

Abstract
Cyclosporin A (CsA) is an immunosuppressant drug that inhibits nitric oxide (NO) synthase induction in vascular smooth muscle cells. Splanchnic artery occlusion (SAO) shock is a lethal type of shock characterized by a marked vascular dysfunction in which the L-arginine/nitric oxide pathway plays an important role. We investigated whether CsA exerts protective effects in SAO shock by interfering with the L-arginine/nitric oxide pathway.
Male anaesthetized rats (n=156) were subjected to clamping of the splanchnic arteries for 45 min. This surgical procedure resulted in an irreversible state of shock (SAO shock). Sham operated animals were used as controls. SAO shocked rats had a decreased survival (86±6 min, while sham shocked rats survived more than 240 min), marked hypotension, increased serum levels of TNF-α, enhanced plasma nitrite/nitrate concentrations (75±7.1 μM; sham shocked rats=1.6±0.5 μM) and enhanced inducible NO synthase (iNOS) protein induction and activity in the aorta. Moreover aortic rings from shocked rats showed a marked hyporeactivity to phenylephrine (PE, 1 nM–10 μM).
CsA (0.25, 0.5 and 1 mg kg−1, 5 min after reperfusion) increased survival rate (SAO+CsA=236±9 min following the highest dose), reverted the marked hypotension, reduced plasma nitrite/nitrate concentration (11±5.2 μM following the highest dose), restored to control values the hyporeactivity to PE, and blunted iNOS protein induction and activity in aortic rings.
The present data indicate that in an experimental rat model CsA may have antishock properties related to inhibition of L-arginine/nitric oxide pathway.
Keywords: Cyclosporin A, iNOS, splanchnic artery occlusion shock, vascular dysfunction
Introduction
Occlusion of the major splanchnic arteries followed by reperfusion in anaesthetized rats results in an irreversible circulatory failure and shock (splanchnic artery occlusion shock; SAO shock) (Squadrito et al., 1994b). It has been demonstrated that in SAO shock a marked and composite vascular dysfunction is present in which the L-arginine/nitric oxide (NO) pathway plays an important role (Squadrito et al., 1994a; 1996).
Indeed aortic rings from shocked rats show a marked hyporeactivity to phenylephrine and removal of endothelium does not restore the phenylephrine-induced contractile response to the values of the sham animal, thus suggesting that smooth muscle cells are involved in the hyporesponsiveness to phenylephrine. This complex dysfunction is probably the result of an increase in the endogenous NO produced by the inducible NO synthase (iNOS).
Cyclosporin A (CsA), is an immunosuppressant drug that blocks T-cell proliferation in response to ligation of the T-cell receptor (Kahan, 1989; Fruman et al., 1994). Type 1 T helper cells appear to be preferentially suppressed compared with type 2 T helper cells (Schreiber & Crabtree, 1992). It binds with high affinity to a family of cytoplasmatic immunosuppressant binding proteins called immunophilins. The drug exerts its effect principally through impairment of gene expression in target cells.
The immunophilin-drug complex inhibits calcineurin phosphatase (Clipstone & Crabtree, 1993) and therefore the drug blocks calcium-dependent events, such as cytokine gene expression, nitric oxide synthase activation, cell degranulation and apoptosis (Wiederrecht et al., 1993; Muhl et al., 1993; Fast et al., 1993).
The aim of our study was to investigate the efficacy of cyclosporin A in a rat model of circulatory shock. The drug, reduced iNOS activation, increased blood pressure, decreased the plasma levels of nitrite/nitrate and significantly improved survival in rats subjected to splanchnic artery occlusion shock.
Methods
Animal preparation
Male Sprague-Dawley rats (n=156) weighing 200–250 g were permitted access to food and water ad libitum. The rats were anaesthetized with urethane (1.3 g kg−1, i.p.). After midline laparotomy, the coeliac and superior mesenteric arteries were isolated near their aortic origins. During this procedure, the intestinal tract was maintained at 37°C by placing it between gauze pads soaked with warmed 0.9% NaCl solution. Rats were given heparin (1000 u kg−1, i.v.) and were observed for a 30 min stabilization period prior to either splanchnic ischaemia or sham ischaemia. Splanchnic artery ischaemia-reperfusion injury (SAO) was induced by clamping both the superior mesenteric artery and the coeliac trunk so as to produce a total occlusion of these arteries for 45 min. The clamps were then removed. Following reperfusion the rats were observed for 240 min. Sham-operated rats were subjected to the same surgical procedures as SAO rats except the arteries were not occluded.
