

Abstract
Troglitazone lowers blood glucose levels in Type II diabetic patients. To evaluate the insulin sensitizing action of troglitazone on glycogen synthesis we have used dexamethasone-treated 3T3 adipocytes as an in vitro model.
Differentiated 3T3 adipocytes were incubated with 100 nM dexamethasone for 6 days. Troglitazone (1.0 μM) or metformin (1.0 mM) with or without 200 nM insulin was added during the last 4 days. At the end, insulin (100 nM) stimulated glycogen synthesis was determined using 14C-glucose.
Dexamethasone caused a 50% reduction in glycogen synthesis. Troglitazone caused an approximately 3 fold increase in glycogen synthesis from 43.9±3.4 to 120±16.2 nmols h−1. Under identical conditions metformin had no significant effect.
When cells were incubated with troglitazone and dexamethasone simultaneously for 6 days, troglitazone but not metformin completely prevented dexamethasone-induced insulin resistance. RU 486 (1.0 μM) also completely prevented the insulin resistance.
Chronic incubation with dexamethasone and insulin resulted in a 73% reduction in glycogen synthesis. In these adipocytes, troglitazone was partially active with glycogen synthesis rising from 23.1±3.0 to 44.4±4.5 nmol h−1, P<0.01 while metformin was inactive.
Troglitazone stimulated 2-deoxyglucose uptake by 2–3 fold in dexamethasone-treated adipocytes. Metformin also increased glucose uptake significantly. Troglitazone did not affect insulin binding while a 2 fold increase was observed in normal adipocytes where it exhibited a modest effect.
Since the effect of troglitazone was greater in dexamethasone-treated adipocytes, troglitazone is likely to act by preventing dexamethasone-induced alterations which may include (i) binding to glucocorticoid receptor and (ii) effect on glucose uptake.
These data demonstrate the direct insulin sensitizing action of troglitazone on glycogen synthesis and suggest a pharmacological profile different from metformin.
Keywords: Glycogen synthesis, troglitazone, dexamethasone, 3T3 adipocytes, metformin, in vitro model, insulin resistance
Introduction
Insulin resistance is a major pathophysiological abnormality in patients with non-insulin-dependent diabetes mellitus (NIDDM) and obesity. Resistance to insulin action develops in skeletal muscle, adipose tissue and liver contributing to hyperglycaemia (De Fronzo et al., 1992). These impairments in insulin action play an important role not only in the development of hyperglycaemia of NIDDM but also in the pathogenesis of long term complications. Therefore, treatments that improve peripheral insulin resistance would be beneficial in the long term management of these patients. Thiazolidinediones are a new class of agents with potent insulin sensitizing action in NIDDM patients (Saltiel & Olefsky, 1996). Troglitazone, an extensively studied member of the thiazolidinedione class, lowers blood glucose levels in various animal models of insulin resistance and in NIDDM patients (Reginato & Lazer, 1999). At the cellular level, troglitazone seems to act by improving insulin action on glucose transport and utilisation in peripheral tissues. Recently, it has been reported that thiazolidinediones, including troglitazone, bind with high affinity to the peroxisome proliferator activated receptor gamma (PPAR-γ), a nuclear receptor expressed exclusively in the adipose tissue (Lehmann et al., 1995). However, the mechanism by which this binding of troglitazone to adipocyte PPAR-γ, could bring about overall improvement in insulin action in NIDDM patients is not clearly established.
To understand the mechanism of action studies have been carried out in cultured cells. Troglitazone induced differentiation of 3T3 preadipocytes (Sandouk et al., 1993; Tafuri, 1996) and promoted insulin action in the preadipocyte cell line C3HIOT½ (Lenhard et al., 1997) and in 3T3-L1 cells (Ranganathan & Kern, 1998). These studies have demonstrated the effect of troglitazone added during differentiation. By contrast, the effect on mature adipocytes have not been fully investigated. In this regard, fully differentiated 3T3 adipocytes may be considered as in in vitro model of adipose tissue suitable for studying the action of troglitazone. 3T3-L1 cells are a well established cell line that responds to physiological doses of insulin with increases in glucose uptake, glucose oxidation, glycogen synthesis and lipogenesis under in vitro condition (Knutson et al., 1995; Knutson & Balba, 1997; Shepherd et al., 1995). Another advantage is that 3T3 adipocytes can be rendered insulin resistant by chronic incubation with dexamethasone (Turnbow et al., 1994; Grunfeld et al., 1981). Although these studies have characterized insulin signalling events leading to glucose uptake in 3T3 adipocytes, impairments in glycogen synthesis have not been investigated so far. Insulin dependent glycogen synthesis represents an important pathway of nonoxidative glucose disposal in man. In subjects with a family history of NIDDM, insulin resistance on glycogen synthesis occurs before the development of overt diabetes (Schalin-Jantti et al., 1992; Vaag et al., 1992). Previous studies have shown that impairment of glycogen synthesis in NIDDM, is independent of impaired glucose uptake (Henry et al., 1996; Thorburn et al., 1990) and these defects persist even after correcting hyperglycaemia by oral agents (Damsbo et al., 1991). Thus impaired glycogen synthesis represents a fundamental defect characterizing insulin resistance rather than hyperglycaemia (Damsbo et al., 1998). Therefore agents that improve insulin resistance on glycogen synthesis are likely to improve overall insulin sensitivity in NIDDM patients.
Metformin is a biguanide antidiabetic agent that has been in clinical use for a long time (Bailey, 1992). Besides having other effects on glucose utilization, it has been reported to enhance insulin action on glucose transport in rat adipocytes (Matthaei et al., 1991). However, neither metformin nor troglitazone has been investigated and compared in the insulin resistant adipocytes. The present study was carried out to evaluate the direct insulin sensitizing action of troglitazone on glycogen synthesis in normal and (dexamethasone-induced) insulin resistant 3T3 adipocytes and to compare with metformin.
