

Abstract
The goal of this work was to test the role of nitric oxide synthase (NOS) and its substrate L-arginine in development of tolerance to nitroglycerin's (GTN) vasodilator actions.
GTN's effects on NOS activity and NO formation were tested in cultured bovine aortic endothelial cells (BAECs). The arginine to citrulline conversion assay showed that GTN stimulated NOS basal activity in BAECs by ∼40%, comparable with acetylcholine (ACh)-treated controls. Both effects were blocked by L-NMMA. Photometric assays showed that both GTN and ACh-stimulated NO formation. Both effects were potentiated by L-arginine and inhibited by L-NAME. L-NAME inhibited ACh responses ∼80% compared with ∼40% for GTN responses.
The aortic ring assay showed that 2 h pretreatment with GTN caused substantial tolerance to GTN's vasodilating effects as evidenced by a 38 fold rightward shift of the concentration-relaxation curve. In contrast to D-arginine, addition of L-arginine substantially inhibited this effect, reducing the rightward shift to 4.4 fold of control values. GTN tolerance was associated with a 40% reduction in L-arginine tissue levels. GTN had a biphasic effect on BAEC uptake of L-arginine, stimulating uptake at 5 and 15 min, and suppressing uptake after 1 and 4 h
In summary, acute GTN treatment stimulates endothelial NOS activity in producing NO and increases cellular uptake of L-arginine. Prolonged GTN exposure reduces GTN's vasodilator actions, decreases L-arginine tissue levels and depresses BAECs uptake of L-arginine. Supplementation of L-arginine reduces development of GTN tolerance. These data indicate that GTN tolerance depends in part on activation of the NOS pathway.
Keywords: Nitroglycerin, tolerance, L-arginine, nitric oxide synthase
Introduction
The mechanism of GTN's vasodilator action is generally agreed to involve denitration and release of NO from GTN which in turn activates smooth muscle guanylyl cyclase, causing vasorelaxation (Ignarro et al., 1981; Murad, 1986; Salvemini et al., 1992). However, evidence has accumulated to support an additional, endothelium-dependent mechanism of GTN-induced vasodilation (Ahlner et al., 1987; 1988; Malta, 1989a,1989b; Abou-Mohamed et al., 1995).
Mechanisms of tolerance to GTN have been intensely investigated and are thought to involve both vascular and humoral mechanisms (Parker & Parker, 1998). Well established vascular mechanisms include desensitization of smooth muscle guanylyl cyclase (Molina et al., 1987), enhanced cyclic GMP breakdown due to increased phosphodiesterase activity (Pagani et al., 1993), and impaired GTN biotransformation in the blood (Chung & Fung, 1990). Humoral mechanisms include increases in plasma levels of catecholamines, vasopressin, angiogensin II and aldosterone which elevate vascular resistance and volume (Stewart et al., 1986; Parker, 1989).
Another mechanism of GTN tolerance may involve enhancement of superoxide production. Chronic treatment with known NOS agonists such as acetylcholine or Ca2+ ionophore has been found to induce vascular tolerance (Gold et al., 1989; 1990). This effect was associated with depletion of the NOS substrate, L-arginine, and was reversed upon L-arginine repletion. When L-arginine is limited, superoxide anion is produced from O2 by nitric oxide synthase (NOS) (Pritchard et al., 1995; Kurz et al., 1996; Presta et al., 1997). In this context, blood vessels made tolerant to GTN showed a 2 fold increase in superoxide formation in response to GTN (Münzel et al., 1995). Endothelium denudation from these vessels reduced superoxide formation, an observation implicating a role of endothelium in GTN tolerance. Our preliminary clinical data have shown that the combination of nitrates with supplemental L-arginine can be useful in reversing nitrate tolerance (Kaesemeyer et al., 1997). Thus, it is possible that GTN tolerance involves effects on the endothelium leading to NOS activation and L-arginine depletion. Based on these collective observations, we have hypothesized that tolerance to GTN results in part from diminished L-arginine availability due to activation of NOS pathway by GTN and reduction of L-arginine uptake by endothelial cells.
Methods
NOS activation and NO production by bovine aortic endothelial cells
Effect of GTN on nitric oxide synthase activity
BAECs (passages 2–5) were grown to confluency in M-199 medium in 6-well plates. After 12 h incubation in serum free medium, the cell layer was incubated for additional 2 h in L-arginine-free medium. Cells were washed with HEPES buffer and incubated with 1 ml of the same buffer at 37°C. The buffer had the following composition (mM): NaCl, 125; KCl, 5; NaHCO3, 25; MgSO4, 1.2; KH2PO4·H2O, 1.19; CaCl2·2H2O, 2.54; glucose, 11 and HEPES, 10 (pH 7.4). [3H]-L-arginine (2 μCi) and 10 μM cold L-arginine were added to each well. Two minutes later, cells received 10−6 M of either ACh or GTN. In some experiments, 10−6 M of Ca+2 ionophore A23187 was also used as a positive control. In parallel experiments, 10−3 M of L-NMMA, a NOS inhibitor, was added and allowed to act for 15 min prior to test agents application. Twenty minutes later, the reaction was stopped by a wash of 2 ml of cold phosphate buffer containing 5×10−6 M L-arginine and 4×10−3 M EDTA. Cell-lysate was applied to 2-ml columns of Dowex AG50WX-8 (Na+ form), and eluted with 2 ml of washing buffer. The radioactivity corresponding to [3H]-citrulline content in the eluent was measured by liquid scintillation spectroscopy (Beckman Instruments). Basal formation of [3H]-citrulline was reduced by 71.7±5.4% in the presence of 10−3 M of L-NMMA.
Estimation of nitric oxide production by GTN
The direct effects of ACh and GTN on NO production in BAECs were determined using a highly sensitive photometric assay for conversion of oxyhaemoglobin to methaemoglobin. NO oxidizes oxyhaemoglobin (HbO2) to methaemoglobin (metHb) in the following reaction: HbO2+NO→metHb+NO3−. Therefore, the amount of NO produced by endothelial cells can be quantified by measuring the change in absorbance as HbO2 oxidizes to metHb. Oxyhaemoglobin has an absorbance peak at 415 nm, while metHb has a 406 nm absorbance peak. By subtracting the absorbance of metHb from that of HbO2, the concentration of NO can be assessed. Our general method is patterned after Feelisch et al. (1995).
For this assay, early passages (2–5) of BAECs (107) were suspended in culture medium with 0.5 g of microcarrier beads (Cytodex #3). Cells, beads and medium were transferred to a spinner flask (Wheaton) where the culture was left undisturbed at 37°C with 95%O2 and 5% CO2 for 29 min and then spun (20 r.p.m.) in this same environment for 1 min. This sitting cycle allows for cell adherence to the beads while the spinning creates an even distribution of cells and beads. After 4 h of alternate sitting and spinning cycles in the attachment phase, the spinner flask was left on the stirrer for 2–3 days for uniform cellular coating of beads.