Survival evaluation and arterial blood pressure monitoring
A first group of animals (n=80) was used to study survival and arterial blood pressure. The animals were previously implanted with cannulae (PE 50) into the left common carotid artery and right jugular vein, as described elsewhere (Caputi et al., 1980). Cyclosporin A (CsA; 0.25; 0.5 and 1 mg kg−1), 1400 W (20 mg kg−1), a highly selective inhibitor of iNOS (Garvey et al., 1997), NG-nitro-L-arginine methyl-ester (L-NAME; 5 mg kg−1) a non selective inhibitor of NOS or vehicle (DMSO-NaCl 0.9% 1 : 1, v v−1; 1 ml kg−1) were injected intravenously 5 min following the onset of reperfusion. Survival was evaluated for 240 min after the onset of reperfusion and expressed as survival time. The arterial catheter was connected to a pressure transducer. The pressure pulse triggered a cardiotachometer, and arterial blood pressure was displayed on a polygraph. Arterial blood pressure is reported as mean arterial pressure (MAP) in mmHg. MAP was measured at several time points: 0 min before occlusion (basal), 45 min after occlusion of the splanchnic arteries (end of occlusion) and 70 min following the onset of reperfusion (end of reperfusion).
Isolated aortic rings
A second group of rats (n=56) was used to study vascular reactivity, plasma nitrite/nitrate concentration, serum TNF-α levels and iNOS protein induction and activity. In this group of rats Cyclosporin A (CsA; 0.25; 0.5 and 1 mg kg−1), 1400 W (20 mg kg−1), L-NAME (5 mg kg−1) or vehicle (NaCl 0.9% 1 ml kg−1) were injected intravenously 5 min following the onset of reperfusion. Thoracic aortae were removed 70 min after reperfusion and placed in cold Krebs' solution of the following composition (nM): NaCl 118.4, KCL 4.7, MgSO4 1.2, CaCl2 2.5, KH2PO4 1.2, Na HCO3 25.0 and glucose 11.7; then aortae were cleaned of adherent connective and fat tissue and cut into rings of approximately 2 mm in length. In some rings, the vascular endothelium was removed mechanically by gently rubbing the luminal surface with a thin wooden stick. Rings were then placed under 1 g of tension in an organ bath containing 10 ml of Krebs' solution at 37°C and bubbled with 95% O2 and 5% CO2 (pH 7.4). All experiments were carried out in the presence of indomethacin (10 μM) in order to exclude the involvement of prostaglandins and their metabolites. Developed tension was measured with an isometric force transducer and recorded on a polygraph (Ugo Basile, Varese, Italy). After an equilibration period of 60 min during which time the rings were washed with fresh Krebs' solution at 15–20 min intervals and basal tension was readjusted to 1 g, the tissue was exposed to phenylephrine (PE, 100 nM). When the contraction was stable, the functional integrity of endothelium was assessed by a relaxant response to acetylcholine (ACh, 100 nM). The tissue was then washed occasionally for 30 min. Concentration-response curves were obtained by cumulative concentrations of PE (1 nM–10 μM) to intact or endothelium denuded aortic rings. The results (mean±s.e.mean) are expressed as g of tension mg−1 tissue.
Nitrite/nitrate measurement
Nitric oxide release was determined spectrophotometrically by measuring the accumulation of both nitrite and nitrate (the latter is reduced to nitrite) in plasma. Plasma samples were taken before occlusion (basal) and at the end of reperfusion (70 min following the onset of reperfusion). Nitrate was stoichiometrically reduced to nitrite by incubation of sample (100 μl plasma) for 2 h at 37°C, in the presence of 0.1 unit ml−1 nitrate reductase (NAD[P]H: nitrate oxidoreductase, EC 1.6.6.2; Aspergillus species; Sigma Chemical Co., St. Louis, MO, U.S.A.) 120 μM, NADPH and 5 μM FAD (flavinadenine dinucleotide, Sigma Chemical Co., St. Louis, MO, U.S.A.) in final volume of 103 μl. After nitrate had been reduced to nitrite, NADPH which interfered with the subsequent nitrite determination was oxidized with 10 units ml−1 L-lactic dehydrogenase (EC 1.1.127; type XI; from rabbit muscle; Sigma Chemical Co., St. Louis, MO, U.S.A.) and 10 mM sodium pyruvate for 30 min at 37°C in a final volume of 114 μl. Sodium nitrate was used as a standard. Nitrite/nitrate concentration in plasma was assayed by a standard Griess reaction (Ding et al., 1988). Briefly, 100 μl plasma was incubated with an equal volume of Griess reagent (1% sulphanilamide/0.1% naphtylenediamine dihydrochloride/2.5% H3PO3) at room temperature for 10 min. The absorbance of the chromophore formed was determined at 540 nm using a microtiter plate reader. Nitrite concentrations were calculated by comparison with a standard curve with sodium nitrite and control baseline plasma as a blank.