Methods
Cell culture
3T3-L1 cells were cultured in 25 cm2 flasks in Dulbecco's modified Eagle's medium (DMEM) with high glucose (25 mM) containing 10% calf serum in a humidified atmosphere of 5% CO2 at 37°C in a CO2 incubator. The medium had 60 μg ml−1 penicillin and 100 μg ml−1 streptomycin sulphate. Differentiation was induced by the method of Frost & Lane (1985) as modified by Shepherd et al. (1995). Briefly, two days after confluence, cells in 35- or 60-mm dishes were treated with 0.25 μM dexamethasone, 0.5 mM isobutyl methyl xanthine, IBMX and 5 μg ml−1 bovine insulin in DMEM containing 10% foetal calf serum (FCS). After 48 h, dexamethasone and IBMX were withdrawn and only insulin containing medium was continued for the next 3 days. Thereafter, cells were maintained in insulin free DMEM with 10% FCS till the cells are completely differentiated into adipocytes (7–9 days after initiation).
Effect of troglitazone
Effect of troglitazone was studied both in normal and in insulin resistant adipocytes. Fully differentiated mature 3T3 adipocytes grown in 35- or 60 mm dishes (7–10 days after initiation of differentiation) were incubated with standard medium (DMEM+10% FCS) containing troglitazone (1.0 μM) or metformin (1.0 mM) or vehicle for 6 days at 37°C in 5% CO2. Medium was changed every 2 days until the time of experimentation.
Insulin resistance was induced in differentiated 3T3 adipocytes by the method of Grunfeld et al. (1981). Briefly, differentiated adipocytes (in 35- or 60-mm dishes) were exposed to 100 nM dexamethasone in standard medium for a maximum of 6 days. Troglitazone (10−7–10−5 M) or metformin (10−3 M) or vehicle was added 48 h after starting dexamethasone treatment and the drugs were incubated together with dexamethasone (100 nM) for 4 days. In some experiments, troglitazone or metformin or RU 486 was added simultaneously with dexamethasone for 3–6 days. In another set of experiments, troglitazone or metformin was added along with insulin (10 or 200 nM) and dexamethasone (100 nM) for 4 days to cells that were pretreated with dexamethasone for 48 h. In all experiments medium was changed every 2 days. Dexamethasone, was dissolved in ethanol. RU 486 and troglitazone were dissolved in dimethyl sulphoxide, DMSO. Metformin was dissolved in sterile distilled water. All drugs were added to DMEM containing 10% FCS and control cells received appropriate vehicle. The final concentration of the vehicle was less than 0.20% and at this concentration no effect on cell morphology or metabolic activity was observed.
14C-Glucose incorporation into glycogen
Glycogen synthesis from 14C-Glucose was assayed by the modified methods of Chan & Krebs (1985) and Shepherd et al., 1995. After incubating the cells (in 35- or 60-mm petri dishes) with appropriate drugs, the medium was changed to serum free DMEM and incubated for 1 h at 37°C. Later, they were incubated in Kreb's Ringer phosphate buffer, KRP (pH 7.4) containing glucose (5.5 mM), 2% bovine serum albumin, BSA and 1.0 μCi ml−1 of 14C-Glucose in the presence of (0–100 nM) porcine insulin for 30 min in a CO2 incubator at 37°C. The reaction was terminated by removing labelled medium and washing the cell layer twice with ice cold phosphate buffered saline, PBS. The cell layer was finally dissolved in 1.0 ml of 0.5 N NaOH, and heated at 70°C for 30 min. Carrier glycogen (25 μl of 50 mg ml−1) was added and glycogen was precipitated with ethanol (70% final concentration). The precipitate was washed twice with ethanol (70%) and dried. The final pellet was dissolved in deionized water and the radioactivity was determined in a liquid scintillation counter (LKB Wallac, Finland). Under these conditions 14C-Glucose incorporation into glycogen was linear for 60 min.
Insulin binding
125I-labelled insulin binding was determined by the modified method of Gamou et al. (1990) using 0.20 ng ml−1 125I-labelled porcine insulin (∼30,000 c.p.m. ml−1) and 0–1 μg ml−1 unlabelled porcine insulin in 50 mM HEPES, pH 7.8 (referred to as the binding buffer). The incubation was carried out at 4°C for 18 h after which the cell layer was washed with ice cold binding buffer. Later, the monolayer was dissolved in 0.5 N NaOH (0.5 ml) and the radioactivity was determined in a gamma counter (Electronic Corporation of India Ltd. ECIL, Mumbai, India). Radioactivity in the presence of 1 μg ml−1 unlabelled insulin was considered as nonspecific binding and was subtracted from other values. At this incubation temperature of 4°C, >95% of the label is considered to represent cell surface bound insulin with negligible internalization.
Deoxyglucose uptake
Deoxyglucose uptake was determined essentially by the method of Knutson & Balba (1997). After appropriate incubation with various agents, the cell monolayer was washed twice with glucose free KRP buffer, pH 7.4 containing 0.1% BSA and incubated with 25 nM porcine insulin for 20 min at 37°C in a CO2 incubator. An aliquot of 14C-deoxyglucose was added to the dishes such that each received 0.5 μCi in 1.5 ml at a final concentration of 2.0 mM and incubation was continued for 20 min at 37°C. Uptake was terminated by aspiration of the radioactive buffer and washing the monolayers with ice cold glucose free PBS. The cell layer was finally solubilized in 1% sodium dodecylsulphate and the radioactivity was determined in a liquid scintillation counter. Under these conditions the uptake was linear for 60 min. Cytochalasin B (50 μM) was included in some dishes during the later 20 min to determine the cytochalasin B inhibitable transport.
