

Abstract
Binding of the 5-HT7 receptor antagonist radioligand [3H]-SB-269970 to human 5-HT7(a) receptors expressed in HEK293 cell membranes (h5-HT7(a)/293) and to guinea-pig cerebral cortex membranes, was characterized and compared with [3H]-5-CT binding.
[3H]-SB-269970 (1 nM) showed full association with h5-HT7(a)/293 membranes after 40 min. Specific binding at equilibrium represented >90% of total binding and was fully reversible by methiothepin (10 μM), full dissociation occurring by 100 min. The association (k+1) and dissociation (k−1) rate constants were 0.05 nM−1min−1 and 0.05 min−1 respectively, giving a KD (k−1/k+1) of 1.0 nM.
[3H]-SB-269970 bound saturably and apparently monophasically to both h5-HT7(a)/293 and guinea-pig cortex membranes, with KD values of 1.25±0.05 and 1.7±0.3 nM respectively. The Bmax for [3H]-SB-269970 to both h5-HT7(a)/293 and guinea-pig cortex membranes (5780±380 and 125±8.2 fmoles mg protein−1 respectively) was similar to that for [3H]-5-CT (6190±940 and 143±19 fmoles mg protein−1 respectively). These data suggest that, in each tissue, both radioligands labelled the same population of receptors, which appear to be present in an agonist high affinity state.
The profile of compound inhibition of [3H]-SB-269970 binding to h5-HT7(a)/293 and guineapig cortex membranes correlated well (corr. coeff. 0.98) with those for [3H]-5-CT binding and were consistent with the profiles reported previously for the human 5-HT7(a) and guinea-pig cortex 5-HT7 receptors using [3H]-5-CT. Hill slopes for inhibition of [3H]-SB-269970 and [3H]-5-CT binding were close to 1, consistent with binding to a single receptor population in both tissues.
[3H]-SB-269970 represents the first selective 5-HT7 antagonist radioligand, which should aid further characterization of 5-HT7 receptors in recombinant and native tissues and help establish their role in brain function.
Keywords: [3H]-5-CT binding, [3H]-SB-269970 binding, human cloned 5-HT7 receptors, guinea-pig cortex
Introduction
Seven major classes of 5-HT receptor (5-HT1–7) have been identified to date, based on structural, functional and pharmacological criteria (Hoyer et al., 1994). The 5-HT7 receptor, which has been cloned from mouse (Plassat et al., 1993), rat (Lovenberg et al., 1993; Ruat et al., 1993), guinea-pig (Tsou et al., 1994) and human (Bard et al., 1993), is positively coupled to adenylyl cyclase (AC) when expressed in cell lines (e.g. Bard et al., 1993) and displays a unique pharmacological profile which is consistent across species. A number of splice variants of both the human (5-HT7a/b/d) and rat (5-HT7a/b/c) receptor have been identified, which display similar pharmacological and functional characteristics when expressed in cell lines (Jasper et al., 1997; Heidman et al., 1997).
5-HT7 receptor mRNA has been shown to be present in smooth muscle from cardiovascular and gastrointestinal tissues (Bard et al., 1993; Schoeffter et al., 1996; Hagan et al. in press). Comparative in situ hybridization and autoradiography studies in guinea-pig and rat brain have revealed a similar distribution pattern for 5-HT7 mRNA and receptor binding sites, with the highest receptor density in thalamic and limbic regions (Branchek et al., 1994; To et al., 1995; Gustafson et al., 1996). As seen in recombinant systems, 5-HT7 receptors are positively coupled to AC in guinea-pig brain, since the 5-carboxamidotryptamine (5-CT) stimulation of AC in guinea-pig hippocampus is antagonized by the selective 5-HT7 receptor antagonist, SB-258719 (Thomas et al., 1999, 1999). Based on brain localization and pharmacological studies with non-selective compounds, it has been suggested that 5-HT7 receptors may play a role in the control of circadian rhythms (Lovenberg et al., 1993; Tsou et al., 1994; Ying & Rusak, 1997) and may have potential therapeutic applications in depression (Sleight et al., 1995; Hagan et al. in press) and schizophrenia (Roth et al., 1994).