For experiments, beads/cells were rinsed twice and then suspended in a HEPES-buffered Krebs-Ringer solution containing all necessary co-factors. To prevent a reaction between NO and superoxide (O2.−), superoxide dismutase (200 u ml−1) was added to the buffer. Catalase (100 u ml−1) was added to decompose hydrogen peroxide, keeping the hemoglobin active. This basic buffer also contained 5×10−5 M of L-arginine. Two-ml of BAEC/beads were placed into a water-jacketed chromatography column (Pharmacia) and superfused at a rate of 2 ml min−1 with HEPES-buffered Krebs-Ringers solution containing 3×10−6 M oxyhaemoglobin. The perfusate was then directed into a flow-through cuvette in a dual wavelength spectrophotometer and absorbance was measured to determine the basal and stimulated NO release. A parallel column circuit was filled with only beads (no cells) to determine basal and spontaneous release of NO in this system without cells. Vehicle (buffer w/o agent) did not cause a change in absorbance when infused into the cell/bead column.
Experimental stimulation was carried out by 3 min infusion periods of ACh or GTN added to buffer perfusion system via a 3-way stopcock using a microsyringe pump at a rate of 45 μl min−1 to yield a final concentration of 10−6 and 10−5 M for each agent in the buffer. The effects of buffer containing L-NAME (10−3 M) in blocking the actions of these drug agents and then a buffer without L-NAME but with excess L-arginine (10−3 M) in reversing any L-NAME effect were also examined. Each drug agent concentration was given twice for each of the three buffer systems; a period of 10 min was allowed between infusions of agents. Our data demonstrate that this cell perfusion and monitoring system remains stable for at least 4–6 h. At the end of each experiment, cell viability was then checked using trypan blue exclusion.
For analysis, we determined the area under the curve for the change in absorbance per min caused by each agent above baseline levels and calculated metHb production using an extinction coefficient of 39 mM−1 cm−1. During the 3 min infusion of agents absorbance rose rapidly. Changes in absorbance to these agents persisted from 2–8 min depending on the size of the response before returning to baseline levels. We assumed a one to one correspondence for NO and metHb production, the known stoichiometric balance for this reaction.
We also determined the change in basal NO production during perfusion with each of the buffer systems. Basal levels of NO in our cell/bead chamber perfusion system varied between 32 and 167 nmol min−1 mg protein−1. Perfusion of BAECs with buffer containing L-NAME (10−3 M) reduced basal NO formation by 38±5%. Subsequent perfusion with L-arginine containing buffer (10−3 M) raised the basal levels back to those observed before L-NAME treatment. Basal NO values were subtracted from drug-induced responses to determine NO production caused solely by each drug.
Measurement of vascular tone
Male Sprague-Dawley rats (200–250 g) were killed by decapitation. The descending thoracic aorta was removed, cleaned from the adjacent tissues and cut into ring segments of approximately 4 mm in length. Two metal hooks were passed carefully through the lumen of each ring and the tissues were then mounted under 2×10−3 Newtons of tension in 25-ml organ baths containing Krebs solution. Tissues were left to equilibrate for 90 min with intermittent changing of the solution every 15 min. Krebs solution had the following composition (mM): NaCl, 118; KCl, 4.75; NaHCO3, 25; MgSO4, 1.2; KH2PO4·H2O, 1.19; CaCl2·2H2O, 2.54; glucose 11. The bathing solution was kept at 37°C and was continuously aerated with 5% CO2 and 95% O2 gas mixture. After equilibrium, a predetermined submaximal dose of phenylephrine, PE (3×10−7 M, producing ∼86% of maximum contraction), was added to the tissue bath and the full response allowed to develop. A cumulative concentration effect curve for GTN was then constructed. The bathing solution was drained and replaced with fresh solution. The tissues were allowed to return to the baseline. Tolerance to GTN was developed by incubating the tissues with 5×10−4 M of GTN for 1 h followed by a replenishment of the same dose for a second hour. In one series of experiments, L-arginine (5×10−4 M) was present in half of the tissue baths throughout the incubation time, while the other half had no L-arginine present. In a similar series of experiments, D-arginine (5×10−4 M) was used instead of L-arginine. Tissues were washed extensively every 15 min for 1 h post-incubation. Rings were reconstricted with 3×10−7 M PE and a second cumulative concentration effect curve for GTN was established. The pD2 values for GTN (negative log of the dose producing 50% of the maximal response) were determined for each treatment. In another set of experiments, rings were contracted as above with PE in the presence of 5×10−4 M of L-arginine or D-arginine to determine the effect of L-arginine or D-arginine alone on GTN responses.
A separate series of experiments was conducted to investigate the response of GTN-tolerant vessels to ACh (NOS activator) and SNAP (NO donor). In these experiments, aortas were contracted with 3×10−7 M of PE. After maximal responses to PE were fully developed, vessels were relaxed with by stepwise addition of ACh or SNAP. Subsequently, vessels were made tolerant to GTN as above and concentration-relaxation curves (CRC) for ACh or SNAP were reconstructed.
In some aortic rings, the effect of 30 min pretreatment of L-NMMA (10−4 M) on the vascular responses to ACh was investigated. In these experiments, CRCs for ACh were constructed in the absence and the presence of the NOS inhibitor, L-NMMA.
For each experiment, four ring segments were obtained from the rat and randomly assigned to one of our experimental treatments. All responses were expressed as a percentage of the response produced by a time control to PE alone.
L-arginine content determination
Thoracic aortas isolated from rats were cut into two equal halves (∼20 mg). Each half was incubated in 25-ml organ bath containing Krebs solution at 37°C and was continuously aerated with 5% CO2 and 95% O2 gas mixture. The aorta from one group of rats was kept in the bath for only 45 min, the period for the first concentration response curve (initial control). Another group was made tolerant to GTN using the long exposure protocol described above. For the third group, aortas were incubated only in Krebs solution for this same total 2 h-period. After the given periods, tissues in each group were washed extensively and frozen in liquid nitrogen. L-arginine was separated according to Gold et al. (1989). Briefly, aortic segments (30–50 mg) were pooled per sample. Tissues were homogenized in 6% trichloroacetic acid and centrifuged. The supernatant was extracted with ether. Samples were lyophilized and reconstituted in 4 ml of 0.1 M citrate buffer, pH 5.5. Samples were chromatographed on columns of resin (AG50WX8, sodium salt) and washed with citrate buffer to remove citrulline and ornithine. The columns were eluted with 4 ml of 0.2 N NaOH. Samples were assayed for L-arginine spectrophotometrically by a modified Sakaguchi reaction using L-arginine as a standard.