Drugs and chemicals
3T3-L1 cells were obtained from the National Centre for Cell Sciences (Pune, India). T-25 flasks, 60- and 35-mm dishes were purchased from NUNC. Calf serum and foetal calf serum (FCS) were purchased from Hyclone (U.S.A.). Dexamethasone, 1-Methyl-3-isobutylxanthine (IBMX), bovine insulin, DMEM, BSA, glycogen (from bovine liver), trypsin (1:250). Penicillin G, and streptomycin sulphate were purchased from the Sigma Chemical Co., MO, U.S.A. Porcine insulin, 2-deoxyglucose, and cytochalasin B were purchased from ICN Pharmaceuticals, U.S.A. RU 486 was a gift from Dr Leighton (Oxford, U.K.). Troglitazone was supplied by Dr Reddy's research foundation (Hyderabad, India). Troglitazone, GR 92132X was obtained from Glaxo Wellcome, Stevenage, U.K. Metformin hydrochloride was a gift from Dr Sudarsanam (B.V. Patel Pharm. Education & Research Centre, Ahmedabad, India). U-14C-Glucose (specific activity 310 mCi mmol−1) and 125I-labelled porcine insulin (100 μCi μg−1) were obtained from the Board of Radiation and Isotope Technology (BRIT, Mumbai, India). 2-Deoxy-D-glucose [1-14C] (specific activity 57.2 mCi mmol−1) was purchased from ICN Pharmaceuticals, U.S.A.
Statistics and data analysis
Data are expressed as mean±s.e.mean per dish. Each dish had 1–2×106 cells in various experiments. Glycogen synthesis was expressed either as c.p.m. h−1 or nmols h−1. Statistical significance of the difference between means in two different groups was assessed by the unpaired t-test. A P value of less than 0.05 was considered significant.
Results
Effect of troglitazone on insulin-resistant adipocytes
Fully differentiated 3T3 adipocytes exhibited highly insulin sensitive glycogen synthesis. Insulin caused a dose-dependent stimulation of glucose incorporation into glycogen (Figure 1) leading to a 7–8 fold increase from basal at a maximal concentration of 100 nM. This corresponds to 99.3±15.3 nmols h−1 in terms of glucose incorporated into glycogen. Chronic (3 days) incubation with dexamethasone caused a reduction in glycogen synthesis which was significant at high insulin concentrations. Maximal insulin concentration of 100 nM caused only a 3–4 fold stimulation amounting to nearly a 50% reduction compared to control adipocytes. Dexamethasone treatment had no effect on basal glycogen synthesis and on the insulin dose response whereas responsiveness to insulin was affected. When the dexamethasone incubation was continued for 6 days, insulin resistance on glycogen synthesis was maintained at the same level except that the data were more consistent. Based on these results, all experiments on troglitazone were carried out using cells treated for 6 days with dexamethasone and only maximal insulin-(100 nM) stimulated glycogen synthesis was investigated. Expressing the data as c.p.m. h−1 or nmols h−1 did not make any difference in the results.
Figure 1.
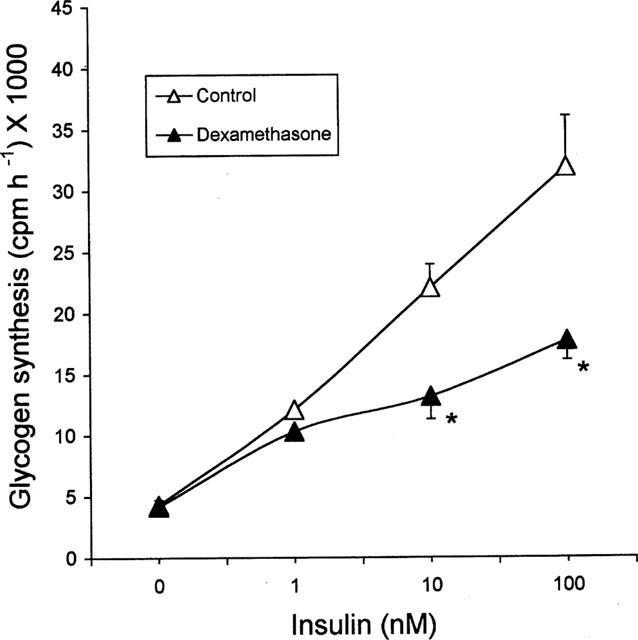
Effect of dexamethasone on glycogen synthesis in 3T3 adipocytes. Fully differentiated 3T3-adipocytes were incubated for 3 days with 100 nM dexamethasone or vehicle and glycogen synthesis was studied using 14C-glucose in the presence of 0–100 nM porcine insulin. Data are mean±s.e.mean of n=4 at each insulin concentration. *P<0.05 compared to vehicle treatment. Data are presented on a per dish basis. Each dish had 1–2×106 cells. Basal glycogen synthesis of vehicle treated cells corresponds to 13.5±1.5 nmols h−1 and maximal insulin stimulated glycogen synthesis corresponds to 99.3±15.3 nmols h−1.
To determine whether troglitazone is able to reverse and/or prevent the insulin resistance caused by dexamethasone, troglitazone was added to 3T3 adipocytes either 2 days after starting dexamethasone or simultaneously with dexamethasone. The effect was termed ‘reversal' when troglitazone was added 2 days after dexamethasone addition, while the effect was termed ‘prevention' when both troglitazone and dexamethasone were added simultaneously to normal insulin sensitive adipocytes. In experiments where both prevention and reversal were evaluated, the cells were exposed to dexamethasone only for the first 2 days and to both troglitazone and dexamethasone for the next four days. A dose-response study to troglitazone was carried out in dexamethasone treated cells where both reversal and prevention of insulin resistance could be determined. Troglitazone caused dose-dependent stimulation of glycogen synthesis reaching a maximum at 1.0 μM after which a decline was observed (Figure 2a). Therefore, troglitazone was used at this concentration (of 1.0 μM) in all studies comparing troglitazone and metformin. In a similar experiment as above, troglitazone (1.0 μM) treatment caused a nearly 3 fold increase in glycogen synthesis compared to dexamethasone treated cells (Figure 2b). In this experiment troglitazone not only reversed the dexamethasone effect completely but also attenuated the insulin response to levels (about 50%) above the control levels. Under identical conditions, metformin (1.0 mM) caused a 62% increase in glycogen synthesis although it did not enhance the levels significantly. In these dexamethasone-induced insulin-resistant adipocytes, troglitazone exhibited unique anti-insulin-resistant action different from metformin.
Figure 2.