Tissue preparation
Thoracic aortae were used for determination of iNOS. Rats were killed by cardiac puncture 70 min following the onset of reperfusion and thoracic aortae were immediately excised, cleaned with PBS, frozen in liquid nitrogen, and stored at −70°C. Homogenates (25% w v−1) were prepared in 10 mM HEPES buffer, pH 7.4, containing (mM) sucrose 320, EDTA 1, DTT 1, 10 μg ml−1 leupeptin, and 2 μg ml−1 aprotinin at 0°C to 4°C with the aid of a tissue grinder fitted with a motor-driven ground glass pestle. Homogenates were centrifuged at 12,000×g for 5 min at 4°C to remove tissue debris without precipitation of plasma membrane fragments. The supernatant was used for determination of iNOS activity and protein mass. Protein concentration was determined with a Bio-Rad kit.
iNOS activity assay
To determine calcium-independent iNOS activity in the homogenates, production of L-[3H]-citrulline from L-[3H]-arginine (7.4 kBq per tube) was measured in the presence of 10 μM L-arginine/1 mM NADPH/300 units of calmodulin per ml/5 μM tetrahydrobiopterin/50 mM L-valine/1 mM EGTA for 30 min at room temperature. Reactions were stopped by adding 1 ml of ice cold 20 mM HEPES buffer, pH 5.5, containing (mM) EDTA 2 and EGTA 2. After separation by using Dowex 50 W (sodium form), L-[3H]-citrulline activity was measured by counting scintillation (Beckman analytical instruments S.p.A., Milan, Italy). Experiments performed in the absence of NADPH determined the extent of L-[3H]-citrulline formation independent of iNOS activity.
Western blot analysis
Aorta tissue preparation (50 μg of protein) were size-fractionated on 4–12% Tris Glycine gel (Novex) at 120 V for 3 h. In preliminary experiments, we found that the given protein concentration was within the linear range of detection of our Western blot technique. After electrophoresis, proteins were transferred onto Hybond-ECL membrane (Amersham Life Science Inc.) at 400 mA for 120 min using the Novex transfer system. The membrane was prehybridized in 10 ml of buffer A (mM): (Tris-HCL 10 (pH 7.5), NaCl 100, 0.1% Tween 20, and 10% nonfat milk powder) for 1 h and hybridized for an additional 1-h period in the same buffer containing 10 μl of the given anti iNOS monoclonal antibody (1 : 1000). The membrane was then washed for 30 min in a shaking bath, with the buffer (buffer A without nonfat milk) changed every 5 min before 1 h of incubation in buffer A plus goat anti-rat IgG-horseradish peroxidase at the final titer of 1 : 1000. Experiments were carried out at room temperature. The washes were repeated before the membrane was developed with a light-emitting non radioactive method using ECL reagent (Amersham Inc.). The membrane underwent autoluminography in a darkroom with a fixed camera for 1–5 min. The captured image, sent to an image analysis software (BIO-PROFIL Celbio, Milan, Italy), was subjected to a densitometric analysis and then printed on VPN-120 printer.
Biological assay for TNF-α activity
Killing of L929 mouse tumour cells were used to measure TNF-α levels in serum on the basis of a standard micro-ELISA assay (Altavilla et al., 1989). L929 cells in RPMI 1640 medium containing 5% foetal calf serum were seeded at 3×10−4 cells per well in 96-well microdilution plates and incubated overnight at 37°C in an atmosphere of 5% CO2 in air. Serial 1 : 2 dilutions of serum (drawn at different time points: 0, 10, 20 and 45 min of occlusion and 10, 20, 30, 40, 50, 60 and 70 min of reperfusion) were made in the above-described medium containing 1.0 μg of actinomycin D per ml and 100 μl volumes of each dilution were added to the wells. One TNF-α unit was defined as the amount giving 50% cell cytotoxicity. The TNF-α content in the sample was calculated by comparison with a calibration curve obtained with recombinant murine TNF-α.
Drugs
Acetylcholine chloride, phenylephrine hydrochloride, indomethacin, EGTA, EDTA, NADPH, nitrate reductase, sodium nitrate, L-arginine and Dowex 50 W anion exchange were obtained from Sigma Chemical Co., St. Louis, MO, U.S.A. Cyclosporin A (Sandimmun) was obtained from Sandoz, S.p. A. Milan, Italy. 1400 W was a kind gift from APHOGEPHA Arzneimittel GmbH, (Dresden, Germany) and L-NAME was purchased from SIGMA.
