

Abstract
The effects of the phosphatidylinositol 4-kinase inhibitor, phenylarsine oxide (PAO), on acetylcholine (ACh) release and on prejunctional Ca2+ currents were studied at the frog neuromuscular junction using electrophysiological recording techniques.
Application of PAO (30 μM) increased both spontaneous ACh release reflected as miniature end-plate potential (mepp) frequencies and evoked ACh release reflected as end-plate potential (epp) amplitudes with a similar time course.
Following the initial increase in epp amplitudes produced by PAO, epps slowly declined and were eventually abolished after approximately 20 min. However, mepp frequencies remained elevated over this time period.
PAO (30 μM) also inhibited the perineural voltage change associated with Ca2+ currents through N-type Ca2+ channels (prejunctional Ca2+ currents) at motor nerve endings. Addition of British anti-lewisite (BAL, 1 mM), an inactivator of PAO, partially reversed both the inhibition of epps and the inhibition of the prejunctional Ca2+ current.
The effects of PAO on N-type Ca2+ channels were investigated more directly using the whole cell patch clamp technique on acutely dissociated sympathetic neurons. Application of PAO (30–40 μM) to these neurons decreased the voltage-activated calcium currents through N-type Ca2+ channels, an effect that was partially reversible by BAL.
In combination, these results suggest that inhibition of neurotransmitter release by PAO occurs as a consequence of the inhibition of Ca2+ entry via N-type calcium channels. The relationship between the effects of PAO on N-type Ca2+ channels in motor nerve endings and in neuronal soma is discussed.
Keywords: Acetylcholine, nerve terminal, neuromuscular junction, neurotransmitter release, N-type calcium channels, phenylarsine oxide, phosphatidylinositol 4-kinase, synaptic vesicles
Introduction
Phosphatidylinositol 4-kinase (PtdIns 4-kinase), an enzyme associated with synaptic vesicles, has been suggested to play an essential role both in the release of neurotransmitters from nerve terminals (Wiedemann et al., 1998; Bennett, 1998; Khvotchev & Südhof, 1998) and the stimulated secretion of catecholamines from chromaffin granules (Wiedemann et al., 1996). Indeed, inhibition of PtdIns 4-kinase by 30 μM phenylarsine oxide (PAO) inhibited the formation of phosphoinositides in synaptosomal preparations and this effect was coincident with the complete inhibition of Ca2+-dependent neurotransmitter release (Wiedemann et al., 1998). These authors also found that the dithiol compound British anti-lewisite (BAL), an agent that inactivates PAO, reversed the inhibition of PtdIns 4-kinase activity by PAO and partial restored neurotransmitter release (Wiedemann et al., 1998). These observations led to the suggestion that PtdIns 4-kinase activity represents the basis for an ATP dependent step in vesicle priming, a step that would be mandatory for neurotransmitter release. In addition, it has been suggested that phosphoinositides may also play important roles in the endocytosis and the recycling of synaptic vesicles through interactions with dynamin (De Camilli et al., 1996).
Given the degree of current interest in the roles of the various synaptic proteins in regulating and mediating the release of neurotransmitter substances, we decided to investigate the effects of PAO on the release of the neurotransmitter acetylcholine (ACh) at the frog neuromuscular junction. The results, whilst demonstrating an inhibitory effect of PAO on ACh release, suggest that this effect of PAO at motor nerve endings is due to an action on N-type Ca2+ channels rather than a direct effect on the secretory apparatus.
Methods
Electrophysiological recordings at frog neuromuscular junctions
Frogs (Rana pipiens) were humanely killed by anaesthesia with 5% ether, followed by double pithing. Experiments were performed on the isolated cutaneous pectoris muscle preparation of the frog at 20–23°C. Normal Ringer solution contained (mM) NaCl 115, KCl 2, Ca2Cl 1.8, HEPES 2, pH 7.2–7.4). For studies of evoked release, the Ringer solution was supplemented with tubocurarine (6–10 μM) to reduce end-plate potentials (epps) below threshold for muscle action potential generation. Epps were evoked in response to supramaximal electrical stimulation (0.1 Hz) of the motor nerve. Intracellular recordings from the end-plate region of the muscle were made using 3 M KCl filled glass microelectrodes (5–15 MΩ). Miniature end-plate potentials (mepps) were also recorded in separate experiments in the absence of tubocurarine.