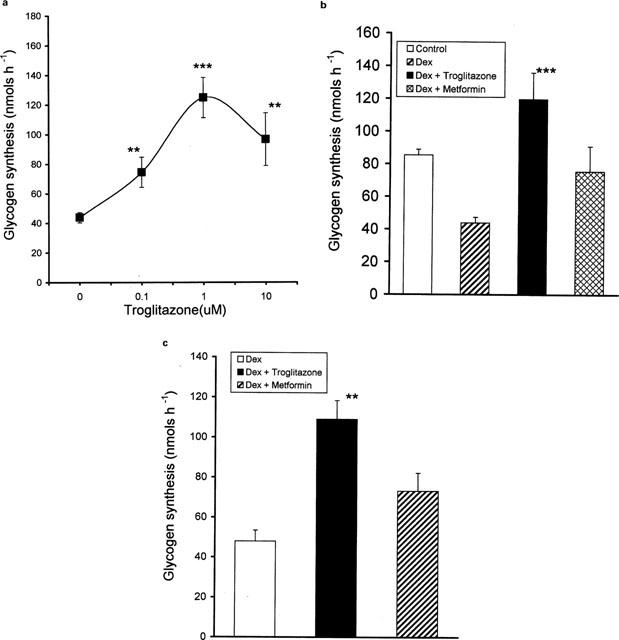
(a) Dose response of troglitazone on dexamethasone treated adipocytes. 3T3 adipocytes were incubated with 100 nM dexamethasone for 2 days. After 2 days troglitazone 0–10 μM and dexamethasone, 100 nM were added and incubated for 4 days. At the end of 6 days, cells were removed and glycogen synthesis from 14C-glucose was assayed in the presence of 100 nM insulin. Data are mean±s.e.mean of n=4, **P<0.02, ***P<0.001 compared to dexamethasone-treated adipocytes. (b) Effect of troglitazone and metformin on dexamethasone-treated adipocytes. 3T3 adipocytes were incubated with or without 100 nM dexamethasone for 2 days. After 2 days troglitazone (1.0 μM) or metformin (1.0 mM) was added to dishes incubated with dexamethasone along with 100 nM dexamethasone for the next 4 days. At the end of 6 days, the dishes were removed and glycogen synthesis was studied in the presence of 100 nM insulin. Data are mean±s.e.mean, n=4, ***P<0.001 compared to dexamethasone-treated cells. (c) Effect of troglitazone and metformin incubated simultaneously with dexamethasone. 3T3 adipocytes were incubated with troglitazone (1.0 μM) or metformin (1.0 mM) or vehicle along with 100 nM dexamethasone for 6 days. At the end of 6 days glycogen synthesis was studied in the presence of 100 nM insulin. Data are mean±s.e.mean, n=8 for troglitazone and n=6 for other groups. **P<0.01 compared to dexamethasone-treated cells.
In another series of experiments designed to investigate whether these drugs prevented the development of insulin resistance, troglitazone added simultaneously with dexamethasone was able to completely prevent the dexamethasone effect (Figure 2c) resulting in a more than 2 fold increase in glycogen synthesis (P<0.01), while metformin was unable to prevent this effect. Thus, troglitazone but not metformin was effective in preventing the development of insulin resistance.
Effect of troglitazone in the presence of chronic insulin
When insulin resistant (by treatment with dexamethasone for 6 days) adipocytes were incubated with 200 nM insulin during the last 4 days, insulin resistance on glycogen synthesis was aggravated (Figure 3). In these cells, glycogen synthesis was reduced to 55% of dexamethasone-treated cells (P<0.05). In other words, dexamethasone alone caused a 50% reduction in glycogen synthesis compared to control while cotreatment with 200 nM insulin caused a 73% reduction in glycogen synthesis. Under these experimental conditions, troglitazone was still able to stimulate glycogen synthesis (by 2 fold, P<0.01) to the level in dexamethasone-treated adipocytes. Metformin did not exhibit any significant effect on this system. In a similar experiment carried out using 10 nM insulin instead of 200 nM (as given in Figure 3) troglitazone caused a modest (36%) but significant (P<0.05) increase in insulin-mediated glycogen synthesis. In these doubly insulin resistant (by dexamethasone and insulin) adipocytes, troglitazone was able to reverse insulin resistance on glycogen synthesis only partially.
Figure 3.
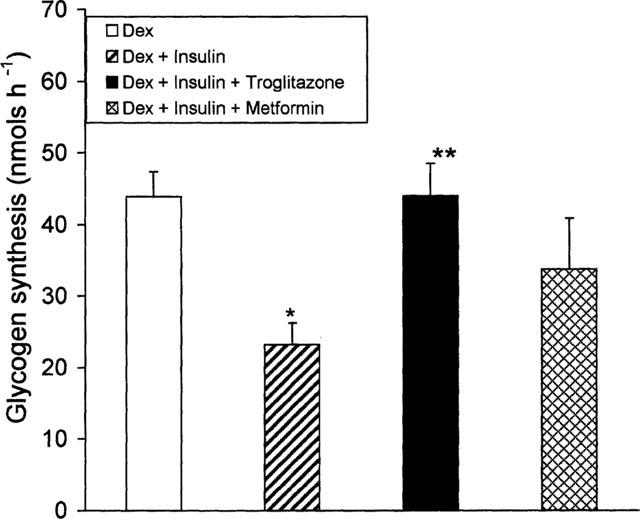
Effect of troglitazone or metformin on 3T3 adipocytes incubated with dexamethasone and insulin. 3T3 adipocytes were incubated for 2 days with 100 nM dexamethasone. After 2 days, troglitazone (1.0 μM) or metformin (1.0 mM) was added and incubated along with 200 nM insulin and 100 nM dexamethasone for a further 4 days. At the end of 6 days, cells were removed and glycogen synthesis was assayed in the presence of 100 nM insulin. Data are mean±s.e.mean n=4, *P<0.05 versus dexamethasone alone **P<0.01 versus cells treated with chronic insulin and dexamethasone.