Statistical analysis
Data are expressed as means±s.e.mean and were analysed by analysis of variance for multiple comparison of results; Duncan's multiple range test was used to compare group means. In all cases, a probability error of less than 0.05 was selected as criterion for statistical significance. For survival data, statistical analysis was done with Fisher's exact probability test.
Results
Survival
Table 1 shows survival time for the groups of rats subjected to splanchnic artery occlusion shock or sham shock. All sham rats survived the entire 240 min observation period. In contrast in rats treated with the vehicle, occlusion and reperfusion of the splanchnic region produced a profound shock state characterized by a high lethality: no rat survived at 2 h of reperfusion (survival time=86±6 min). Administration of cyclosporin A increased survival in shocked rats. Surviving animals were still alive 24 h following the induction of splanchnic artery occlusion shock. The highest dose of cyclosporin A (1 mg kg−1) produced the highest degree of protection.
Table 1.
Effect of Cyclosporin A (CsA), 1400 W and L-NAME on survival in splanchnic artery occlusion (SAO) shocked rats
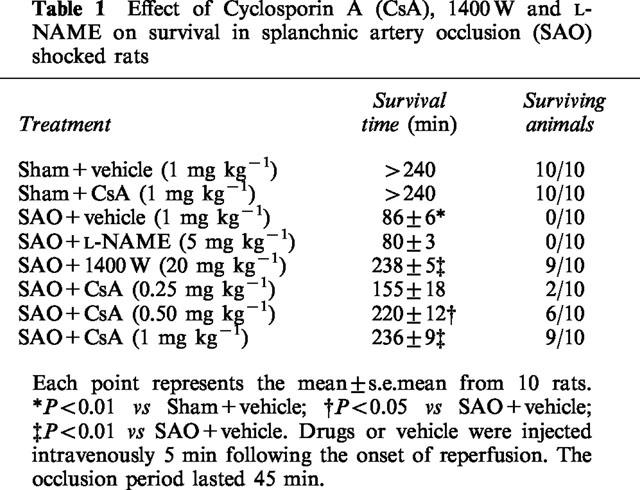
Treatment with the specific iNOS inhibitor 1400 W also resulted in marked protection against the lethality induced by splanchnic artery occlusion shock. In contrast the non-selective inhibitor of NOS L-NAME did not affect survival in rats subjected to splanchnic artery occlusion shock.
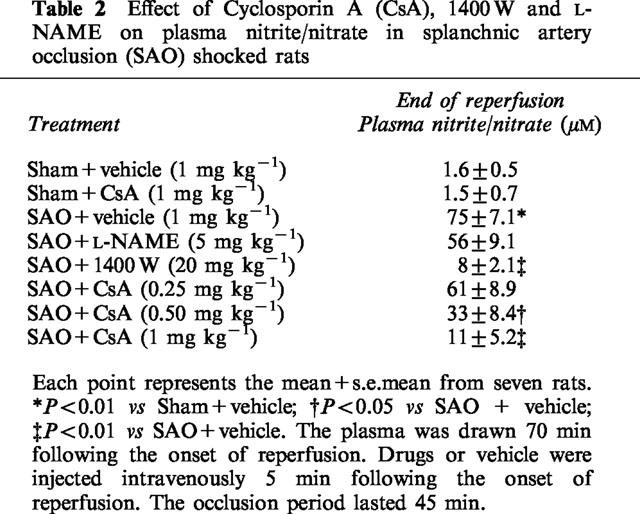
Plasma nitrite/nitrate
Plasma levels of nitrite/nitrate were very low in sham operated rats treated either with vehicle or the highest dose of cyclosporin A (Table 2). Nitrite/nitrate levels were significantly increased in plasma collected from SAO rats at the end of reperfusion period (70 min following the onset of reperfusion). Treatment with cyclosporin A or 1400 W decreased plasma nitrite/nitrate concentrations (Table 2). In contrast L-NAME did not significantly affect the circulating levels of nitrite/nitrate (Table 2).
Table 2.
Effect of Cyclosporin A (CsA), 1400 W and L-NAME on plasma nitrite/nitrate in splanchnic artery occlusion (SAO) shocked rats
Vascular reactivity of aortic rings
In intact aortic rings prepared from splanchnic artery occlusion shocked rats, the contractile response to phenylephrine (1 nM–10 μM) was significantly reduced (Figure 1). The maximum force of contraction induced by 10 μM phenylephrine in aortic rings from sham rats was 1.8±0.1 g mg−1 tissue, whereas it was 0.6±0.2 g mg−1 tissue in rings from splanchnic artery occlusion shocked rats. Removal of the endothelium did not increase the constrictor response elicited by phenylephrine in rat aortic rings obtained from both sham operated animals and splanchnic artery occlusion shocked rats (1.9±0.2 g mg−1 and 0.5±0.09 g mg−1, respectively). The contractile response to phenylephrine in endothelium denuded aortic rings was also in the absence of endothelium significantly smaller in splanchnic artery occlusion shocked rats than in sham operated animals. Administration of cyclosporin A or 1400 W improved the impaired contractile response to phenylephrine in shocked rats (Figure 1). In contrast L-NAME administration did not cause any significant change in the reduced contractile response to phenylephrine of aortic rings collected from shocked rats (Figure 1).