Local potential changes associated with nerve terminal ionic currents were recorded in response to motor nerve stimulation (0.05 Hz) using the perineural recording technique (Gundersen et al., 1982; Mallart, 1984; Anderson & Harvey, 1988; Silinsky & Solsona, 1992; Silinsky, 2000). These perineural deflections are due to the voltage change produced across the resistance of the perineural sheath surrounding several small axon bundles by currents flowing between the myelinated portion of the axons and the nerve terminals. These currents are proportional to the difference in potential between the nerve endings and the nodes of Ranvier. Whilst these waveforms are not strictly membrane currents, they are highly related to the membrane conductance changes that occur both at nerve endings and at the last nodes of Ranvier. The conductance change that underlies the Ca2+ current through the N-type Ca2+ channel is localized to the nerve endings and is responsible for the initiation of neurotransmitter release (Mallart, 1985; Robitaille et al., 1990). The Ca2+ current in the nerve endings generates a proportional current in the perineural space; this current flows upstream to the recording site where it is detected as an outward (upward-going) deflection. For the sake of clarity, therefore, we will term the upward-going voltage change that is antagonized by N-type Ca2+ channel blockers the ‘prejunctional Ca2+ current' as this voltage change is generated by the Ca2+ current emanating from the nerve endings flowing across the perineural resistance. For similar reasons, perineural voltage changes attributable to Na+ currents in the presynaptic element will be termed perineural Na+ currents as they are reflective of the membrane permeability changes associated with Na+ channels. Perineural deflections are quantified as the magnitude of the extracellular voltage change as measured from the baseline to the peak of the current (for a more complete discussion of these currents and its possible contaminants, see Silinsky & Solsona, 1992; Silinsky, 1999). The perineural recording solution had the same composition as normal Ringer solution with the addition of 3,4-diaminopyridine (100 μM) and tetraethylammonium (1 mM) to block K+ channels. With this solution, the repetitive firing that occurs in response to nerve stimulation serves as an additional index of Ca2+ entry as it is due to depolarization near the last node of Ranvier resulting from prolonged Ca2+ currents at the nerve terminal (Molgo & Mallart, 1985; Silinsky & Solsona, 1992). Perineural recordings were amplified with an Axoclamp 2B amplifier and collected on computer (DigiData 1200 interface, Axon Instruments Inc.) Signals were analysed using SCAN and CDR software (J. Dempster, University of Strathclyde).
Electrophysiological recordings of whole cell N-type calcium currents in acutely-dissociated sympathetic neurones
N-type Ca2+ channels have been previously demonstrated in a number of autonomic ganglia (e.g. Hille, 1992; Barajas-Lopez et al., 1996). In these experiments we used acutely-dissociated neurons from young adult guinea-pig coeliac ganglia, employing dissociation and recording procedures previously described (Matsumoto et al., 1993; Silinsky & Gerzanich, 1993). Cells were used 18–96 h after dissociation.
Patch electrodes (2–5 MΩ) were fabricated using a four stage pull on a horizontal microelectrode puller (Flaming-Brown P87). The internal solution used to fill the patch electrodes contained (mM) caesium gluconate, 140 (to block K+ channels); HEPES, 10; EGTA, 10. We purposely omitted ATP and GTP in our internal recording solution, to reduce the possibility that any effects of PAO were G-protein mediated. The external solution contained (mM) NaCl, 140; glucose, 10; HEPES, 10; CaCl2, 2.5 and KCl, 3 and was gravity fed into the recording chamber (1–2 ml min−1). In some experiments, (e.g. Figure 4b), 2 mM BaCl2 was added to the external solution.
Figure 4.
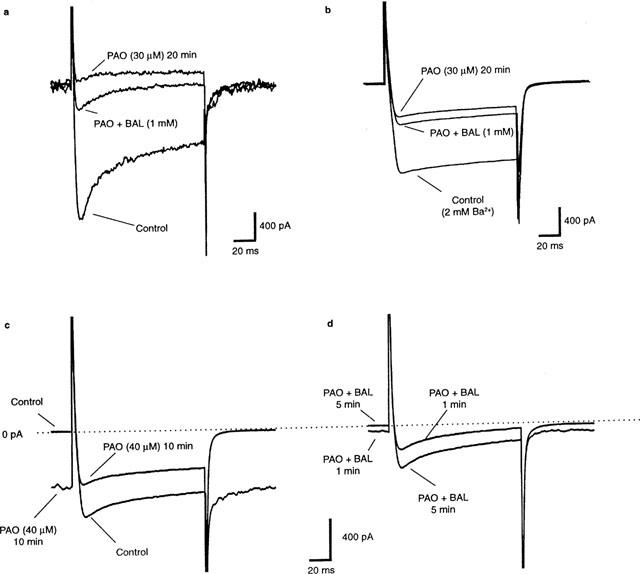
Effects of PAO on N-type Ca2+ currents and its reversal by BAL (1 mM). (a) shows individual traces depicting the inhibitory effect of a 20 min exposure to PAO (30 μM) and its partial reversal by BAL in normal Ca2+ solutions. (b) shows the averaged data from another experiment in which Ba2+ was used as the charge carrier. In this experiment, the mean peak Ba2+ current through N-type Ca2+ channels (−1.25±0.01 nA) was reduced by PAO (−0.47±0.01 nA) and reversed by coapplication of BAL (0.58±0.01 nA, n=4 for all). An analysis of variance followed by a multiple comparison using Bonferroni's method revealed highly statistically-significant differences between the three conditions (P<0.05). (c) shows that higher concentrations of PAO (40 μM), in addition to inhibiting Ca2+ currents, also increased the inward holding current observed before the depolarizing step. In the same experiment, BAL reversed both effects in a time dependent manner (d). Cells were held at −80 mV. Voltage-activated Ca2+ currents were triggered by voltage steps of 120 ms duration from the holding potential (−80 mV) to 0 mV. This depolarizing step was applied once every 20 s. For further details, see text.