Effect of RU 486
To determine whether dexamethasone-induced insulin resistance on glycogen synthesis involved binding to the glucocorticoid receptor, 3T3 adipocytes were incubated with the glucocorticoid receptor antagonist, RU 486 (1.0 μM) and dexamethasone (100 nM) together for 3 days and insulin dose response on glycogen synthesis was determined. In this study, RU 486 completely blocked the dexamethasone-induced insulin resistance at all insulin concentrations (Figure 4) and brought the glycogen synthesis to the level of control adipocytes.
Figure 4.
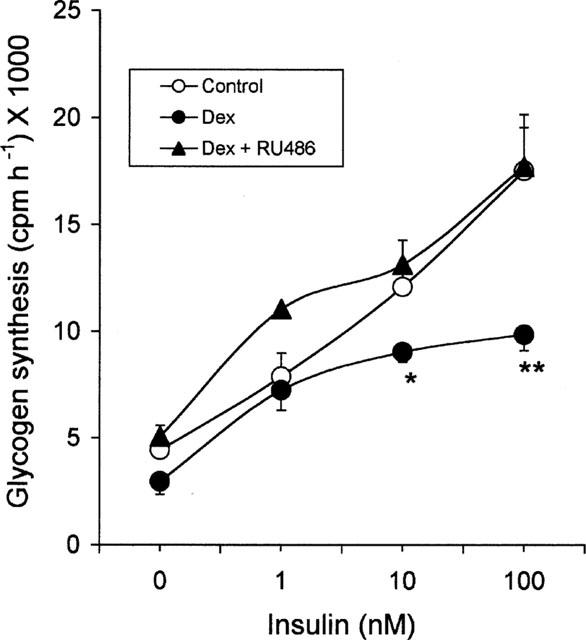
Effect of RU 486 on insulin dose response on glycogen synthesis in dexamethasone treated cells. 3T3 adipocytes were incubated with 100 nM dexamethasone or vehicle or 100 nM dexamethasone and 1.0 μM RU 486 for 72 h. After 72 h, cells were removed and glycogen synthesis was assayed. Data are mean±s.e.mean for n=4, *P<0.05 and **P<0.02 for dexamethasone-treated cells compared to vehicle treatment.
Effect on normal adipocytes
In the fully differentiated insulin-sensitive normal adipocytes, troglitazone (1.0 μM) treatment for 6 days resulted in a modest but significant increase in glycogen synthesis (∼1.5 fold, P<0.05 (Figure 5). Metformin (1.0 mM) was inactive in these cells.
Figure 5.
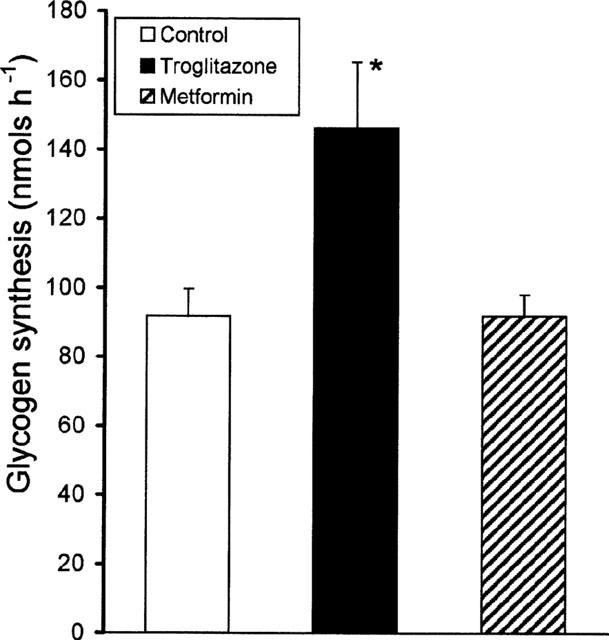
Effect of troglitazone on normal 3T3 adipocytes. 3T3 adipocytes were treated with troglitazone (1.0 μM), or metformin (1.0 mM) or vehicle for 6 days. At the end of 6 days, the cells were removed and glycogen synthesis was assayed in the presence of 100 nM insulin. Data are mean±s.e.mean for n=6, *P<0.05 for troglitazone treated cells compared to vehicle treatment.
In the studies presented so far, troglitazone was found to be effective in enhancing glycogen synthesis in both normal and insulin-resistant adipocytes, while metformin was ineffective. Among the effects of troglitazone on glycogen synthesis, its effect in the insulin-resistant adipocytes was greater than in the insulin-sensitive adipocytes. This demonstrates the unique anti-insulin resistant property of troglitazone. Among the two systems of insulin resistance (dexamethasone alone or dexamethasone and insulin together) the effect was higher in the dexamethasone-treated cells than that seen in cells treated with both agents.
125I-labelled insulin binding
Normal mature adipocytes exhibited specific insulin binding of 4.4±0.37% (per cent of label added, n=8). Chronic dexamethasone treatment did not affect insulin binding significantly (4.77±0.45%). Troglitazone, when added to dexamethasone treated cells had no effect on insulin binding (4.95±0.31%, n=8) while it caused a marginal increase from 4.10±0.45% (n=7) to 5.28±0.45% (n=8) in the presence of chronic insulin. Contrary to this effect, troglitazone stimulated insulin binding from 3.60±0.90 to 6.55±0.01%, n=3 (P<0.001) in the normal insulin sensitive adipocytes. Metformin did not cause any change in insulin binding in the normal adipocytes (3.11±0.43%, n=3 of metformin treated cells compared to 3.60±0.90%, n=3 in the control adipocytes).
Deoxyglucose uptake
The effect of troglitazone on glucose uptake was determined using the non-metabolizable glucose analogue, 2-Deoxy-D-glucose (1-14C) in the insulin resistant adipocytes. The effect on deoxyglucose uptake was studied in cells treated with dexamethasone for 6 days, during which troglitazone 1.0 μM (with or without 200 nM insulin) was present for the last 4 days. This experimental condition is similar to that presented previously (Figures 2b and 3) wherein troglitazone exhibited a maximal effect on glycogen synthesis. Troglitazone enhanced glucose uptake by 3–4 fold, both in the presence and absence of chronic insulin with the highest effect observed in chronically insulin treated cells (Figure 6). Metformin treatment stimulated glucose uptake nearly 2 fold contrary to the lack of effect seen on glycogen synthesis. Unlike the inhibitory effect of chronic insulin on glycogen synthesis, glucose uptake was stimulated by nearly 2 fold in these cells compared to dexamethasone-treated cells. In this upregulated state, troglitazone stimulated the transport even further representing additive effects of insulin and troglitazone on glucose uptake. Glucose uptake in all these dishes were more than 90% inhibited by 50 μM cytochalasin B, a specific inhibitor of glucose transport. In this study troglitazone exhibited greater stimulatory effect compared to metformin which was also active.