Figure 1.
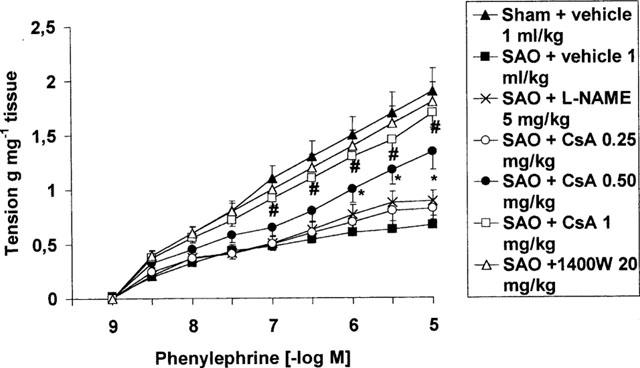
Contractile response to cumulative doses of phenylephrine (PE) in endothelium denuded aortic rings from sham-operated rats and rats subjected to splanchnic ischaemia-reperfusion injury (SAO) treated with vehicle (1 ml kg−1 i.v, 5 min after the onset of reperfusion), Cyclosporin A (CsA 0.25, 0.50 and 1 mg kg−1 i.v. 5 min following the onset of reperfusion), L-NAME (5 mg kg−1, 5 min following the onset of reperfusion) and 1400 W (20 mg kg−1, 5 min following the onset of reperfusion). Aortae were removed 70 min following the onset of reperfusion. The occlusion period lasted 45 min. Each point represents the mean±s.e.mean of seven experiments. *P<0.05 vs SAO+vehicle. #P<0.01 vs SAO+vehicle.
Mean arterial blood pressure
Occlusion of the splanchnic arteries produced a marked increase in mean arterial blood pressure. Subsequently mean arterial blood pressure decreased upon the release of the occlusion (Table 3). The administration of both cyclosporin A and 1400 W significantly blunted the reduction in mean arterial blood pressure at the end of reperfusion (Figure 3). In contrast L-NAME did not affect this delayed hypotension.
Table 3.
Effect of Cyclosporin A (CsA), 1400 W and L-NAME on mean arterial blood pressure (MAP; mmHg) in splanchnic artery occlusion shocked rats
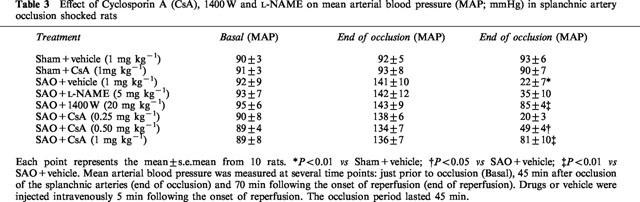
Figure 3.

Effect of vehicle (1 ml kg−1 i.v., 5 min after the onset of reperfusion) or Cyclosporin A (CsA, 1 mg kg−1 5 min after the onset of reperfusion) on the induction of iNOS protein in aortae collected 70 min following the onset of reperfusion in splanchnic artery occlusion (SAO; 45 min of occlusion) shocked rats.
Serum TNF-α
Serum levels of TNF-α were undetectable in sham-operated rats treated either with vehicle or cyclosporin A (results not shown). Figure 2 shows the time course of the cytokine during splanchnic artery occlusion shock. The serum levels of TNF-α promptly rose 10 min following the onset of occlusion and remained sustained for the entire duration of the experimental protocol (Figure 2). Administration of cyclosporin A reduced the serum levels of the inflammatory cytokine (Figure 2). In contrast treatment with 1400 W or L-NAME did not change the serum levels of TNF-α (results not shown).
Figure 2.