The majority of radioligand binding studies to characterize 5-HT7 receptors in recombinant and native tissue systems have used the agonist radioligands [3H]-5-HT (Bard et al., 1993; Tsou et al., 1994; Sleight et al., 1995; Clemett et al., 1999a) or [3H]-5-CT (To et al., 1995; Boyland et al., 1996; Thomas et al., 1998; Stowe & Barnes, 1998; Thomas et al., 1999, 1999). These radioligands have proved useful in characterizing the pharmacological profile of cloned and native 5-HT7 receptors, as well as providing information on their native tissue distribution, particularly in brain. However, neither [3H]-5-HT nor [3H]-5-CT selectively labels 5-HT7 receptors and in native tissues it has therefore been necessary to include selective blocking compounds to prevent labelling of non-5-HT7 binding sites. In the presence of such blocking compounds to prevent binding to 5-HT1A/1B/1D receptors, the pharmacological profile of [3H]-5-CT binding to both rat and guinea-pig brain was found to correlate well with that reported for recombinant 5-HT7 receptors (Boyland et al., 1996; Stowe & Barnes, 1998). Similarly, [3H]-8-OH-DPAT has been reported to label 5-HT7 receptors in hamster brain, when tested in the presence of pindolol to block 5-HT1A receptors (Duncan et al., 1999). However, it is important to note that the use of such blocking compounds could, depending on their selectivity profile, produce incomplete blockade of non-5-HT7 sites. [3H]-5-HT, in the presence of pindolol to block 5-HT1A/1B sites, has been reported to label 5-HT7 receptors in rat hypothalamus, but also appears to label an additional binding site under these assay conditions, revealed by shallow competition curves for some competing ligands (Sleight et al., 1995; Gobbi et al., 1996; Clemett et al., 1999). Similarly, [3H]-5-CT binding studies in rat brain, using cyanopindolol and sumatriptan to inhibit binding to 5-HT1A/1B and 5-HT1D receptors respectively, have also revealed shallow competition curves, suggesting that, under these conditions, [3H]-5-CT may label a heterogenous receptor population in this tissue (Waeber & Moskowitz, 1995; Boyland et al., 1996). An additional complication is the lack of selectivity of the blocking compounds used, such that partial blockade of 5-HT7 receptors can occur, thereby resulting in an underestimation of the 5-HT7 binding component. This possibility has been discussed previously by To et al. (1995).
A further potential complicating factor, relating to the use of agonist radioligands, is that, if multiple agonist affinity states of the receptor are present, only a fraction of the total 5-HT7 receptor population may be labelled. In order to avoid this possibility, Hemedah et al. (1999) have recently reported the use of an antagonist radioligand, [3H]-mesulergine, to label 5-HT7 receptors in rat brain and guinea-pig ileum. However, [3H]-mesulergine is a non-selective 5-HT7 receptor antagonist and the studies of Hemedah et al. were carried out in the presence of a variety of blocking agents, namely, cinanserin, RS-102221, raclopride, prazosin and yohimbine, to inhibit [3H]-mesulergine binding to 5-HT2A, 5-HT2C, dopamine D2, alpha1 and alpha2 receptors respectively.
There is, therefore, a need for a selective, antagonist radioligand to aid characterization of 5-HT7 receptors in both recombinant and native tissues. SB-269970-A ((R)-3-(2-(2-(4-Methyl-piperidin-1-yl)ethyl)-pyrrolidine-1-sulphonyl)-phenol), an analogue of SB-258719 (Forbes et al., 1998), has recently been identified and shown to be a potent 5-HT7 receptor antagonist (Lovell et al., 2000; Thomas et al., 1999b; Hagan et al., in press), displaying at least 100 fold selectivity versus all other 5-HT receptor subtypes except the human 5-HT5A receptor (50 fold, Lovell et al., 2000). SB-269970-A has therefore been tritiated (specific activity 49 Ci mmol−1) and the present study was carried out in order to characterize [3H]-SB-269970 binding to the human cloned 5-HT7(a) receptor expressed in HEK293 cells (h5-HT7(a)/293) and guinea-pig brain 5-HT7 receptors in comparison with the profile for [3H]-5-CT binding. These data have been presented previously in abstract form (Price et al., 1999; Atkinson et al., 1999).