Cells were held at −80 mV. Voltage-activated Ca2+ currents were triggered by voltage steps of 120 ms duration from the holding potential (−80 mV) to 0 mV. This depolarizing step was applied once every 20 s. All potentials were corrected for liquid junction potentials of −11 mV. Ca2+ currents through N-type Ca2+ channels were confirmed by demonstrating their sensitivity to inhibition by either ω-conotoxin GVIA (ω-CTx, see Figure 4a) or Cd2+ (see Figure 4b) and their insensitivity to 10 μM nifedipine (see Garcia et al., 1998; Barajas-Lopez et al., 1996 for justification). Tetrodotoxin (TTX, 3–10 μM) was used to block voltage-gated Na+ currents. Experiments were performed at 20–23°C.
Whole cell currents were recorded using an Axopatch 1D amplifier (Axon instruments). Signals were recorded on computer using the pCLAMP software suite (Axon Instruments) in conjunction with a DigiData 1200 interface. Records were sampled at a rate of 2–10 kHz after filtering at 2–5 kHz.
Due to the small size of the currents and the low pipette resistance, series resistance compensation was used only in a few experiments. Such compensation did not significantly modify the characteristics of the currents. For simplicity, statistical analysis was carried out using the peak amplitudes of the individual voltage activated currents.
Statistical methods
Statistical methods were similar to those described previously (Silinsky, 1984). Epps and perineural waveforms were averaged to reduce the coefficient of variation to less than 5% and to make statistically significant differences generally at P<<0.01 (Silinsky, 1984; 1999; Silinsky & Solsona, 1992). In most experiments, the responses to 4–20 stimuli were averaged. When smaller numbers (4–10) of stimuli were averaged, the individual data traces were first tested for normality and then comparisons between control and drug-treated neuromuscular junctions made by either parametric statistics (e.g. Student's paired t-test) or non-parametric statistics (Mann-Whitney rank sum test, see Glantz, 1992). In experiments in which more than two groups were compared, an analysis of variance for the normally distributed data was followed by multiple comparisons using the Bonferroni inequality (see legend to Figure 4b). This method is reasonable when three to four groups are compared and is the most conservative of the multiple comparisons procedures (see Glantz, 1992). In all neuromuscular junction experiments, n=number of preparations whilst in all whole-cell voltage clamp experiments, n=number of individual cells (each cell from a separate, naïve cover slip). Data are presented as mean values±1 s.e.mean. The statistical methods are illustrated more fully in the legends to Figures 3 and 4.
Figure 3.
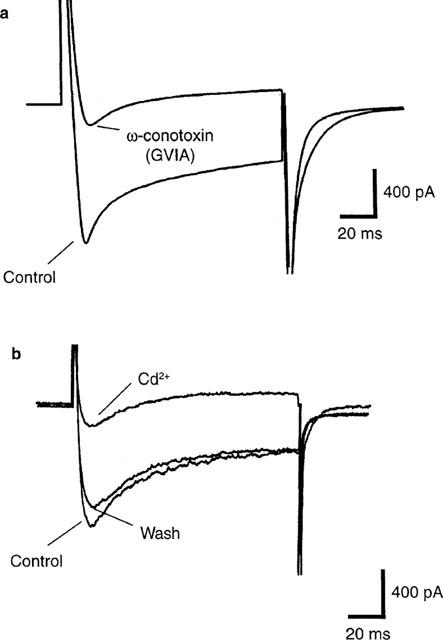
Inhibition of Ca2+ currents in acutely dissociated-sympathetic neurons by the N-type Ca2+ channel blockers ω-CTX (a) and Cd2+ (b). Cells were held at −80 mV. Voltage-activated Ca2+ currents were triggered by voltage steps of 120 ms duration from the holding potential (−80 mV) to 0 mV. This depolarizing step was applied once every 20 s. In (a), the individual records that comprise the averaged peak current amplitudes (−1.16±0.05 nA, n=6 stimuli) ranged from −1.27 to −0.95 nA in control conditions, whilst the individual records comprising the averaged peak response made after treatment with ω-CTX ranged from −0.18 to −0.15 nA. In the Cd2+ experiments (b), an analysis of variance followed by multiple comparisons using Bonferroni's method revealed highly statistically-significant differences between control (−1.16±0.03 nA, n=6) and Cd2+ treatment (−0.18±0.02 nA, n=6) and between Cd2+ exposure and the wash period (−1.14±0.03 nA, n=3; P<0.05) but not between the control and wash periods. For further details of the N-type Ca2+ currents in acutely-dissociated autonomic neurons in guinea-pig (see Barajas-Lopez et al., 1996).
Chemicals
All chemicals used in this study were purchased from Sigma Chemicals (St Louis, MO, U.S.A.). Stock solutions of 10−1 or 10−2 M PAO or 1 M BAL (which structurally is 2,3-dimercaptopropanol) in DMSO (0.003–0.03%) were used. These concentrations of DMSO had no effect on neuromuscular junctions or acutely dissociated neurons.