L-arginine transport system in bovine aortic endothelial cells
Confluent cultures of BAECs (passages 2–5) were prepared in 24-well plates. The predominant transporter system for L-arginine is the Na+-independent y+ system (Cynober et al., 1995; Ogonowski et al., 2000). In order to determine the contribution of the y+ transporter for L-arginine supplied to the cells, BAECs were incubated in an uptake buffer containing 20 nM [3H]-L-arginine for periods of 5, 15 and 60 min. The incubation buffer contained (mM): HEPES, 25; CaCl2, 1.8; KC, 5.4; choline (rather than sodium) chloride, 140; MgSO4, 0.8 and glucose, 5. Uptake of L-arginine was terminated by addition of ice-cold buffer and cells were washed three times with 1 ml of buffer. After final washing, cells were lysed by adding 1 ml 0.5% sodium dodecyl sulphate in 0.1 N NaOH. Cellular lysates (850 μl) were added to 15 ml Ecoscint-A scintillation fluid, the amount of [3H]-L-arginine was determined by scintillation spectroscopy and represented cellular transport of L-arginine. Our earlier studies of cellular uptake of L-arginine in BAECs indicate that the Na+-dependent B0,+ system contributes only 5–10% of total uptake (Ogonowski et al., 2000). In addition, experiments were performed in which the y+ transporter was characterized for cellular uptake of [3H]-L-arginine for 5, 15, and 60 min in the presence of excess (10−3 M) unlabelled L-arginine. The results of these experiments allowed us to determine the extent of non-specific binding and non-saturable uptake of L-arginine to the BAECs contributed to total L-arginine uptake; this was subtracted from experimental values. The BAECs were incubated with or without GTN at a concentration of 10−6 M for 5, 15, 60 and 240 min for determination of the effect of GTN on L-arginine-uptake. For the 240 min exposure with GTN, the media containing 10−6 M GTN was replaced every 1 h and the uptake of [3H]-L-arginine for this experiment was measured during the fourth hour. Uptake of [3H]-L-arginine for individual wells was normalized by calculating uptake per mg of cell protein.
Drugs and chemicals
The following materials were used: preservative free glyceryl trinitrate (Perlinganit®, Schwartz Pharma, Germany), L-[2,3-3H]-arginine (Du Pont, NEN, Boston, MA, U.S.A.), and Micro BCA protein assay kit (Pierce, IL, U.S.A.). Nitro L-arginine methyl ester (L-NAME), NG-monomethyl-L-arginine (L-NMMA), Dowex AG50WX-8, S-nitroso-N-acetyl penicillamine (SNAP), Acetylcholine (ACh), Ca2+ ionophore A23187, D-arginine, and L-arginine were purchased from Sigma Chemical Co., St. Louis, MO, U.S.A. Other chemicals were of analytical grade.
Statistical analysis
Data are presented as mean±s.e.mean of the indicated number of observations (n) and the difference between groups was assessed using the paired t-test or analysis of variance, when appropriate. A probability value (P) less than 0.05 was considered to be statistically significant.
Results
NOS activation and NO production by bovine aortic endothelial cells
Effect of GTN on nitric oxide synthase activity
The potential stimulatory action of GTN on NOS activity was investigated by assaying the conversion of [3H]-L-arginine into [3H]-citrulline and NO. In these experiments, the effects of GTN were compared with the known NOS agonist ACh. The data showed that the basal activity of NOS in non-stimulated BAECs was 4.11±0.6 pmol mg protein−1 min−1. ACh (10−6 M) increased the activity of the enzyme by 49±12% above the baseline. Incubation of cells with GTN (10−6 M) for 20 min increased eNOS activity to the same extent, 46±18% above the baseline (Figure 1). L-NMMA treatment reduced the responses of ACh and GTN to 3.5 and 4.2% above baseline, respectively. The observed increases in activity of eNOS were almost totally blocked by prior incubation of cells for 15 min with 10−3 M of L-NMMA.
Figure 1.
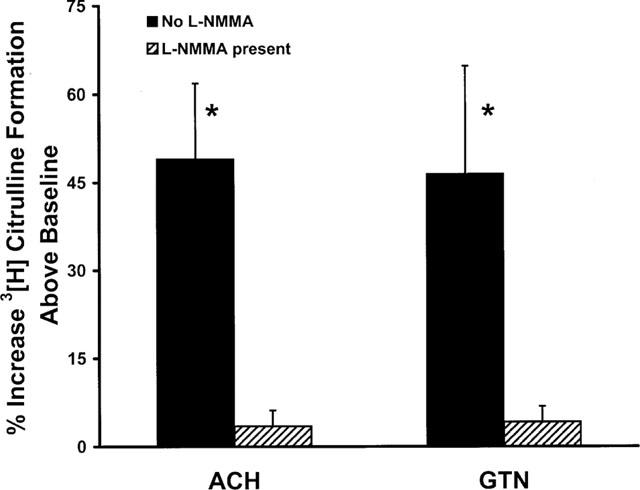
Effect of ACh and GTN (1 μM each) on eNOS activity in BAECs as measured by the conversion of [3H]-L-arginine into [3H]-citrulline. The basal activity of eNOS was 4.11±0.6 pmol mg protein−1 min−1. Treatment with L-NMMA (10−3 M) reduced basal 3H-citrulline formation by 71.7±5.4%. This treatment reduced the effects of ACh and GTN by over 90%. Data are presented as a mean±s.e.mean of six experiments run in duplicates. *Indicates a significant difference from the baseline value.
Estimation of nitric oxide production by GTN
This series of experiments was designed to determine the effects of GTN on activity of endothelial cells in producing NO and to assess the relative contribution of NOS activity to the total NO production by GTN. The results demonstrated that ACh produced a transient, concentration-related increase in NO formation above baseline levels (Figure 2). In control buffer containing 5×10−5 M L-arginine, there was approximately a 2 fold increase in NO production between 10−5 and 10−6 M ACh. Subsequent treatment of these cells with buffer containing 10−3 M of L-NAME, markedly reduced ACh-induced production of NO by 80%. When this L-NAME buffer was replaced with another containing L-arginine (10−3 M), ACh-elicited production of NO returned to control levels.
Figure 2.
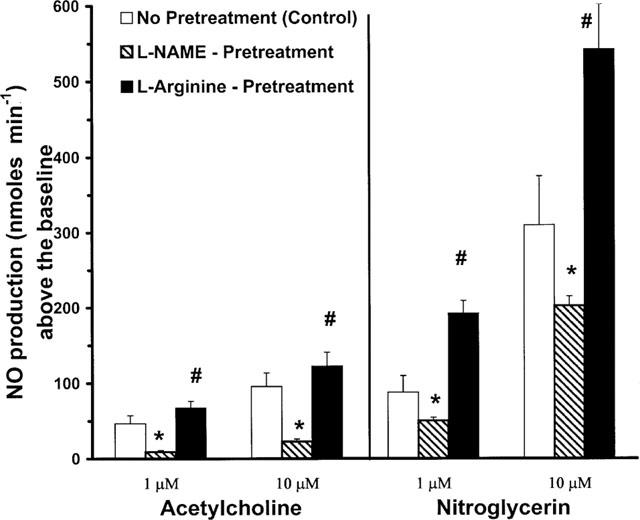
Effects of acetylcholine and nitroglycerin (10−6 and 10−5 M) on NO production. Responses to drugs are transient elevations in NO production above basal levels of NO production in control buffer with 10−5 M L-arginine. Subsequent perfusion of the cell/bead system with 10−3 M L-NAME in buffer, and then with 10−3 M of L-arginine in the buffer system. Production of NO is displayed in nmol min−1 (mean±s.e.mean). #Denotes difference between responses in L-NAME buffer vs both basic and L-arginine added buffers (P<0.05). *Indicates difference vs control basic buffer responses (P<0.05).