Methods
Radioligand binding to h5-HT7(a)/293 membranes
For investigation of [3H]-5-CT and [3H]-SB-269970 binding to h5-HT7(a)/293 membranes, washed cell membranes were prepared from HEK293 cells stably expressing the human cloned 5-HT7(a) receptor using the method described by Thomas et al. (1998). Membranes (8–10 μg protein tube−1) were incubated in Tris HCl buffer (50 mM, pH 7.4 at 37°C) containing CaCl2 (4 mM), pargyline (0.1 mM) and ascorbic acid (1 mM). MgCl2 (5 mM) was also included for experiments to investigate the effect of guanosine-5′-O-(3-thiotriphosphate) (GTPγS) and 5′-guanylylimidodiphosphate (GppNHp) on binding. For [3H]-SB-269970 association rate studies, incubation was carried out in the presence of [3H]-SB-269970 (1 nM) for between 0 and 100 min in the absence (total binding) or presence (non-specific binding) of 10 μM 5-HT. Dissociation of [3H]-SB-269970 was initiated by addition of 10 μM methiothepin and specific binding measured from 0 to 100 min. For saturation experiments, h5-HT7(a)/293 membranes were incubated (60 min, 37°C) with 0.05–5 nM [3H]-5-CT or 0.1–12 nM [3H]-SB-269970. For competition studies, membranes were incubated (60 min, 37°C) with 0.5 nM [3H]-5-CT or 1 nM [3H]-SB-269970 in the absence or presence of test compounds. For both saturation and competition experiments, non-specific binding was defined in the presence of 10 μM 5-HT. Incubation was stopped by rapid filtration through Whatman GF/B grade filters (pre-soaked with 0.3% polyethyleneimine) followed by 5×1 ml ice-cold buffer washes. Bound radioactivity was determined by liquid scintillation counting.
Radioligand binding to guinea-pig cerebral cortex membranes
Guinea-pigs (male, Dunkin Hartley, 300–450 g) were killed by cervical dislocation, and cerebral cortices removed and homogenized (Polytron, 15 s, setting 5) in 20 volumes (based on wet weight of tissue) of 50 mM Tris (pH 7.4 at 37°C) containing 0.5 mM EDTA. Following centrifugation (50,000×g, 12 min 4°C) and resuspension in the same medium, the membranes were incubated at 37°C for 20 min. After three further centrifugation and resuspension steps, membranes were stored at −80°C prior to use. Membranes (40–55 μg protein tube−1) were incubated in Tris HCl buffer (50 mM, pH 7.4 at 37°C) containing CaCl2 (4 mM), MgCl2 (5 mM), pargyline (0.1 mM) and ascorbic acid (0.5 mM). [3H]-5-CT and [3H]-SB-269970 saturation and competition binding studies using guinea-pig cortical membranes were performed as for the human cloned 5-HT7(a) receptor binding studies, except that for [3H]-5-CT binding, WAY 100635 (1 μM) (Fletcher et al., 1996) and GR-127935 (10 μM) (Skingle et al., 1995) were included to inhibit binding to 5-HT1A and 5-HT1B/5−HT1D receptors respectively. Non-specific binding was defined in the presence of 10 μM 5-HT.
Data analysis
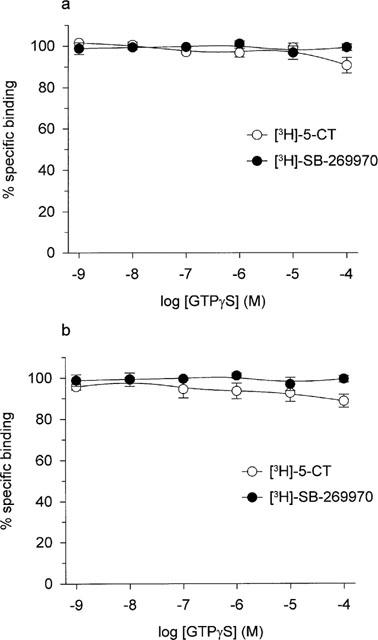
Analysis of both saturation and kinetic data was performed by non-linear curve fitting using KELL (Biosoft). Saturation binding data were analysed according to the equation:
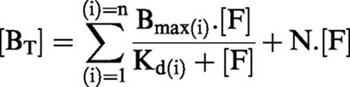 |
where [BT]=total observed binding, and N is the ratio of bound/free at infinite free concentration. This curve-fitting procedure provided estimates for KD (equilibrium dissociation constant) and Bmax (maximal binding density) for each radioligand, while kinetic analysis of [3H]-SB-269970 binding provided estimates for the observed association (Kobs) and dissociation rate constants. These were in turn used to derive values for the association rate constant and KD. The concentration of drug inhibiting specific [3H]-5-CT or [3H]-SB-269970 binding by 50% (IC50) and Hill coefficient (nH) was determined by iterative curve fitting (Bowen & Jerman 1995). pKi values (−log of the inhibition constant) were then calculated from the IC50 values as described by Cheng & Prusoff (1973). Correlation plots were analysed by linear regression (Grafit, Erithacus Software). Data represent the mean±s.e.mean of at least three separate experiments each performed using duplicate (inhibition experiments) or triplicate (saturation and kinetic experiments) determinations.