Results
General observations on the effect of PAO on neurotransmitter release
Application of PAO to isolated neuromuscular junctions produced substantial effects on both spontaneous ACh release (reflected as mepp frequency) and evoked ACh release (reflected as epp amplitudes). With respect to spontaneous neurotransmitter secretion, application of PAO (30 μM) resulted in pronounced increases in mepp frequency from the control level of 4.2±0.7 to 32.4±9.0 s−1 at 15 min after the beginning of exposure (mean±1 s.e.mean, n=4). These increases in mepp frequencies are well-sustained for over 60 min (45±11.6 s−1; n=4 at 60 min). Mepp frequencies were also accelerated by PAO in the absence of extracellular Ca2+ (from 1.4±0.8 to 51.3±19 at 10 min; n=3 experiments). However, such increases in spontaneous ACh release were only briefly sustained in the absence of extracellular Ca2+, with peak mepp frequencies subsiding after about 15 min. When PAO was applied in the presence of BAL (1 mM), mepp frequencies were unaffected (n=4 experiments).
With respect to evoked ACh release, the effects of PAO on epps were bi-phasic. Firstly application of PAO (30 μM) resulted in a rapid initial increase in epp amplitudes (192.7±33.2% of control; n=4 after 7.5 min: see Figure 1, filled circles, and traces a, b) when compared to control preparations bathed in normal Ringer (Figure 1, open circles). The initial increase in epp amplitudes had an onset of similar time course to the increase in mepp frequency.
Figure 1.
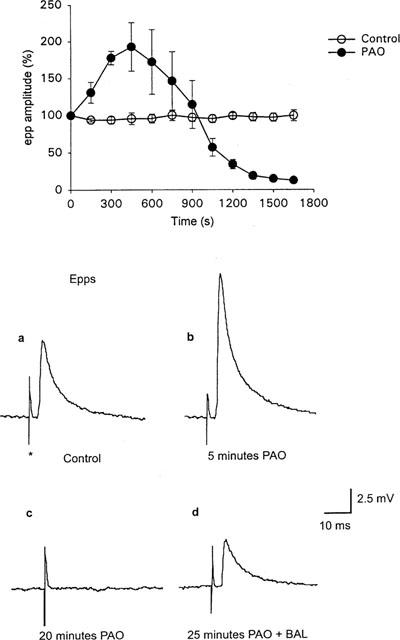
The effect of PAO on epp amplitudes. Open circles in the graph are epps recorded in the absence of PAO and filled circles are in the presence of PAO (30 μM). Each point represents the mean±s.e.mean from four separate preparations. In each experiment the amplitudes of eight consecutive epps (0.1 Hz) corresponding to each time period were expressed as a percentage of the first eight epps. Note the initial increase in epp amplitudes following application of PAO and the subsequent inhibition of epp amplitudes. (a), (b), (c) and (d) show averages of four consecutive epps evoked at 0.1 Hz, in (a) control, (b) following 5 min application of PAO, (c) 20 min application of PAO and (d) following the addition of BAL (1 mM) in the continued presence of PAO. See text for further details.
After this increase, PAO produced a time-dependent reduction in epp amplitudes and eventually, complete abolition of epps (elimination of epps occurred at 25±7.5 min; n=6; see Figure 1 and trace c). The abolition of epps by PAO was independent of nerve stimulation (i.e. epps were also abolished after 30 min of PAO exposure in the absence of nerve stimulation, n=2 experiments). In addition, simple removal of PAO (for up to 45 min) failed to have any effect on epps previously abolished by PAO (n=2). We also investigated the effects of a lower concentration of PAO (15 μM) on epps. In these experiments similar inhibitory effects of PAO on epps were observed but the time course was much slower, with epps peaking in amplitude after about 20 min and complete inhibition occurring at about 1 h incubation in 15 μM PAO (n=2).
As shown in Figure 1d, abolition of epps by 30 μM PAO was partially reversed by the co-application of BAL (1 mM) with PAO (43±6% control epp amplitude; n=5). In contrast with the effects of PAO alone on epps, co-application of PAO (30 μM) and BAL (1 mM) had no significant effects on epp amplitudes (n=5).
These complex effects of PAO are consistent with those attributed to actions on the secretory apparatus, but could also be due to effects on voltage-gated ionic channels in the motor nerve ending. We thus examined the effects of PAO on the electrophysiological correlates of the Ca2+ currents in motor nerve endings, namely, the prejunctional Ca2+ current and its repetitive firing as recorded from the perineurium (see Methods for justification of terminology).