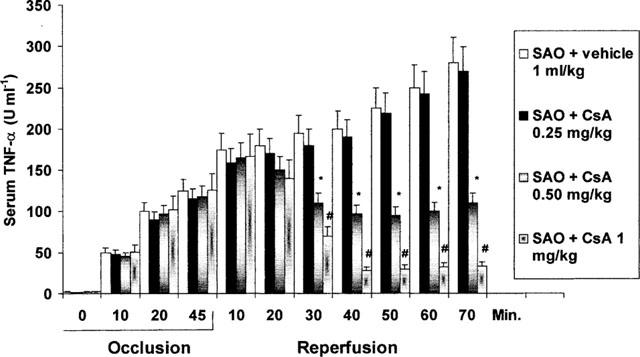
Effects of vehicle (1 ml kg−1 i.v., 5 min after the onset of reperfusion) or Cyclosporin A (CsA 0.25, 0.50 and 1 mg kg−1 i.v., 5 min following the onset of reperfusion) on serum TNF-α in rats subjected to splanchnic ischaemia-reperfusion injury (SAO). The occlusion period lasted 45 min. Each point represents the mean±s.e. mean of seven experiments. *P<0.01 vs SAO+vehicle; #P<0.001 vs SAO+vehicle.
iNOS protein
Splanchnic artery occlusion shock resulted in a significant increase in iNOS protein induction and activity in thoracic aortae removed 70 min following the onset of reperfusion (Figure 3 and Table 4). Administration of cyclosporin A reduced the increased iNOS induction and activity in shocked animals (Figure 3 and Table 4)
Table 4.
Effect of Cyclosporin A (CsA) on iNOS activity in aortae collected 70 min following the onset of reperfusion in splanchnic artery occlusion (SAO) shocked rats

Discussion
Cyclosporin A is a potent immunosuppressive agent used clinically to prevent tissue reject. Interest in the understanding of its biochemical mechanism of action has led to the discovery of some unexpected protective effects in experimental models of ischaemia-reperfusion injury and in toxic shock (Siesjo & Siesjo, 1996; Massoudy et al., 1997; Miethke et al., 1993). However the effect of cyclosporin A in splanchnic artery occlusion shock has not yet been tested.
Our results show that Cyclosporin A administration was able to increase the resistance of rats to the pathophysiological consequence of splanchnic artery occlusion shock. The effect was remarkable in terms of survival rate and improvement in vascular failure. In fact this latter parameter was positively affected by Cyclosporin A treatment: in fact the immunosuppressive agent, injected 5 min after the onset of reperfusion, produced a marked increase in blood pressure and furthermore aortic rings of Cyclosporin A treated rats exhibited a greater contractile response to phenylephrine.
The mechanisms underlying the irreversible circulatory failure have been the subject of intense investigations.
It has been proposed that the L-arginine/nitric oxide (NO) pathway plays an important role in the pathogenesis of circulatory shock. In fact the production of large amount of NO by the inducible isoform of NOS (iNOS) contributes to the delayed vascular decompensation and to the hyporeactivity of the vasculature to vasoconstrictor agents observed in several experimental models of circulatory shock (Szabo' & Thiemermann, 1994; Moncada & Higgs, 1993, Thiemermann, 1994).
As far as splanchnic artery occlusion shock is concerned, previous findings have indicated that an increase in NO likely derived by the iNOS might play an important role in the pathophysiology of this type of experimental circulatory shock (Squadrito et al., 1996). Indeed this hypothesis is confirmed by our experiments showing that 1400 W, a selective iNOS inhibitor (Garvey et al., 1997), was able to protect against the pathological sequelae associated with splanchnic artery occlusion shock. In contrast L-NAME a non-selective inhibitor of NOS failed to exert beneficial effects in splanchnic artery occlusion shock. This latter result is not surprising but confirms our previous data indicating also the presence of a reduced production of endothelial NO in splanchnic artery occlusion shock (Squadrito et al., 1996). Indeed this endothelial dysfunction is reversed by antioxidant agents (Squadrito et al., 1995), thus suggesting that oxidative stress occurring during ischaemia with reperfusion of the splanchnic region may be involved in determining the impairment in endothelial derived NO release. L-NAME, by inhibiting endothelial NOS, further worsens endothelial dysfunction and does not influence the outcome of rats subjected to splanchnic artery occlusion shock.
Furthermore the present paper extends the previous observations: in fact our shocked rats showed a marked increase in iNOS protein induction and activity in the thoracic aortae collected following 70 min of reperfusion.
The increased formation of NO by the iNOS implies that this inducible form of NOS must be expressed in response to certain immunological stimuli. TNF-α induces the expression of iNOS in a number of cells including fibroblasts, glial cells, cardiac myocytes and more specifically smooth muscle cells (Busse & Mulsch, 1990).
iNOS requires at least 2 h to be expressed in vivo (Fukuto & Chaudhuri, 1995). In agreement with this pattern of activation, increased serum levels of TNF-α were already observed 10 min following the onset of occlusion. Therefore we suggest that TNF-α, at least in SAO shock, may represent one of the stimuli involved in the priming of iNOS protein induction.