Drugs
5-hydroxytryptamine HCl (5-HT), 5-carboxamidotryptamine (5-CT), 8-hydroxy-dipropylaminotetralin (8-OH-DPAT), risperidone, methiothepin mesylate, mesulergine HCl, clozapine, ketanserin tartrate, ritanserin, mianserin hydrochloride, pindolol, guanosine-5′-O-(3-thiotriphosphate) (GTPγS) and 5′-guanylylimidodiphosphate (GppNHp) were obtained from Sigma-Aldrich (Poole, U.K). SB-258719 ((R)-3,N-Dimethyl-N-[1-methyl-3-(4-methylpiperidin-1-yl)propyl]benzene-sulphonamide), SB-269970-A ((R)-3-(2-(2-(4-methyl-piperidin-1-yl)ethyl)-pyrrolidine-1-sulphonyl)-phenol), GR 127935 and WAY 100635 were synthesized at SmithKline Beecham (Harlow, U.K.). [3H]-5-CT was obtained from Amersham (U.K.). [3H]-SB-269970 was obtained from the Synthetic Isotope Chemistry Unit, SmithKline Beecham (Upper Merion, U.S.A.). Stock drug solutions were prepared fresh on the day of assay in DMSO (the final assay concentration of DMSO not exceeding 0.4%). Drug dilutions were prepared in 5 mM Tris buffer (pH 7.4 at 37°C) containing 0.5 mM ascorbic acid.
Results
Association and dissociation kinetics of [3H]-SB-269970 binding to h5-HT7(a)/293 membranes
[3H]-SB-269970 (1 nM) showed full association with h5-HT7(a)/293 membranes after 40 min (Figure 1a). Specific binding at equilibrium represented >90% of total binding and was fully reversible by methiothepin (10 μM), with full dissociation occurring by 100 min (Figure 1b). The profile of association and dissociation of [3H]-SB-269970 was consistent with interaction with a single population of binding sites in the h5-HT7(a)/293 membranes. The association (k+1) and dissociation (k−1) rate constants were 0.049±0.003 nM−1 min−1 and 0.050±0.003 min−1 respectively (mean data from three experiments), giving a KD (k−1/k+1) of 1.04±0.1 nM, comparable to that derived from saturation analysis (see below).
Figure 1.
Kinetics of association (a) and dissociation (b) of 1 nM [3H]-SB-269970 binding to h5-HT7/293 membranes. Dissociation was initiated by addition of 10 μM methiothepin. Data points represent the mean±s.e.mean of three separate experiments each performed using triplicate determinations.