The effects of PAO on perineural currents from motor nerve endings
Figure 2a–d depicts the effects of PAO on the perineural currents. Note that, in the presence of potassium channel blockade, both the Na+ current underlying the propagation of the action potential in the nerve and the prejunctional Ca2+ current can be measured (Gundersen et al., 1982; Mallart, 1984; Anderson & Harvey, 1988; Silinsky & Solsona, 1992). Application of PAO resulted in both a highly significant reduction in the prejunctional Ca2+ currents and the abolition of repetitive firing associated with the increased calcium influx produced by the presence of K+ channel blockers (n=4, Figure 2b,c). In contrast, PAO had no significant effect on prejunctional Na+ currents (n=8, see e.g. Figure 2a–d). These inhibitory effects of PAO on prejunctional Ca2+ currents developed steadily over a period of 30–45 min in the presence of PAO and did not occur in control preparations (Figure 2). We and others have shown that neurotransmitter release from frog motor nerve endings is mediated by N-type Ca2+ channels (Kerr & Yoshikami, 1984; Silinsky & Solsona, 1992; Redman & Silinsky, 1995; Katz et al., 1995). Specifically both evoked ACh release and the upward going component of the perineural waveform are inhibited by the N-type channel blockers ω-conotoxinGVIA and Cd2+, but not by blockers of L-type Ca2+ channels or P-type Ca2+ channels (e.g. Redman & Silinsky, 1995; Katz et al., 1995). Indeed PAO had no effect on the waveform in the presence of the Ca2+ channel blocker Cd2+ (n=3, data not shown), suggesting that the effect of PAO is indeed due to an inhibition of Ca2+ entry via N-type Ca2+ channels in motor nerve endings. As with the epps, BAL also resulted in a partial reversal of the reduction in the prejunctional Ca2+ currents produced by PAO. Specifically, PAO reduced the calcium current to 44.2±8.0% of control (n=4) after 27.5 min and this was reversed by co-application of BAL (76.1±5.5% of control, n=4). This partial reversal of both the peak Ca2+ current and the restoration of repetitive firing by BAL is illustrated in Figure 2d.
Figure 2.
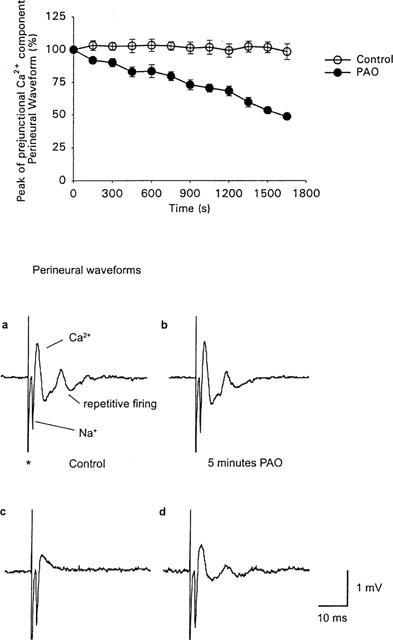
The effect of PAO on the prejunctional Ca2+ current. Open circles in the graph were recorded in the absence of PAO and filled circles are in the presence of PAO (30 μM). Each point represents the mean amplitude of the positive component of the perineural wave form±s.e.mean from four separate preparations (see Figure 1 and Methods for further details). In each experiment the amplitudes of the eight consecutive prejunctional Ca2+ components corresponding to each time period (evoked at 0.05 Hz) were expressed as a percentage of the first eight perineural recordings made in the absence of PAO. Note the gradual reduction in the prejunctional Ca2+ current occurs during the initial increase in epp amplitude (compare with Figure 1). (a), (b), (c) and (d) are averages of eight consecutive perineural wave-forms (recorded from a separate experiment) evoked at 0.05 Hz. (a) is in control perineural recording solution (see Methods). (b) is following 5 min and (c) 20 min in the continued presence of PAO. (d) shows the perineural waveform following the addition of BAL (1 mM) in the continued presence of PAO. *Indicates the stimulation artefact. Note that both the abolition of the epp by PAO, shown in Figure 1, and the reduction in the prejunctional Ca2+ current (including a decrease in the degree of repetitive firing) by PAO occur with a similar time course. In addition both effects are partially reversed following the addition of BAL in the presence of PAO. See text for further experimental details.
Separate studies on mouse phrenic nerve motor nerve endings showed that PAO (30 μM) applied for as long as 2.5 h, had no statistically significant effect on EPPs (n=5 experiments) or perineural Ca2+ currents (n=2 experiments; data not shown); mouse motor nerves produce evoked neurotransmitter release using P-type but not N-type Ca2+ channels (Xu & Atchison, 1996).
Given the apparent inhibitory effects of PAO on N-type Ca2+ currents coupled to the secretory apparatus at intact neuromuscular junctions (Figure 2a–d), it was of interest to examine the effects of PAO directly on N-type Ca2+ currents using whole cell patch clamp recording techniques. The soma of acutely-dissociated autonomic neurons provide an ample supply of N-type Ca2+ channels that are unlikely to be coupled to the transmitter release process. We therefore investigated the effects of PAO and BAL on N-type Ca2+ currents in acutely-dissociated autonomic neurons.
The effects of PAO and BAL on N-type calcium currents in acutely dissociated sympathetic ganglion cells
Voltage activated Ca2+ currents were evoked from neurons acutely dissociated from guinea-pig coeliac ganglia in the presence of TTX (3–10 μM) using procedures previously described for N-type Ca2+ currents in other autonomic neurons (for details of N-type current in guinea-pig enteric neurons, which behave similarly to coeliac neurons, see Barajas-Lopez et al. 1996).