Figure 6.
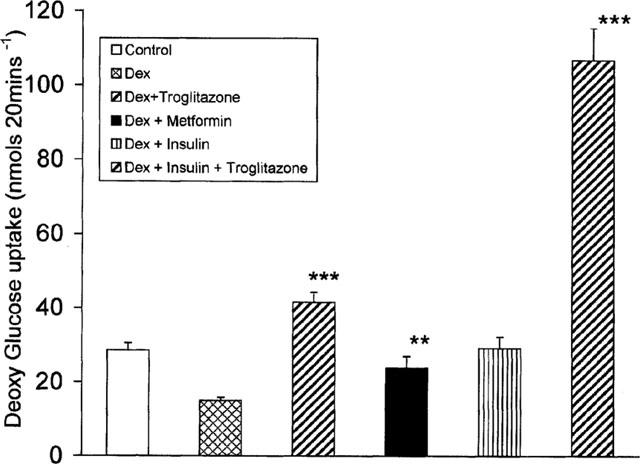
Effect of troglitazone or metformin on deoxyglucose uptake. 3T3 adipocytes were incubated with 100 nM dexamethasone for 2 days. On day 3, the cells were incubated with troglitazone (1.0 μM) in the presence or absence of 200 nM insulin, or metformin (1.0 mM) and 100 nM dexamethasone for the next 4 days. At the end of 6 days, the cells were removed and 14C-deoxyglucose uptake was measured in the presence of 25 nM porcine insulin for 20 min. Data are mean±s.e.mean of n=8. **P<0.02 for metformin compared to dexamethasone-treated cells. ***P<0.001 for troglitazone treatments comparing cells incubated with dexamethasone and insulin or dexamethasone alone.
Discussion
Troglitazone has been shown to counteract insulin resistance in NIDDM patients and in various animal models of obesity. To understand the direct effect of troglitazone on insulin resistant glucose metabolism, we have used dexamethasone treated 3T3 adipocytes as an in vitro model. We studied glycogen synthesis since (i) various studies have established insulin-dependent glycogen synthesis (although in muscle) as the hallmark of insulin resistance (Damsbo et al., 1998) and (ii) 3T3 adipocytes exhibit a robust insulin response under in vitro conditions (Knutson et al., 1995; Shepherd et al., 1995) unlike muscle cells which respond poorly to insulin. Using glycogen synthesis as an index of insulin sensitivity, we found that troglitazone acted as a potent insulin sensitizing agent and exhibited a different and superior pharmacological profile to metformin. To the best of our knowledge this is the first report of a direct action of troglitazone as an insulin sensitizer of glycogen synthesis in the dexamethasone-induced insulin-resistant 3T3 adipocytes.
Dexamethasone treatment markedly reduced maximal insulin-stimulated glycogen synthesis similar to the reports of Dimitriadis et al. (1997) in animals. Chronic insulin-induced insulin resistance on glycogen synthesis has been well established (Knutson et al., 1995; 1997; Thomson et al., 1997; Rice et al., 1993). We have used a combination of chronic dexamethasone and insulin which produced a severely desensitized glycogen synthesis. We evaluated the insulin sensitizing action in (i) reversing the already existing insulin resistance, resembling the pathophysiology of NIDDM, and (ii) in preventing the development of new insulin resistance. In these studies troglitazone but not metformin improved glycogen synthesis in both the above pathways. Based on the marked effect of troglitazone on insulin-resistant cells compared to normal insulin-sensitive adipocytes it is inferred that troglitazone has potent insulin sensitizing action.
The mode of action of troglitazone as an insulin sensitizer is of interest. The greater effect seen in dexamethasone-treated cells suggests that troglitazone might be acting largely by preventing the dexamethasone effect on glycogen synthesis rather than having an independent action. Accordingly troglitazone could inhibit: (i) dexamethasone binding to the glucocorticoid receptor or (ii) dexamethasone action on various insulin signalling events leading to glycogen synthesis. In the present study we have attempted to address some of these issues.
In our study, RU 486, a glucocorticoid receptor antagonist (Moguilewsky & Philibert, 1984) completely prevented the dexamethasone effect. This suggests that the dexamethasone action on glycogen synthesis involves the glucocorticoid receptor and troglitazone might be acting analogous to RU 486 in preventing dexamethasone binding to the glucocorticoid receptor. Another site of troglitazone action might involve inhibition of dexamethasone action on glycogen synthesis. Glycogen synthesis consists of several insulin mediated processes such as insulin binding, glucose uptake and activation of glycogen synthase, the key enzyme involved in glycogen synthesis. Troglitazone had no effect on insulin binding in the dexamethasone-treated cells. Consistent with our data is the finding of a lack of effect by Ciaraldi et al. (1990) in cultured HepG2 and BC3H1 cells.