GTN also caused a concentration-related increase in NO production above baseline levels. There was a larger increment in response to the 10−5 vs 10−6 M concentrations of GTN (∼3 fold) compared with that of ACh. Superfusion of the cell suspension with L-NAME (10−3 M), also blunted NO production in response to GTN, but to a lesser extent than was seen with ACh. The reduction was only about 40% as compared with the 80% reduction seen with ACh. These observations suggest that in addition to the well-known effect of GTN as NO donor, its stimulation of NO production is also due in part to NOS activity. Additionally, subsequent perfusion of the cells with a buffer supplemented with L-arginine (10−3 M) resulted in a return in NO production to a level above the amount induced by the GTN in control buffer. This restoration of responses to GTN after L-arginine addition was about three times greater than that observed for ACh. Administration of GTN or ACh into a perfusion system containing only beads without cells did not induce metHb/NO production.
Vascular tone
The isolated aortic rings contracted with 3×10−7 M of PE produced 1.75±0.15 gm of tension. Experiments using these rings demonstrated that GTN induces a concentration-dependent (10−10 to 10−5 M) vasorelaxation with a pD2 value of 7.75±0.08 (Figure 3). Exposure of the ring segments to a high concentration of GTN (5×10−4 M) for 2 h reduced their sensitivity to subsequent relaxation by GTN. Another concentration-response curve constructed on the GTN-exposed vessels, without L-arginine in the bath, was shifted to the right by ∼38 fold (pD2 value of 6.17±0.22, P<0.001). These vessels were tolerant to GTN. However, in the other vessels exposed to the high concentration of GTN, but with L-arginine (5×10−4 M) in the bath, there was a significant inhibition of the rightward shift of the concentration-response curve. The pD2 value for this curve after GTN exposure with L-arginine in the bath was 7.13±0.12 (P<0.05 vs tolerant); this is only a 4.4 fold difference from the control concentration-response curve (P<0.05). Thus, the presence of supplemental L-arginine in the bath substantially reduced the rightward shift of the concentration-response curve in the GTN treated vessels (by ∼8 fold, P<0.05). In experiments in which tolerance was induced in the presence of D-arginine (5×10−4 M), there was no prevention of GTN-induced tolerance. D-arginine appears to shift the concentration-response curve to the right, but the difference was not statistically significant from GTN tolerant group.
Figure 3.
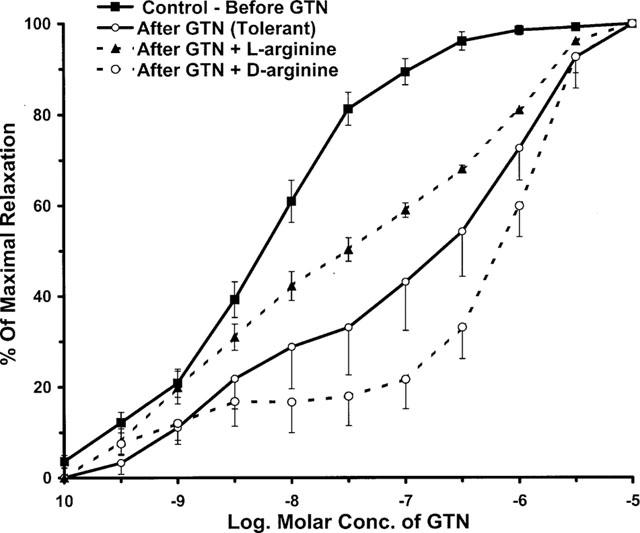
Induction of tolerance to GTN in isolated rat aortic segments and the effects of L-arginine and D-arginine thereon. Tissues were incubated for 2 h with GTN (5×10−4 M) without or with 5×10−4 M of L-arginine or D-arginine (n=8–20). The Initial pD2 value for GTN before tolerance is 7.75±0.08. After incubation for 2 h with GTN, the pD2 values were 7.13±0.12 and 6.17±0.22, respectively in the presence and absence of L-arginine (P<0.05). The presence of 5×10−4 M of D-arginine did not significantly affect the development of tolerance of GTN (pD2 value is 5.71±0.3).
In control experiments, we determined that a 2 h-period of incubation without GTN exposure did not affect the pD2 value for GTN. In a similar way, incubation of aortic rings with either L- or D-arginine for 2 h had no effect on the pD2 values for GTN.
In experiments to investigate the possible development of cross-tolerance between GTN and the NOS agonist ACh, aortic rings were made tolerant to GTN as described above and vasorelaxation responses to ACh were determined (Figure 4). The data showed that relaxation responses to low concentrations of ACh were reduced. The EC20 values were significantly less in GTN tolerant vessels than in the controls. The negative log. of the EC20 values were 9.48±0.21 and 8.70±0.18 (P<0.05) for non-tolerant and tolerant vessels, respectively. Responses were different at concentrations up to 10−8 M. Experiments testing effects of the NO donor, SNAP, were done to check for possible cross-tolerance to other NO donors in GTN tolerant vessels. In these experiments, GTN-tolerant vessels showed the same sensitivity to SNAP as the non-tolerant vessels. The pD2 values for SNAP on non-tolerant and tolerant vessels were 7.24±0.30 and 7.45±0.32, respectively.
Figure 4.
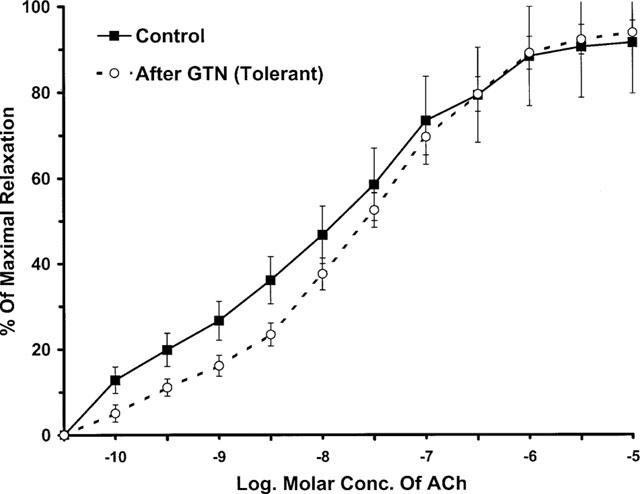
ACh-induced vasorelaxant responses of GTN-tolerant and non-tolerant aortic segments. The pD2 value for ACh in non-tolerant vessels is similar to the tolerant vessels (7.72±0.31 vs 7.63±0.33, respectively). However, the curves are significantly different at concentrations up to 10−8 M of ACh (P<0.05). Data are represented as the mean of eight experiments±s.e.mean.