Figure 3a shows the control Ca2+ current is blocked by the N-type Ca2+ channel blocker ω-CTX (100 nM, n=6). In addition, the N-type Ca2+ channel blocker Cd2+ (300 μM) also reversibly inhibits the inward current in these cells (Figure 3b, n=8, see also Garcia et al., 1998). In contrast, these currents were not affected by the L-type Ca2+ channel blocker nifedipine (10 μM, n=3, data not shown).
Figure 4 depicts the results of several different experiments that demonstrate that the application of PAO to autonomic neurons inhibits the N-type Ca2+ current, an effect that was partially reversible by co-application of BAL. Figure 4a shows the raw data traces from an experiment made in normal Ca2+ solutions. Note the complete elimination of the Ca2+ currents by PAO (30 μM) and its partial reversal following co-application of BAL (1 mM). These effects do not appear to be dependent on an increase in Ca2+-dependent K+ conductances as similar results were obtained using Ba2+ as the permeant ion (Figure 4b, which shows averaged data–see Figure legend). The experiment of Figure 4b also illustrates that co-application of BAL with PAO both stopped the reduction in Ca2+ current and produced a partial reversal of its inhibition. Similar results were observed in a total of six experiments (Ba2+, n=2; Ca2+, n=4).
In a number of experiments, the prolonged application of 30 μM PAO (>20 min) or brief exposure to higher concentrations of PAO (40–100 μM) produced an additional effect. As shown in Figure 4c, 40 μM PAO in addition to inhibiting the N-type Ca2+ current, also produced a dramatic increase in the steady-state inward current seen here before the depolarizing step (from 0.028±0.005 to 1.94±0.43 nA; n=6). Co-application of BAL in the same experiment both reversed the increase in steady-state inward current and partially restored the depolarization-evoked Ca2+ current (Figure 4d). In addition, the N-type Ca2+ channel blockers ω-CTX (100 nM) and Cd2+ (300 μM) prevented the increase in holding current produced by PAO (300 μM, n=3, data not shown).
In combination, these results demonstrate that the inhibitory effects of PAO are mediated via an action on N type Ca2+ channels at both neuromuscular junctions and autonomic neurons.
Discussion
In their study of the release of glutamate from rat cerebrocortical synaptosomes, Wiedemann et al. (1998) found that, following 10 min incubation in the presence of PAO, glutamate release was substantially reduced in response to either KCl depolarization or 4-aminopyridine stimulation (see also Khvotchev & Sudhof, 1998). The observation that PtdIns 4-kinase associated with synaptic vesicles was concomitantly inhibited led to the suggestion that PtdIns 4-kinase plays a critical role in the events preceding the exocytosis of neurotransmitters from synaptic vesicles.
In this study we found that PAO (30 μM) produced several effects on transmitter release at the frog neuromuscular junction all of which were prevented when PAO was coapplied with an agent that inactivates PAO (BAL, 1 mM). PAO had a biphasic effect on the evoked epps recorded in tubocurarine treated preparations, with an initial rapid increase in epp amplitude followed by stimulus-independent reduction and eventual abolition of the epps. In the absence of tubocurarine, PAO caused an initial increase in mepp frequency with a similar time course in onset as seen with the increase in epp amplitudes. Increases in epp amplitudes were not accompanied by any measurable changes in the prejunctional Ca2+ current. It seems possible that this transient increase in transmitter release was not observed in previous studies using PAO due to the incubation protocols employed (e.g. Wiedemann et al., 1998).
The increase in mepp frequency occurred in the absence of extracellular Ca2+ (although it was not as well sustained), suggesting that part of the increases in epp amplitude and mepp frequency was due to an action downstream from Ca2+ entry. In support of this notion, it has previously been shown that PAO can release Ca2+ from mitochondria by opening the permeability transition pore (e.g. Lenartowicz et al., 1991; Schweitzer et al., 1994). The observation that in normal extracellular Ca2+ concentrations, mepp frequencies were elevated well beyond the time that evoked epps were abolished suggests that the exocytotic machinery is not directly inhibited by PAO at motor nerve endings.
The other effect of PAO, namely, abolition of evoked epps, is consistent with the observations of Wiedermann et al. (1998) and Khvotchev & Südhof (1998). However, we found that the abolition of epps by PAO was associated with an inhibition of prejunctional Ca2+ currents in motor nerve endings. It thus seemed likely that an inhibition of Ca2+ entry into the nerve terminal alone could account for the abolition of the epp as both events which occurred roughly at the same point in time. We therefore investigated this possibility more directly, using whole-cell recording methods to record N-type Ca2+ channels from acutely dissociated sympathetic neurons. In these experiments, application of PAO also produced a reduction in the voltage-activated calcium current, which was partially reversed by BAL.