Our results also showed the presence of a reduced vascular sensitivity to vasoconstrictor stimuli in aortic rings collected 70 min following the onset of reperfusion. This impaired vascular reactivity is not due to an increased production of eicosanoids since experiments were carried out in the presence of indomethacin but, as suggested for other models of experimental shock (Thiemermann et al., 1993), may be a consequence of an overproduction of NO by the inducible NO synthase (iNOS): in agreement with this finding 1400 W, a specific iNOS inhibitor markedly reverted the vascular impairment and blunted the increased levels of plasma nitrite/nitrate, thus confirming that NO derived by the inducible NOS is likely to be involved in the vascular derangement of splanchnic artery occlusion shock.
In the present paper aortic rings collected from rats subjected to ischaemia-reperfusion injury and treated with Cyclosporin A exhibited a greater contractile response to phenylephrine when compared to vehicle treated rats. Furthermore the in vivo administration of the immunosuppressant agent blunted iNOS expression and activity in thoracic aortae. In agreement with these findings, it has been shown that the Cyclosporin A inhibits nitric oxide synthase induction in vascular smooth muscle cells (Akita et al., 1994; Marumo et al., 1995) rather than its activity (Hattori & Nakanishi, 1995).
Collectively, these results would suggest that Cyclosporin A inhibits the expression of iNOS protein. This could explain why the drug caused a reduction in plasma nitrite/nitrate levels, ameliorated vascular tone and protected against splanchnic artery occlusion shock.
In conclusion we have shown that Cyclosporin A inhibits iNOS protein expression and activity. The Cyclosporin A-induced inhibition of NO production, at least in splanchnic ischaemia-reperfusion injury, enhances blood pressure, increases survival, and improves vascular dysfunction. These findings would suggest that iNOS inhibition may contribute, at least in part, to the acute vasoprotective effects of Cyclosporin A during low flow states such as circulatory shock.
Abbreviations
- CsA
Cyclosporin A
- iNOS
inducible nitric oxide synthase
- SAO shock
splanchnic artery occlusion shock
- TNF-α
tumour necrosis factor-α
References
- AKITA K., DUSTING G.J., HICKEY H. Suppression of nitric oxide production by cyclosporin and FK506 in rat vascular smooth muscle cells. Clin. Exp. Pharmacol. Physiol. 1994;21:231–233. doi: 10.1111/j.1440-1681.1994.tb02503.x. [DOI] [PubMed] [Google Scholar]
- ALTAVILLA D., BERLINGHIERI M.C., SEMINARA S., IANNELLO D., FOCA' A., MASTROENI P. Differential effects of bacterial lipopolysaccharide on superoxide anion production by macrophages from normal and tumor bearing rats. Immunopharmacology. 1989;17:99–105. doi: 10.1016/0162-3109(89)90055-6. [DOI] [PubMed] [Google Scholar]
- BUSSE R., MULSCH A. Induction of nitric oxide synthase by cytokines in vascular smooth muscle cells. FEBS. Lett. 1990;275:87–90. doi: 10.1016/0014-5793(90)81445-t. [DOI] [PubMed] [Google Scholar]
- CAPUTI A.P., ROSSI F., CARNEY K., BREZENOFF H.E. Modulatory effect of brain acetylcholine on reflex-induced bradycardia and tachycardia in conscious rats. J. Pharmacol. Exp. Ther. 1980;215:309–316. [PubMed] [Google Scholar]
- CLIPSTONE N.A., CRABTREE G.R. Calcineurin is a key signaling enzyme in lymphocyte activation and the target of immunosuppressive drugs cyclosporin A and FK506. Ann. NY. Acad. Sci. 1993;696:20–30. doi: 10.1111/j.1749-6632.1993.tb17138.x. [DOI] [PubMed] [Google Scholar]
- DING A.H., NATHAN C.F., STUEHR D.J. Release of reactive nitrogen intermediates from mouse peritoneal macrophages. J. Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- FAST D.J., LYNCH R.C., LEU R.W. Cyclosporin A inhibits nitric oxide production by L929 cells in response to tumor necrosis factor and interferon-gamma. J. Interferon Res. 1993;13:235–240. doi: 10.1089/jir.1993.13.235. [DOI] [PubMed] [Google Scholar]
- FRUMAN D.A., BURAKOFF S.J., BIERER B.E.Molecular actions of cyclosporin A, FK 506 and rapamycin Immunosuppressive Drugs: Developments in Anti-rejection Therapy 1994London: Edward Arnold; 15–35.ed. Thomson, A.W. & Starzl, T.E. pp [Google Scholar]