As an additional control experiment, we also determined the ability of L-NMMA, a NOS inhibitor, to affect vasorelaxant responses to ACh. The purpose of this experiment was to assess the possible contribution of NOS-independent processes to ACh-induced responses. Treatment of aortic rings for 30 min with L-NMMA (10−4 M) substantially shifted the CRC to the right and blunted ACh-induced maximal relaxation by about 55% (data not shown). The residual 45% relaxation response to ACh supports the existence of a NOS-independent component.
GTN effects on L-arginine levels and uptake
The results of the vascular tone assays are consistent with the hypothesis that a lack of L-arginine may contribute to GTN tolerance. In order to test this idea more directly, we measured tissue levels of L-arginine in vessels that had been previously rendered tolerant by GTN treatment as compared with non-tolerant controls. One group was exposed to GTN briefly (45 min in tissue bath) for construction of concentration-response curves; another group was made tolerant by exposure to GTN (5×10−4 M) for 2 h; and a third group was exposed to Krebs solution for 2 h. As shown in Figure 5, L-arginine levels in the non-tolerant control vessels were similar for the initial and 2 h-vehicle control group (698±18 and 689±34 nmol gm−1 wet wt., respectively). In contrast, the tissue arginine level in GTN-tolerant vessels was reduced to 435±35 nmol gm−1 wet wt. Thus, prolonged exposure to GTN reduced tissue L-arginine by about 40%. This effect on tissue L-arginine levels was correlated with biphasic time-dependent changes in cellular uptake of L-arginine. GTN (10−6 M) treatment initially stimulated transport of [3H]-L-arginine into endothelial cells at 5 and 15 min of exposure by 24 and 40%, respectively. However, longer exposure to GTN, 1 and 4 h, resulted in a reduction of L-arginine-uptake by 22 and 35%, respectively (Figure 6).
Figure 5.
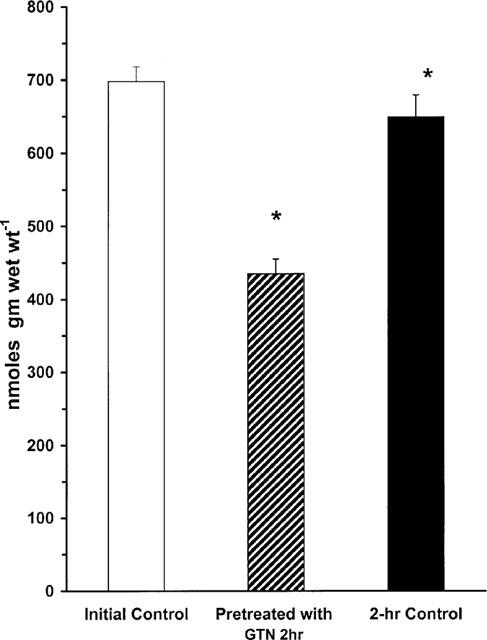
L-arginine content in rat aortic segments pre-constricted with phenylephrine. The figure shows L-arginine levels in control buffer initial (n=11), presence of GTN for 2 h (5×10−4 M, n=7), and control buffer for 2 h (n=8). *Indicates a difference from values for both initial control buffer and control buffer after 2 h (P<0.05).
Figure 6.
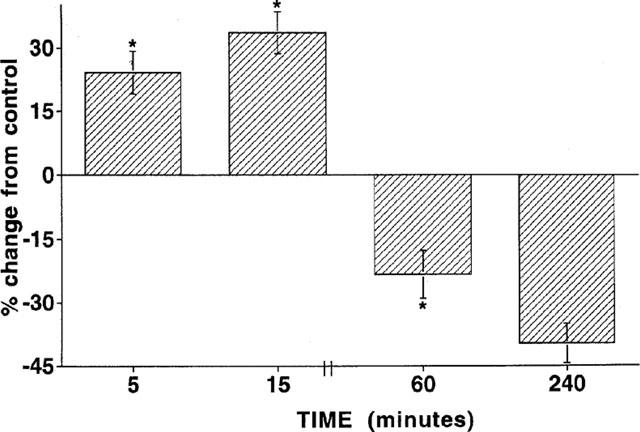
The time course of effects of GTN (10−6 M) on [3H]-L-arginine uptake in isolated bovine aortic endothelial cells. Basel rates of L-arginine uptake are 44.8±10.3, 113.8±34, and 344±68 pmol L-arginine mg protein−1 min−1, respectively after 5, 15 and 60 min, respectively. For the 240 min exposure to GTN, uptake of [3H]-L-arginine was measured during the fourth hour. *Indicates difference from basal uptake of L-arginine at specific time points (P<0.05).
Discussion
The mechanism of tolerance development to GTN is still controversial. While some vascular and humoral mechanisms are well established (Stewart et al., 1986; Molina et al., 1987, Parker, 1989; Chung & Fung, 1990; Pagani et al., 1993), other mechanisms involving the endothelium have also been suggested. Among these latter mechanisms is the augmented production of superoxide free radical (Münzel et al., 1995). It has been shown that vascular tolerance can occur with chronic treatment with NOS agonists (Gold et al., 1989; 1990). This effect appears to be due to a limited L-arginine supply. Under conditions where L-arginine is limiting, the production of superoxide by NOS is elevated (Mayer et al., 1991; Pritchard et al., 1995; Kurz et al., 1996).
Our initial studies on mechanism(s) of GTN's action showed that cyclic GMP production in aortic tissues in response to GTN was augmented by L-arginine and inhibited by L-NAME (Abou-Mohamed et al., 1995). As a result of these data and studies by others suggesting an endothelial-dependent mechanism of GTN's vasodilator actions (Ahlner et al., 1987; 1988; Malta 1989a,1989b), we decided to determine the role of NOS activation and L-arginine availability in tolerance to GTN using in vitro vascular models. Our present experiments show that GTN stimulates endothelial cell NOS, in addition to its action as NO donor. Furthermore, we found that GTN-induced tolerance is associated with a reduction in tissue levels of L-arginine and reduced cellular L-arginine uptake. The developed tolerance can be partially prevented by L-arginine supplementation suggesting that NOS activation and L-arginine depletion have a contributing role in GTN tolerance.
In this study, tolerance was produced in rat aortic rings by incubating them for 2 h in 5×10−4 M GTN, replenished once after the first hour of incubation. The development of tolerance was demonstrated by a large rightward shift of the concentration response curve (∼38 fold). Incubation of vessels with L-arginine (5×10−4 M) during the GTN exposure significantly reduced the concentration-response shift (to only ∼4.4 fold). Treatment with L-arginine alone did not alter vascular tone. Moreover, the use of D-arginine rather than L-arginine had no effect on GTN tolerance. Thus, the reduction of the rightward shift of the concentration response curves to GTN by L-arginine represents a specific effect in reversing tolerance to GTN. The persistent 4.4 fold reduction in GTN sensitivity of the L-arginine-treated tolerant vessels is probably due to other previously described mechanisms of GTN-induced tolerance.