The additional effect of higher concentrations or prolonged exposure to PAO to increase the inward holding current in acutely dissociated neurons merits further discussion. The observation that the increase in the inward holding current and further reduction in the size of the voltage activated calcium current by PAO can be blocked by N-type Ca2+ channel blockers raises the possibility that PAO, in addition to blocking N-type Ca2+ channels, can actually open N-type Ca2+ channels. It is not clear whether these two events, the reduction in Ca2+ current and the increase in ‘steady state' Ca2+ conductance, are linked or not and at present it is not clear whether both effects occur at the frog neuromuscular junction. Indeed, both events would be expected to produce a reduction in the numbers of calcium channels available for reopening in response to the nerve stimulation, as Ca2+ calcium channels opened before the depolarizing pulse would not be available for reopening in response to subsequent depolarization. These additional effects of higher concentrations of PAO might be predicted based upon the fact that, PAO is a relatively unselective agent which acts to cross-link vicinial cysteines. In addition to its actions on PtdIns 4-kinase activity, PAO is known to inhibit the activity of phosphatases (Zhang et al., 1992; Wijetunge et al., 1998), hydroxybutyrate dehydrogenase (Phelps & Hatefi, 1981) rhodanese (Prasad & Horowitz, 1997) and GABA transporters in synaptic vesicles (Robillard et al., 1987). As previously mentioned, PAO also causes the opening of the mitochondrial permeability transition pore (e.g. Lenartowicz et al., 1991; Schweizer et al., 1994). Many of these effects occur at similar concentrations to those we employed in this study (15–60 μM).
In conclusion, the effects of PAO to inhibit N-type Ca2+ currents at both frog motor nerve endings and mammalian sympathetic neurons, and the absence of effects of PAO on neurotransmitter release in the mouse (which is mediated via P-type channels), suggest that PAO inhibits neurotransmitter release via an action on N-type Ca2+ channels rather than through an effect on vesicle-associated PtIns 4-kinase.
Acknowledgments
This work was supported by a grant from the NIH (NS12782). The authors would like to thank Shirley Foster for help with the dissociation of the coeliac ganglia and Dr J. Hirsh for reading the manuscript.
Abbreviations
- BAL
British anti-lewisite
- epps
end-plate potentials
- mepps
miniature end-plate potentials
- ω-CTx
ω-conotoxin GVIA
- PAO
phenylarsine oxide
- PtdIns 4-kinase
phosphatidylinositol 4-kinase
References
- ANDERSON A.J., HARVEY A.L. Effects of the potassium channel blocking dendrotoxins on acetylcholine release and motor nerve terminal activity. Br. J. Pharmacol. 1988;93:215–221. doi: 10.1111/j.1476-5381.1988.tb11424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARAJAS-LOPEZ C., PERES A.L., ESPINOSA-LUNA R. Cellular mechanisms underlying adenosine actions on cholinergic transmission in enteric neurons. Am. J. Physiol. (Cell Physiol 40) 1996;271:C263–C275. doi: 10.1152/ajpcell.1996.271.1.C264. [DOI] [PubMed] [Google Scholar]
- BENNETT M.K. Calcium and the regulation of neurotransmitter secretion. Curr. Opin. Neurobiol. 1998;7:316–322. doi: 10.1016/s0959-4388(97)80058-x. [DOI] [PubMed] [Google Scholar]
- DE CAMILLI P., EMR S.D., MCPHERSON P.S., NOVICK P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- GARCIA D.E., LI B., GARCIA-FERREIRO R.E., HERNANDEZ-OCHOA E.O., YAN K., GAUTAM N., CATTERALL W.A., MACKIE K., HILLE B. G protein beta subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J. Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLANTZ S.A. Primer of Biostatistics. New York: McGraw Hill Inc; 1992. [Google Scholar]
- HILLE B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc; 1992. [Google Scholar]
- GUNDERSEN C.B., KATZ B., MILEDI R. The antagonism between botulinum toxin and calcium in motor nerve terminals. Proc. R., Soc. Lond. (Biol). 1982;216:369–376. doi: 10.1098/rspb.1982.0080. [DOI] [PubMed] [Google Scholar]
- KATZ E., FERRO P.A., CHERKSEY B.D., SUGIMORI M., LLINAS R., UCHITEL O.D. Effects of Ca2+ channel blockers on transmitter release and presynaptic currents at the frog neuromuscular junction. J. Physiol. 1995;486:695–706. doi: 10.1113/jphysiol.1995.sp020845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERR L.M., YOSHIKAMI D.C. A venom peptide with novel presynaptic blocking action. Nature. 1984;308:282–284. doi: 10.1038/308282a0. [DOI] [PubMed] [Google Scholar]
- KHVOTCHEV M., SÜDHOF T.C. Newly synthesised phosphates are required for synaptic norepinephrine but not glutamate or γ-aminobutyric acid (GABA) release. J. Biol. Chem. 1998;273:21451–21454. doi: 10.1074/jbc.273.34.21451. [DOI] [PubMed] [Google Scholar]