Troglitazone completely reversed dexamethasone-induced inhibition of deoxyglucose uptake, suggesting a significant contribution of glucose uptake to troglitazone action. It appears that troglitazone influences both glucose uptake and glycogen synthesis although not to similar extents. Kreutter et al. (1990) have reported a partial effect of another thiazolidinedione, CP68722, on glucose uptake in dexamethasone-treated adipocytes. In animals treated with dexamethasone, troglitazone (Okumura et al., 1998) and metformin (Thomas et al., 1998) have been reported to improve glucose disposal. It is not clear whether these effects are direct or mediated through improvements in blood glucose. Another potential site of troglitazone action could be the activation of the enzyme, glycogen synthase. Unfortunately we have not tested the activity of glycogen synthase although Park et al. (1998) found stimulatory effect in muscle cell cultures. Chronic incubation of 3T3 adipocytes with insulin has been associated with downregulation of the insulin receptor and desensitization of insulin stimulated glycogen synthesis (Knutson et al., 1995; Knutson & Balba, 1997). However, it is not known whether insulin resistance worsens when both insulin and dexamethasone are present together chronically. Our data seem to suggest that insulin resistance, at least on glycogen synthesis, is maximum when both are present simultaneously and troglitazone is unable to overcome this defect completely. Obviously more studies are needed to fully understand the nature of the defects in this system. In the present study adipocytes used were fully differentiated and therefore troglitazone action is unlikely to be due to an increased differentiation of preadipocytes to adipocytes.
In summary, the present study shows that troglitazone has a direct insulin sensitizing action on glycogen synthesis in 3T3 adipocytes treated chronically with dexamethasone. Under similar conditions the biguanide, metformin did not exhibit significant insulin sensitizing properties. Troglitazone completely prevented and reversed insulin resistance on glycogen synthesis in cells treated with dexamethasone alone. In cells incubated chronically with insulin and dexamethasone, troglitazone partially prevented and reversed the insulin resistance. These actions are partly mediated by (a) its action on (i) glucocorticoid receptor displacing dexamethasone binding; and (ii) dexamethasone actions on glucose transport and glycogen synthesis; and (b) unlikely to be due to altered insulin binding to the receptor. In the normal insulin sensitive adipocytes troglitazone appears to act by increasing insulin binding. Using glycogen synthesis as an index of insulin sensitivity, we found that troglitazone exhibited a superior pharmacological profile as an anti-insulin resistant agent in this model. This may be desirable in the long term treatment of NIDDM patients.
Acknowledgments
The authors are grateful to Ms Nair and Ms Sherigar for help in the work and data presentation. The authors are thankful to Dr M.P. Desai for encouragement and support. Financial support was from the Department of Science & Technology, Government of India, DST Project No. SP/SO/B-13/93 of Dr A.R. Marita. Mr K.L. Anil Kumar was a Senior Research Fellow supported by DST.
Abbreviations
- DMEM
Dulbecco's modified eagle's medium
- FCS
foetal calf serum
- KRP
Kreb's ringer phosphate
- PBS
phosphate buffered saline
- PPAR
peroxisome proliferator activated receptor
References
- BAILEY C.J. Biguanides and NIDDM. Diabetes Care. 1992;15:755–772. doi: 10.2337/diacare.15.6.755. [DOI] [PubMed] [Google Scholar]
- CHAN C.P., KREBS E.G. Epidermal growth factor stimulates glycogen synthase activity in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 1985;82:4563–4567. doi: 10.1073/pnas.82.14.4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIARALDI T.P., GILMORE A., OLEFSKY J.M., GOLDBER G.M., HEIDENREICH K.A. In vitro studies on the action of CS-045. A new antidiabetic agent. Metabolism. 1990;39:1056–1062. doi: 10.1016/0026-0495(90)90166-a. [DOI] [PubMed] [Google Scholar]
- DAMSBO P., HERMANN L.S., VAAG A., HOTHER-NEILSEN O., BECK-NEILSEN H. Irreversibility of the defect in glycogen synthase activity in skeletal muscle from obese patients with NIDDM treated with diet and metformin. Diabetes Care. 1998;21:1489–1494. doi: 10.2337/diacare.21.9.1489. [DOI] [PubMed] [Google Scholar]
- DAMSBO P., VAAG A., HOTHER-NIELSEN O., BECK-NEILSEN H. Reduced glycogen synthase activity in skeletal muscle from obese patients with and without type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1991;34:239–245. doi: 10.1007/BF00405082. [DOI] [PubMed] [Google Scholar]
- DE FRONZO R.A., BONADONNA R.C., FERRANNINI E. Pathogenesis of NIDDM: a balanced overview. Diabetes Care. 1992;15:318–368. doi: 10.2337/diacare.15.3.318. [DOI] [PubMed] [Google Scholar]
- DIMITRIADIS G., LEIGHTON B., PARRY BILLINGS M., SASSON S., YOUNG M., KRAUSE U., BEVAN S., PIVA T., WEGENER G., NEWSHOLME E.A. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem. J. 1997;321:707–712. doi: 10.1042/bj3210707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FROST S.C., LANE M.D. Evidence for the involvement of vicinal sulphydryl groups in insulin-activated hexose transport by 3T3-L1 adipocytes. J. Biol. Chem. 1985;260:2646–2652. [PubMed] [Google Scholar]
- GAMOU S., SHIMIZU Y., SHIMIZU N.Adipocytes Methods in Molecular Biology 19905Clifton New Jersey: Humana Press Inc; 197–207.ed. Pollard, J.W., Walker, J.M.M. Vol [DOI] [PubMed] [Google Scholar]
- GRUNFELD C., BAIRD K., OBBERGHEN E.V., KAHN C.R. Glucocorticoid-induced insulin resistance in vitro: evidence for both receptor and postreceptor defects. Endocrinology. 1981;109:1723–1730. doi: 10.1210/endo-109-5-1723. [DOI] [PubMed] [Google Scholar]