Our finding that tissue levels of L-arginine were significantly lower in the GTN-tolerant vessels than in control vessels further supports the role of L-arginine in contributing to the development and reversal of GTN tolerance. Moreover, since L-arginine levels in the 2 h vehicle control vessels were not different to initial levels, experimental manipulation of tissues is excluded as a cause of L-arginine depletion.
Two mechanisms may have contributed to the observed reduction in L-arginine tissue levels: enhanced L-arginine utilization or reduced cellular uptake of L-arginine. Our results suggest that both mechanisms are probably involved. Regarding utilization, L-arginine serves as substrate for a variety of enzymes, including NOS. Using both direct and indirect methods for assessing NOS activity, our results showed that GTN acts as a NOS agonist. Analysis of the formation of 3H-citrulline from 3H-arginine showed that GTN increased NOS activity by 46% – an amount equivalent to that induced by the known NOS agonist, ACh. Actions of both GTN and ACh were almost completely blocked by L-NMMA, demonstrating specificity of their effects. Our experiments using an indirect photometric assay to measure NO formation also showed that GTN activates NOS. Superfusion of BAECs with GTN or ACh resulted in transient, dose related increases in NO production above the baseline levels. Pretreatment with the NOS antagonist L-NAME significantly inhibited the NO production induced by both agents, providing further evidence of their actions as NOS agonists. NO production by GTN or ACh was reduced by approximately 40 and 80%, respectively. This lesser inhibitory effect of L-NAME on NO produced by GTN reflects GTN's well-known action as NO donor due to its denitration by endothelial cells. When cells were subsequently perfused with buffer containing 10−3 M of L-arginine, NO production induced by both ACh and GTN was augmented as compared with the basal responses. These data indicate that our cell assay system remained viable for the period of study.
In addition to GTN's effect in stimulating NOS activity, the present study also showed a significant effect of GTN in altering cellular uptake of L-arginine. While acute exposure to GTN (5 or 15 min) stimulated the uptake, uptake was suppressed after longer treatment (1 and 4 h). These results are consistent with previous work showing that L-arginine uptake is stimulated by acute treatment with NOS agonists and inhibited by prolonged treatment with NO donors (Bogle et al., 1991; Patel et al., 1996; Ogonowski et al., 2000). The suppressive effect of long-term GTN treatment on L-arginine uptake can limit L-arginine availability to NOS, which would increase superoxide production (Prichard et al., 1995; Kurz et al., 1996). Increased superoxide formation may further suppress the L-arginine transporter (Patel et al., 1996). Studies are in progress to directly test these possibilities.
Additional studies are also necessary to determine the specific mechanisms by which GTN interacts with NOS. GTN may directly activate the enzyme. Alternatively, GTN may activate NOS indirectly through the release of endogenous agonists as suggested by the work of Booth et al. (1997).
One may argue that if GTN-induced tolerance is due in part to a reduction of NOS function, then responses of GTN-tolerant vessels to ACh should be reduced compared to the non-tolerant vessels, i.e. that cross tolerance should occur. We tested this by experiments using our aortic ring in vitro model of GTN tolerance. The data showed a reduction in the ACh-induced vasorelaxation responses for the GTN tolerant vessels at the lower end of the concentration range, resulting in a significant increase in EC20. Significant cross-tolerance was not apparent when ACh was tested at concentrations higher than 10−8 M. Cross-tolerance to ACh has been described previously by De la Lande et al. (1999). In their studies of aortic rings from rats made tolerant to GTN in vivo, these workers found small, but statistically significant, increases in the pD2 value for ACh. It should be noted that some investigators have been unable to detect cross-tolerance between GTN and either ACh or bradykinin (Van de Voorde et al., 1987; De Garavilla et al., 1993).
It is possible that the persistent vasorelaxant action of the higher ACh concentrations on the GTN-tolerant vessels is mediated by NO-independent mechanisms. Data supporting this concept have been presented previously by Vanheel et al. (1994). They demonstrated that ACh induces endothelium-dependent hyperpolarization in rat aorta and that this effect was partially resistant to L-NMMA. The notion of a NOS-independent vasorelaxation action of ACh is reinforced by our control experiment showing that L-NMMA was only about 55% effective in blocking the vasorelaxant responses to ACh. This result is in concert with that reported by Chen et al. (1988). These investigators found that haemoglobin and methylene blue only partially blocked ACh-induced relaxation in rat aorta.
The fact that GTN tolerance did not affect responses to SNAP is consistent with existence of an additional mechanism of action of GTN beyond its action as an NO donor. This action may also be involved in the development of tolerance to GTN.
An auxiliary mechanism of GTN as an eNOS agonist apart from its principal therapeutic mechanism is not unique to GTN. Amrinone, a phosphodiesterase inhibitor and cardiotonic agent used in heart failure, has recently been shown to be an agonist of eNOS (Mori et al., 1996). Nebivolol, a β-adrenergic blocker, may also produce vasodilation by an L-arginine-NO dependent mechanism (Cockcroft et al., 1995). Pravastatin, a lipid-lowering agent, is also a vasodilator and a NOS agonist (Kaesemeyer et al., 1999).
Deficiencies in L-arginine supply are strongly implicated in cardiovascular disease and even in alteration of vasoactive responses during normal states. Supplemental arginine restores endothelium-dependent relaxation in humans with heart failure (Drexler et al., 1992) and atherosclerosis (Creager et al., 1990) and in cardiomyopathic hamsters (Mayhan & Rubinstein, 1992), and enhances ACh-induced vasodilation in normal guinea-pigs (Aisaka et al., 1989).
Although the beneficial effects of L-arginine in reversing endothelial dysfunction are well established, the phenomenon is not yet understood. Measurement of L-arginine levels in endothelial cells has shown that the intracellular concentration (0.1–1 mM) greatly exceeds endothelial NOS's Km (∼3 μM) (Pollock et al., 1991). This means that NOS should be saturated with substrate under all but the most extreme conditions of L-arginine deficiency. This contradiction between NOS's low Km for L-arginine and the strong positive effects of supplemental L-arginine in reversing vascular dysfunction and augmenting the actions of eNOS agonists even in normal conditions has been termed the ‘L-arginine paradox'. This seeming conflict may be explained by experimental evidence indicating that the distribution of L-arginine varies greatly within the endothelium due to intracellular compartmentalization and sequestration (Cynober et al., 1995). The work of McDonald et al. (1997) provides further evidence of L-arginine compartmentalization. They demonstrated that a complex exists between eNOS and a major L-arginine transport protein (system y+) in endothelial cells and that both molecules are located within plasma membrane caveolae. These data suggested that eNOS is sequestered from the intracellular L-arginine supply by being located within caveolae and is, therefore, dependent upon the direct transfer of L-arginine into this subcellular compartment by the system y+ transporter. If the transporter function is decreased as may occur with oxidative injury, the L-arginine supply could immediately become limiting and be the basis of vascular dysfunction.