- FUKUTO J.M., CHAUDHURI G. Inhibition of constitutive and inducible nitric oxide synthase: potential selective inhibition. Annu. Rev. Pharmacol. Toxicol. 1995;35:165–194. doi: 10.1146/annurev.pa.35.040195.001121. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J., KNOWLES R.G. 1400 W is a slow, tight binding and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;278:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- HATTORI Y., NAKANISHI N. Effects of cyclosporin A and FK506 on nitric oxide and tetrahydrobiopterin synthesis in bacterial lipopolysaccharide-treated J744 macrophages. Cell Immunol. 1995;165:7–11. doi: 10.1006/cimm.1995.1180. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D. Cyclosporine. N. Engl. J. Med. 1989;321:1725–1738. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- MARUMO T., NAKAKI T., HISHIKAWA K., SUZUKI H., KATO R., SURUTA T. Cyclosporin A inhibits nitric oxide synthase induction in vascular smooth muscle cells. Hypertension. 1995;25:764–768. doi: 10.1161/01.hyp.25.4.764. [DOI] [PubMed] [Google Scholar]
- MASSOUDY P., ZAHLER S., KUPATT C., REDER E., BECKER B.F., GERLACH E. Cardioprotection by Cyclosporine A in experimental ischemia and reperfusion. Evidence for a nitric oxide-dependent mechanism mediated by endothelin. J. Mol. Cell. Cardiol. 1997;29:535–544. doi: 10.1006/jmcc.1996.0297. [DOI] [PubMed] [Google Scholar]
- MIETHKE T., DUSCHEK K., WAL C., HEEG K., WAGNER H. Pathogenesis of the toxic shock syndrome: T cell mediated lethal shock caused by the superantigen TSST-1. Eur. J. Immunol. 1993;23:1494–1500. doi: 10.1002/eji.1830230715. [DOI] [PubMed] [Google Scholar]
- MONCADA S., HIGGS A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- MUHL H., KUNZ D., ROB P., PFEILSCHIFTER J. Cyclosporin derivatives inhibit interleukin 1 beta induction of nitric oxide synthase in renal mesangial cells. Eur. J. Pharmacol. 1993;249:95–100. doi: 10.1016/0014-2999(93)90666-6. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S.L., CRABTREE G.R. The mechanism of action of cyclosporin and FK 506. Immunol. Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- SIESJO B.K., SIESJO P. Mechanism of secondary brain injury. Eur. J. Anaesthesiol. 1996;13:247–268. [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., AMMENDOLIA L., SQUADRITO G., CAMPO G.M., SPERANDEO A., CANALE P., IOCULANO M., SAITTA A., CAPUTI A.P. Improved survival and reversal of endothelial dysfunction by the 21-aminosteroid U-74389G in splanchnic ischaemia-reperfusion injury in the rat. Br. J. Pharmacol. 1995;115:395–400. doi: 10.1111/j.1476-5381.1995.tb16346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., CANALE P., IOCULANO M., CAMPO G.M., AMMENDOLIA L., FERLITO M., ZINGARELLI B., SQUADRITO G., SAITTA A., CAPUTI A.P. Participation of tumor necrosis factor and nitric oxide in the mediation of vascular dysfunction in splanchnic artery occlusion shock. Br. J. Pharmacol. 1994a;113:1153–1158. doi: 10.1111/j.1476-5381.1994.tb17118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., CANALE P., IOCULANO M., CAMPO G.M., AMMENDOLIA L., SQUADRITO G., SAITTA A., CALAPAI G., CAPUTI A.P. Contribution of intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of splanchnic artery occlusion shock in the rat. Br. J. Pharmacol. 1994b;113:912–916. doi: 10.1111/j.1476-5381.1994.tb17079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SQUADRITO F., ALTAVILLA D., SQUADRITO G., CAMPO G.M., IOCULANO M., CANALE P., ROSSI F., SAITTA A., CAPUTI A.P. Effects of S-ethylisothiourea, a potent inhibitor of nitric oxide synthase, alone or in combination with a nitric oxide donor in splanchnic artery occlusion shock. Br. J. Pharmacol. 1996;119:23–28. doi: 10.1111/j.1476-5381.1996.tb15672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABO' C., THIEMERMANN C. Invited opinion: role of nitric oxide in hemorrhagic, traumatic and anaphylactic shock and thermal injury. Shock. 1994;2:145–155. [PubMed] [Google Scholar]
- THIEMERMANN C. The role of the L-arginine-nitric oxide pathway in circulatory shock. Adv. Pharmacol. 1994;28:45–79. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- THIEMERMANN C., SZABO' C., MITCHELL J.A., VANE J.R. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 1993;90:267–271. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIEDERRECHT G., LAM E., HUNG S., MARTIN M., SIGAL N. The mechanism of action of FK506 and cyclosporin A. Ann. NY. Acad. Sci. 1993;696:9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x. [DOI] [PubMed] [Google Scholar]