Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- SINGH J.P., ROTHFUSS K.J., WIERNICKI T.R., LACEFIELD W.B., KURTZ W.L., BROWN R.F., BRUNE K.A., BAILEY D., DUBE G.P. Dipyridamole directly inhibits vascular smooth muscle cell proliferation in vitro and in vivo: implications in the treatment of restenosis after angioplasty. J. Am. Coll. Cardiol. 1994;23:665–671. doi: 10.1016/0735-1097(94)90752-8. [DOI] [PubMed] [Google Scholar]
- SOUNESS J.E., HASSALL G.A., PARROTT D.P. Inhibition of pig aortic smooth muscle cell DNA synthesis by selective type III and type IV cyclic AMP phosphodiesterase inhibitors. Biochem. Pharmacol. 1992;44:857–866. doi: 10.1016/0006-2952(92)90116-z. [DOI] [PubMed] [Google Scholar]
- STRAUSS B.H., CHISHOLM R.J., KEELEY F.W., GOTLIEB A.I., LOGAN R.A., ARMSTRONG P.W. Extracellular matrix remodeling after balloon angioplasty injury in a rabbit model of restenosis. Circ. Res. 1994;75:650–658. doi: 10.1161/01.res.75.4.650. [DOI] [PubMed] [Google Scholar]
- SUDO T., TACHIBANA K., TOGA K., TOCHIZAWA S., INOUE Y., KIMURA Y., HIDAKA H. Potent effects of novel anti-platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem. Pharmacol. 2000;59:347–356. doi: 10.1016/s0006-2952(99)00346-9. [DOI] [PubMed] [Google Scholar]
- TSUCHIKANE E., FUKUHARA A., KOBAYASI T., KIRINO M., YAMASAKI K., KOBAYASHI T., IZUMI M., OTSUJI S., TATEYAMA H., SAKURAI M., AWATA N. Impact of cilostazol on restenosis after percutaneous coronary balloon angioplasty. Circulation. 1999;100:21–26. doi: 10.1161/01.cir.100.1.21. [DOI] [PubMed] [Google Scholar]
- TSUCHIKANE E., KATO O., SUMITSUJI S., FUKUHARA A., FUNAMOTO M., OTSUJI S., TATEYAMA H., AWATA N., KOBAYASI T. Impact of cilostazol on intimal proliferation after directional coronary atherectomy. Am. Heart. J. 1998;135:495–502. doi: 10.1016/s0002-8703(98)70327-8. [DOI] [PubMed] [Google Scholar]
- UMEKAWA H., TANAKA T., KIMURA Y., HIDAKA H. Purification of cyclic adenosine monophosphate phosphodiesterase from human platelets using new-inhibitor sepharose chromatography. Biochem. Pharmacol. 1984;33:3339–3344. doi: 10.1016/0006-2952(84)90103-5. [DOI] [PubMed] [Google Scholar]
- VEMULAPALLI S., WATKINS R.W., CHINTALA M., DAVIS H., AHN H.S., FAWZI A., TULSHIAN D., CHIU P., CHATTERJEE M., LIN C.C., SYBERTZ E.J. Antiplatelet and antiproliferative effects of SCH51866, a novel type 1 and type 5 phosphodiesterase inhibitor. J. Cardiovasc. Pharmacol. 1996;28:862–869. doi: 10.1097/00005344-199612000-00018. [DOI] [PubMed] [Google Scholar]
- WOJENSKI C.M.I., SILVER M.J. Model system to study interaction of platelets with damaged arterial wall. II. inhibition of smooth muscle cell proliferation by dipyridamole and AH-P719. Exp. Mol. Pathol. 1988;48:116–134. doi: 10.1016/0014-4800(88)90050-0. [DOI] [PubMed] [Google Scholar]
- WU J., DENT P., JELINEK T., WOLFMAN A., WEBER M.J., STURGILL T.W. Inhibition of the EGF-activated MAP-kinase signaling pathways by adenosine 3′, 5′-monophosphate. Science. 1993;262:1065–1069. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- XIONG Y., WESTHEAD E.W., SLAKEY L.L. Role of phosphodiesterase isoenzymes in regulating intracellular cyclic AMP in adenosine-stimulated smooth muscle cells. Biochem. J. 1995;305:627–633. doi: 10.1042/bj3050627. [DOI] [PMC free article] [PubMed] [Google Scholar]