In summary, our data showing that GTN stimulates NOS activity and NO production by cultured endothelial cells indicate a mechanism for the actions of GTN which involves activation of the NOS pathway. The actions of GTN are partially dependent on L-arginine, tissue levels of which decrease at the point of development of tolerance. Furthermore, L-arginine significantly inhibits the development of tolerance to GTN in isolated aortic segments.
Acknowledgments
We wish to thank Traci Taylor, Liming Jin, Sandra Usry and Drs Jianzhong Huang, Alison A. Ogonowski and Ali Behzadian for their excellent assistance and Dr Andreas Reimann, Schwartz Pharma, Monheim, Germany for supplying nitroglycerin. This work was supported in part by USPHS grants HL52957, EY04618, EY 11766 and the American Heart Association, Georgia Affiliate.
Abbreviations
- ACh
acetylcholine
- BAEC
bovine aortic endothelial cells
- EDNO
endothelium derived nitric oxide
- GTN
nitroglycerin (glyceryltrinitrate)
- HbO2
oxyhaemoglobin
- L-NAME
nitro L-arginine methylester
- L-NMMA
NG-monomethyl-L-arginine
- MetHb
methaemoglobin
- NO
nitric oxide
- NOS
nitric oxide synthase
- O2.−
superoxide anion
- pD2
negative log of dose producing 50% of max response
- SNAP
S-nitroso-N-acetyl penicillamine
References
- ABOU-MOHAMED G., KAESEMEYER W.H., PAPAPETROPOULOS A., CATRAVAS J.D., CALDWELL R.W. Nitroglycerin (NTG), but not sodium nitroprusside (SNP), increases aortic ring cGMP levels via an L-arginine dependent and L-NAME sensitive pathway. FASEB J. 1995;9:A327. [Google Scholar]
- AHLNER J., AXELSSON K.L., EKSTRAM-LJUSEGREN M., FRIEDMAN R.I., GRUNDSTROM N., KARLSSON J.O.G., ANDERSSON R.G.G. Relaxation of bovine mesenteric artery induced by glyceryl trinitrate is attenuated by pertussis toxin. Pharmacol. Toxicol. 1988;62:155–158. doi: 10.1111/j.1600-0773.1988.tb01864.x. [DOI] [PubMed] [Google Scholar]
- AHLNER J., AXELSSON K.L., LJUSEGREN M.K., GRUNDSTROM N., ANDERSSON ROLF G.G. Demonstration of a high affinity component of glyceryl trinitrate induced vasodilatation in the bovine mesenteric artery. J. Cyclic Nucleotide Protein Phosphor Res. 1987;11:445–456. [PubMed] [Google Scholar]
- AISAKA K., GROSS S., GRIFFITH O.W., LEVI R. L-arginine availability determines the duration of acetylcholine-induced systemic vasodialtion in vivo. Biochem. Biophys. Res. Commun. 1989;163:710–717. doi: 10.1016/0006-291x(89)92281-x. [DOI] [PubMed] [Google Scholar]
- BOGLE R.G., COADE S.B., MONCADA S., PEARSON J.D., MANN G.E. Bradykinin and ATP stimulate L-arginine uptake and nitric oxide release in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1991;180:926–932. doi: 10.1016/s0006-291x(05)81154-4. [DOI] [PubMed] [Google Scholar]
- BOOTH B.P., NOLAN T.D., FUNG H.L. Nitroglycerin-inhibited whole blood aggregation is partially mediated by calcitonin gene-related peptide – a neurogenic mechanism. Br. J. Pharmacol. 1997;122:577–583. doi: 10.1038/sj.bjp.0701408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG S.H., FUNG H.L. Identification of a subcellular site for nitroglycerin metabolism to nitric oxide in bovine coronary smooth muscle cells. J. Pharmacol. Exp. Ther. 1990;253:614–619. [PubMed] [Google Scholar]
- COCKCROFT J.R., CHOWIENCZYK P.J., BRETT S.E., CHEN C.P., DUPONT A.G., VAN NUETEN L., WOODING S.J., RITTER J.M. Nebivolol vasodilates human forearm vasculature. Evidence for an L-arginine/NO-dependent mechanism. J. Pharmacol. Exp. Ther. 1995;274:1067–1071. [PubMed] [Google Scholar]
- CREAGER M.A., COOKE J.P., MENDELSOHN M.E., GALLAGHER S.J., COLEMAN S.M., LOSCALZO J., DZAU V.J. Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans. J. Clin. Invest. 1990;86:228–234. doi: 10.1172/JCI114688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CYNOBER L., LE BOUCHER J., VASSON M. Arginine metabolism in mammals. J. Nutr. Biochem. 1995;6:402–413. [Google Scholar]
- DE GARAVILLA L., VOLBERG M.L., PRATT P.F., SILVER P.J., BUCHHOLZ R.A. Lack of cross-tolerance between nitroglycerin and endothelium-derived relaxing factor-mediated vasoactive agents in spontaneously hypertensive rats. Eur. Pharmacol. 1993;234:77–82. doi: 10.1016/0014-2999(93)90708-p. [DOI] [PubMed] [Google Scholar]
- DE LA LANDE I.S., STAFFORD I., HOROWITZ J.D. Tolerance induction by transdermal glyceryl trinitrate in rats. Eur. J. Pharmacol. 1999;374:71–75. doi: 10.1016/s0014-2999(99)00319-2. [DOI] [PubMed] [Google Scholar]
- DREXLER H., HAYES D., MÜNZEL T., HORNIG B., JUST H., BRUNNER H.R. Endothelial function in chronic congestive heart failure. Am. J. Cardiol. 1992;69:1596–1601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., BRANDS F., KELM M. Human endothelial cells bioactivate organic nitrates to nitric oxide: implications for the reinforcement of endothelial defense mechanisms. Eur. J. Clin. Invest. 1995;25:737–745. doi: 10.1111/j.1365-2362.1995.tb01952.x. [DOI] [PubMed] [Google Scholar]
- GOLD M.E., BUSH P.A., IGNARRO L.J. Depletion of arterial L-arginine causes reversible tolerance to endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1989;164:714–721. doi: 10.1016/0006-291x(89)91518-0. [DOI] [PubMed] [Google Scholar]
- GOLD M.E., WOOD K.S., BYRNS R.E., BUGA G.M., IGNARRO L.J. L-arginine-dependent vascular smooth muscle relaxation and cGMP formation. Am. J. Physiol. 1990;259:H1813–H1821. doi: 10.1152/ajpheart.1990.259.6.H1813. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., LIPPTON H., EDWARDS J.C., BARICOS W.H., HYMAN A.L., KADOWITZ P.J., GRUETTER C.A. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of s-nitrosothiols as active intermediates. J. Pharmacol. Exp. Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- KAESEMEYER W., ABOU-MOHAMED G., CRUTE T., CALDWELL W. Nitrates supplemented with L-arginine for the reversal and treatment of nitrate tolerance: Two case reports. Appl. Cardiopul. Pathophysiol. 1997;6:255–262. [Google Scholar]