- HENRY R.R., CIARALDI T.P., ABRAMS-CARTER L., MUDALIAR S., PARK K.S. Glycogen synthase activity is reduced in cultured skeletal muscle cells of non-insulin dependent diabetes mellitus subjects. J. Clin. Invest. 1996;98:1231–1236. doi: 10.1172/JCI118906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNUTSON V.P., BALBA Y. 3T3-L1 Adipocytes as a cell culture model of insulin resistance. In Vitro Cell. Dev. Biol. Animal. 1997;33:77–81. doi: 10.1007/s11626-997-0025-2. [DOI] [PubMed] [Google Scholar]
- KNUTSON V.P., DONNELLY P.V., BALBA Y., LOPEZ-REYES M. Insulin resistance is mediated by a proteolytic fragment of the insulin receptor. J. Biol. Chem. 1995;270:24972–24981. doi: 10.1074/jbc.270.42.24972. [DOI] [PubMed] [Google Scholar]
- KREUTTER D.K., ANDREWS K.M., GIBBS E.M., HUTSON N.J., STEVENSON R.W. Insulin like activity of new antidiabetic agent CP 68722 in 3T3-L1 adipocytes. Diabetes. 1990;39:1414–1419. doi: 10.2337/diab.39.11.1414. [DOI] [PubMed] [Google Scholar]
- LEHMANN J.M., MOORE L.B., SMITH-OLIVER T.A. An antidiabetic thiazolidinedione is a higher affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR-gamma) J. Biol. Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- LENHARD J.M., KLEIWER S.A., PAULIK M.A., PLUNKET K.D., LEHMANN J.M., WEIEL J.E. Effects of troglitazone and metformin on glucose and lipid metabolism. Alterations of two metabolic pathways. Biochem. Pharm. 1997;54:801–808. doi: 10.1016/s0006-2952(97)00229-3. [DOI] [PubMed] [Google Scholar]
- MATTHAEI S., HAMANN A., KLEIN H.H. Association of metformin's effect to increase insulin-stimulated glucose transport with potentiation of insulin-induced translocation of glucose transporters from intracellular pool to plasma membrane in rat adipocytes. Diabetes. 1991;40:850–857. doi: 10.2337/diab.40.7.850. [DOI] [PubMed] [Google Scholar]
- MOGUILEWSKY M., PHILIBERT D. RU 38486: Potent antiglucocorticoid activity correlated with strong binding to the cytosolic glucocorticoid receptor followed by an impaired activation. J. Steroid Biochem. 1984;20:271–276. doi: 10.1016/0022-4731(84)90216-4. [DOI] [PubMed] [Google Scholar]
- OKUMURA S., TAKEDA N., TAKAMI K., YOSHINO K., HATTORI J., NAKASHIMA K., SUGIMOTO M., ISHIMORI M., TAKAMI R., YASUDA K. Effects of troglitazone on dexamethasone-induced insulin resistance in rats. Metabolism. 1998;47:351–354. doi: 10.1016/s0026-0495(98)90270-0. [DOI] [PubMed] [Google Scholar]
- PARK K.S., CIARALDI T.P., ABRAMS-CARTER L., MUDALIAR S., NIKOULINA S.E., HENRY R.R. Troglitazone regulation of glucose metabolism in human skeletal muscle cultures from obese type II diabetic subjects. J. Clin. Endocrin. Met. 1998;83:1636–1643. doi: 10.1210/jcem.83.5.4764. [DOI] [PubMed] [Google Scholar]
- RANGANATHAN S., KERN P.A. Thiazolidinediones inhibit lipoprotein lipase activity in adipocytes. J. Biol. Chem. 1998;273:26117–26122. doi: 10.1074/jbc.273.40.26117. [DOI] [PubMed] [Google Scholar]
- REGINATO M.J., LAZAR M.A. Mechanisms by which thiazolidinediones enhance insulin action. Trends Endocrinol. Metab. 1999;8:187–191. doi: 10.1016/s1043-2760(98)00110-6. [DOI] [PubMed] [Google Scholar]
- RICE K.M., TURNBOW M.A., GARNER C.W. Insulin stimulates degradation of IRS-1 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 1993;190:961–967. doi: 10.1006/bbrc.1993.1143. [DOI] [PubMed] [Google Scholar]
- SALTIEL A.R., OLEFSKY J.M. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- SANDOUK T., REDA D., HOFMANN C. Antidiabetic agent pioglitazone enhances adipocyte differentiation of 3T3-F442 A cells. Am. J. Physiol. 1993;264:C1600–C1608. doi: 10.1152/ajpcell.1993.264.6.C1600. [DOI] [PubMed] [Google Scholar]
- SCHALIN-JANTTI C., HARKONEN M., GROO P.C. Impaired activation of glycogen synthase in people at increased risk for developing NIDDM. Diabetes. 1992;41:598–604. doi: 10.2337/diab.41.5.598. [DOI] [PubMed] [Google Scholar]
- SHEPHERD P.R., NAVE B.T., SIDDLE K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3-L1 adipocytes: evidence for the involvement of phosphoinositide 3-kinase and p70 ribosomal protein-S6 kinase. Biochem. J. 1995;305:25–28. doi: 10.1042/bj3050025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAFURI S.R. Troglitazone enhances differentiation, basal glucose uptake, and Glut 1 protein levels in 3T3-L1 adipocytes. Endocrinology. 1996;137:4706–4712. doi: 10.1210/endo.137.11.8895337. [DOI] [PubMed] [Google Scholar]
- THOMAS C.R., TURNER S.L., JEFFERSON W.H., BAILEY C.J. Prevention of dexamethasone induced insulin resistance by metformin. Biochem. Pharmacol. 1998;56:1145–1150. doi: 10.1016/s0006-2952(98)00151-8. [DOI] [PubMed] [Google Scholar]
- THOMSON M.J., WILLIAMS M.G., FROST S.C. Development of insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 1997;272:7759–7764. doi: 10.1074/jbc.272.12.7759. [DOI] [PubMed] [Google Scholar]
- THORBURN A.W., GUMBINER B., BULACAN F., WALLACE P., HENRY R.R. Intracellular glucose oxidation and glycogen synthase activity are reduced in non-insulin-dependent (type II) diabetes independent of impaired glucose uptake. J. Clin. Invest. 1990;85:522–529. doi: 10.1172/JCI114468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURNBOW M.A., KELLER S.R., RICE K.M., GARNER C.W. Dexamethasone down-regulation of insulin receptor substrate-1 in 3T3-L1 adipocytes. J. Biol. Chem. 1994;269:2516–2520. [PubMed] [Google Scholar]
- VAAG A., HENRIKSEN J.E., BECK-NIELSEN H. Decreased insulin activation of glycogen synthase in skeletal muscles in young nonobese caucasian first degree relatives of patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1992;89:782–788. doi: 10.1172/JCI115656. [DOI] [PMC free article] [PubMed] [Google Scholar]