- LENARTOWICZ E., BERNARDI P., AZZONE G.F. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J. Bioener. Biomembr. 1991;23:679–688. doi: 10.1007/BF00785817. [DOI] [PubMed] [Google Scholar]
- MALLART A. Presynaptic currents in frog motor endings. Pflugers Arch. 1984;400:8–13. doi: 10.1007/BF00670529. [DOI] [PubMed] [Google Scholar]
- MALLART A. A calcium-activated potassium current in motor nerve terminals of the mouse. J. Physiol. 1985;368:577–591. doi: 10.1113/jphysiol.1985.sp015877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUMOTO S.G., GRUENER R.P., KREULEN E.L. Neurotransmitter properties of guinea-pig sympathetic neurons grown in dissociated cell culture-I. Adult Neurons. Neuroscience. 1993;57:1135–1145. doi: 10.1016/0306-4522(93)90055-k. [DOI] [PubMed] [Google Scholar]
- MOLGO J., MALLART A. Effects of Anemonia sulcata toxin II on presynaptic currents and evoked transmitter release at neuromuscular junctions of the mouse. Pflugers Arch. Eur. J. Physiol. 1985;405:349–353. doi: 10.1007/BF00595687. [DOI] [PubMed] [Google Scholar]
- PHELPS D.C., HATEFI Y. Inhibition of D(-)-beta-hydroxybutyrate dehydrogenase by modifiers of disulfides, thiols, and vicinal dithiols. Biochemistry. 1981;20:453–458. doi: 10.1021/bi00506a001. [DOI] [PubMed] [Google Scholar]
- PRASAD A.R., HOROWITZ P.M. Chemical modification of bovine liver rhodanese with tetrathionate: differential effects on the sulfur-free and sulfur-containing catalytic intermediates. Biochim. Biophys. Acta. 1987;911:102–108. doi: 10.1016/0167-4838(87)90275-5. [DOI] [PubMed] [Google Scholar]
- REDMAN R.S., SILINSKY E.M. On the simultaneous electrophysiological measurements of neurotransmitter release and perineural calcium currents from frog motor nerve endings. J. Neurosci. Meth. 1995;57:151–159. doi: 10.1016/0165-0270(94)00133-2. [DOI] [PubMed] [Google Scholar]
- ROBILLARD G.T., SCHAAF J.M., TEELKEN A.W. Dithiols and monothiols are linked with GABA transport in membrane vesicles of rat brain synaptosomes. FEBS Lett. 1987;224:391–395. doi: 10.1016/0014-5793(87)80490-8. [DOI] [PubMed] [Google Scholar]
- ROBITAILLE R., ADLER M., CHARLTON M.P. Strategic location of calcium channel at transmitter release sites of frog neuromuscular junctions. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- SCHWEIZER M., DURRER P., RICHTER C. Phenylarsine oxide stimulates pyridine nucleotide-linked Ca2+ release from rat liver mitochondria. Biochem. Pharmacol. 1994;48:967–973. doi: 10.1016/0006-2952(94)90367-0. [DOI] [PubMed] [Google Scholar]
- SILINSKY E.M. On the mechanism by which adenosine receptor activation inhibits the release of acetylcholine from motor nerve endings. J. Physiol. 1984;346:243–256. doi: 10.1113/jphysiol.1984.sp015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILINSKY E.M. Antagonism of calcium currents and neurotransmitter release by barium ions at frog motor nerve endings. Br. J. Pharmacol. 2000;129:360–366. doi: 10.1038/sj.bjp.0703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILINSKY E.M., GERZANICH V. On the excitatory effects of ATP and its role as a neurotransmitter in coeliac neurons of the guinea-pig. J. Physiol. 1993;464:197–212. doi: 10.1113/jphysiol.1993.sp019630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SILINSKY E.M., SOLSONA C.S. Calcium currents at motor nerve endings: absence of effects of adenosine receptor agonists in the frog. J. Physiol. 1992;457:315–328. doi: 10.1113/jphysiol.1992.sp019380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIEDEMANN C., SCHAFER T., BURGER M.M. Chromaffin granule-associated phosphatidylinositol 4-kinase activity is required for stimulated secretion. EMBO J. 1996;15:2094–2101. [PMC free article] [PubMed] [Google Scholar]
- WIEDEMANN C., SCHAFER T., BURGER M.M., SIHRA T.S. An essential role for a small synaptic vesicle-associated phosphatidylinositol 4-kinase in neurotransmitter release. J. Neurosci. 1998;18:5594–5602. doi: 10.1523/JNEUROSCI.18-15-05594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIJETUNGE S., LYMN J.S., HUGHES A.D. Effect of inhibition of tyrosine phosphatases on voltage-operated calcium channel currents in rabbit isolated ear artery cells. Br. J. Pharmacol. 1998;124:307–316. doi: 10.1038/sj.bjp.0701840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU Y.-F., ATCHISON W.D. Effects of ω-agatoxin-IVA and ω-conotoxin-MVIIC on perineurial Ca2+ and Ca2+-activated K+ currents of mouse motor nerve terminals. J. Pharmacol. Exp. Ther. 1996;279:1229–1236. [PubMed] [Google Scholar]
- ZHANG Z.Y., DAVIS J.P., VAN ETTEN R.L. Covalent modification and active site-directed inactivation of a low molecular weight phosphotyrosyl protein phosphatase. Biochem. 1992;31:1701–1711. doi: 10.1021/bi00121a018. [DOI] [PubMed] [Google Scholar]