- KAESEMEYER W.H., CALDWELL R.B., HUANG J.Z., CALDWELL R.W. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J. Am. Coll. Cardiol. 1999;31:234–241. doi: 10.1016/s0735-1097(98)00514-2. [DOI] [PubMed] [Google Scholar]
- KURZ S., VENEMA R., SAYEGH H., KENT J.D., HARRISON D.G. Flavin-dependent superoxide production by nitric oxide synthase. J. Invest. Med. 1996;44:314A. [Google Scholar]
- MALTA E. Biphasic relaxant curves to glycerol trinitrate in rat aortic rings. Evidence for two mechanisms of action. Arch. Pharmacol. 1989a;339:236–243. doi: 10.1007/BF00165149. [DOI] [PubMed] [Google Scholar]
- MALTA E. Studies on the biphasic relaxant curve of glyceryl trinitrate in rat aorta: role of GTN metabolites. Clin. Exp. Pharmacol. Physiol. 1989b;16:829–835. doi: 10.1111/j.1440-1681.1989.tb01522.x. [DOI] [PubMed] [Google Scholar]
- MAYER B., MATHIAS J., HEINZEL B., WERNER E.R., WACHTER H., SCHULTZ G., BÖHMER E. Brain nitric oxide synthase is a biopterin-and flavin-containing multifunctional oxido-reductase. FEBS Lett. 1991;288:187–191. doi: 10.1016/0014-5793(91)81031-3. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., RUBINSTEIN I. Acetylcholine induces vasoconstriction in the microcirculation of cardiomyopathic hamsters: Reversal by L-arginine. Biochem. Biophys. Res. Commun. 1992;184:1372–1377. doi: 10.1016/s0006-291x(05)80034-8. [DOI] [PubMed] [Google Scholar]
- MCDONALD K.K., ZHARIKOV S., BLOCK E.R., KILBERG M.S. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”. J. Biol. Chem. 1997;272:31213–31216. doi: 10.1074/jbc.272.50.31213. [DOI] [PubMed] [Google Scholar]
- MOLINA C.R., ANDERSEN J.W., RAPOPORT R.M., WALDMAN S., MURAD F. Effect of in vivo nitroglycerin therapy on endothelium-dependent and independent vascular relaxation and cyclic GMP accumulation in rat aorta. J. Cardiovasc. Pharmacol. 1987;10:372–378. doi: 10.1097/00005344-198710000-00001. [DOI] [PubMed] [Google Scholar]
- MORI K., TAKESUCHI S., MORITOKI H., TSUCHIYA K., NAKAYA Y., MATSUOKA S., KURODA Y. Endothelium-dependent relaxation of rat thoracic aorta by amrinone-induced nitric oxide release. Eur. Heart J. 1996;17:308–316. doi: 10.1093/oxfordjournals.eurheartj.a014850. [DOI] [PubMed] [Google Scholar]
- MÜNZEL T., SAYEGH H., FREEMAN B.A., TARPEY M.M., HARRISON D.G. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying nitrate tolerance and cross tolerance. J. Clin. Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAD F. Cyclic guanosine monophosphate as a mediator of vasodilation. J. Clin. Invest. 1986;78:1–5. doi: 10.1172/JCI112536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGONOWSKI A.A., KAESEMEYER W.H., JIN L., LEIBACH F.H., GANAPATHY V., CALDWELL R.W. Effect of nitric oxide donors and synthase agonist peptides on endothelial cell uptake of L-arginine. Am. J. Physiol. 2000;278:C136–C143. doi: 10.1152/ajpcell.2000.278.1.C136. [DOI] [PubMed] [Google Scholar]
- PAGANI E.D., VANALLER G.S., O'CONNOR B., SILVER P.J. Reversal of nitroglycerin tolerance in vitro by the cGMP-phosphodiesterase inhibitor zaprinast. Eur. J. Pharmacol. 1993;243:141–147. doi: 10.1016/0014-2999(93)90373-p. [DOI] [PubMed] [Google Scholar]
- PARKER J.O. Intermittent transdermal nitroglycerin therapy in the treatment of chronic stable angina. J. Am. Coll. Cardiol. 1989;13:794–795. doi: 10.1016/0735-1097(89)90217-9. [DOI] [PubMed] [Google Scholar]
- PARKER J.D., PARKER J.O. Nitrate therapy for stable angina pectoris. New Engl. J. Med. 1998;338:520–531. doi: 10.1056/NEJM199802193380807. [DOI] [PubMed] [Google Scholar]
- PATEL J.M., ABELES A.J., BLOCK E.R. Nitric oxide exposure and sulfhydryl modulation alter L-arginine transport in cultured pulmonary artery endothelial cells. Free Rad. Biol. Med. 1996;20:629–637. doi: 10.1016/0891-5849(95)02146-9. [DOI] [PubMed] [Google Scholar]
- POLLOCK J.S., FORSTERMANN U., MITCHELL J.A., WARNER T.D., SCHMIDT H.H., NAKANE M., MURAD F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRESTA A., LIU J., SESSA W.C., STUEHR D.J. Substrate binding and calmodulin binding to endothelial nitric oxide synthase coregulate its enzymatic activity. NITRIC OXIDE: Biol. Chem. 1997;1:74–87. doi: 10.1006/niox.1996.0110. [DOI] [PubMed] [Google Scholar]
- PRITCHARD K.A., JR, GROSZEK L., SMALLY D., SESSA W., WU M., VILLALON P., WOLIN M., STEMERMAN M. Native-low density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ. Res. 1995;77:510–520. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., MOLLACE V., PISTELLI A., ANGGARD E., VANE J. Metabolism of glyceryl trinitrate to nitric oxide by endothelial cells and smooth muscle cells and its induction by Escherichia coli lipopolysaccharide. Proc. Natl. Acad. Sci. U.S.A. 1992;89:982–986. doi: 10.1073/pnas.89.3.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART D.J., ELSNER D., SOMMER O., HOLTZ J., BASSENGE E. Altered spectrum of nitroglycerin action in long-term treatment: nitroglycerin-specific venous tolerance with maintenance of arterial vasodepressor potency. Circulation. 1986;74:573–582. doi: 10.1161/01.cir.74.3.573. [DOI] [PubMed] [Google Scholar]
- VAN DE VOORDE J., VANHEEL B., LEUSEN I. Influence of vascular tolerance to nitroglycerin on endothelium-dependent relaxation. Arch. Int. Pharmacodyn. Ther. 1987;290:215–221. [PubMed] [Google Scholar]
- VANHEEL B., VAN DE VOORDE J., LEUSEN I. Contribution of nitric oxide to the endothelium-dependent hyperpolarization in rat aorta. J Physiol. (Lond.) 1994;475:277–284. doi: 10.1113/jphysiol.1994.sp020068. [DOI] [PMC free article] [PubMed] [Google Scholar]