Saturation analysis of [3H]-SB-269970 and [3H]-5-CT binding to h5-HT7(a)/293 membranes
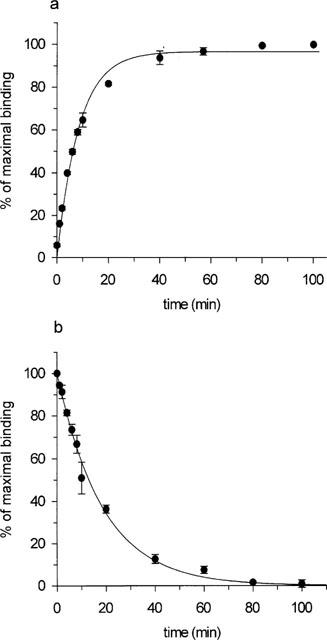
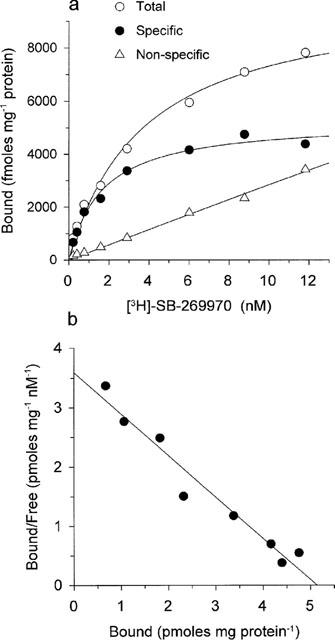
[3H]-SB-269970 (0.1–10 nM) bound saturably and apparently monophasically to h5-HT7(a)/293 membranes, giving a KD value of 1.25±0.05 nM (Table 1). Consistent with this, statistical analysis of the saturation binding data (using KELL) did not significantly favour a multi-site versus a single-site interaction. Figure 2a, b shows saturation binding data for [3H]-SB-269970 (and a Scatchard plot of the same data) from a typical experiment repeated twice. [3H]-5-CT (0.5–5 nM) also bound saturably and apparently monophasically to h5-HT7(a)/293 membranes, with a KD of 0.42±0.04 nM (Table 1, data not shown). The Bmax for [3H]-SB-269970 (5.8±0.4 pmoles mg protein−1, Table 1) was similar to that for [3H]-5-CT (6.2±0.2 pmoles mg protein−1), suggesting that both radioligands labelled the same receptor population. The similar Bmax values also suggested that most sites labelled appeared to be present in the agonist high affinity state. However, the stable GTP analogue, GTPγS (10 nM–100 μM), produced only a weak inhibition of (0.5 nM) [3H]-5-CT binding to h5-HT7(a)/293 membranes (9.0±3.7% inhibition at 100 μM) and had no effect on (1 nM) [3H]-SB-269970 binding to h5-HT7(a)/293 membranes (Figure 5a). Consistent with these data, the GTP analogue GppNHp (0.1–100 μM) failed to significantly inhibit [3H]-5-CT binding (3.6±2.1 inhibition at 100 μM) and did not affect [3H]-SB-269970 binding to h5-HT7(a)/293 membranes (data not shown). In contrast, under the same assay conditions, (1 nM) [3H]-5-CT binding to h5-HT1B/CHO cell membranes (Watson et al., 1996) was markedly inhibited by both GTPγS (51±2% inhibition at 100 μM) and GppNHp (46±0.8% inhibition at 100 μM), confirming that the assay conditions used in the present study were suitable for detecting guanine nucleotide-induced inhibition of agonist binding.
Table 1.
KD and Bmax values from saturation analysis of [3H]-5-CT and [3H]-SB-269970 binding to h5-HT7(a)/293 and guinea-pig cerebral cortex membranes

Figure 2.
(a) Saturation analysis of [3H]-SB-269970 binding to h5-HT7/293 membranes. Data points show total binding, non-specific binding (defined in the presence of 10 μM 5-HT) and specific binding, calculated by subtracting non-specific binding from total binding. Data are from a typical experiment performed using triplicate determinations and repeated twice (n=3). (b) Scatchard plot (bound in pmoles mg protein−1 versus bound/free (pmoles mg protein−1 nM−1)) of the specific binding data shown in (a).
Figure 5.
Effect of GTPγS (1 nM to 100 μM) on 0.5 nM [3H]-5-CT and 1 nM [3H]-SB-269970 binding to (a) h5-HT7(a)/293 membranes and (b) guinea-pig cortex membranes. Data points represent the mean±s.e.mean of at least three separate experiments each performed using duplicate determinations. Results are expressed as per cent of specific binding where non-specific binding was defined using 10 μM 5-HT.
Saturation analysis of [3H]-SB-269970 and [3H]-5-CT binding to guinea-pig cerebral cortex membranes
[3H]-SB-269970 (0.1–10 nM) bound saturably and apparently monophasically to guinea-pig cortex membranes. Consistent with this profile, statistical analysis of the saturation binding data (using KELL) significantly favoured a single site versus a multi-site interaction. Figure 3a, b shows saturation binding data for [3H]-SB-269970 (and Scatchard plot of the same data) from a typical experiment repeated twice. Specific binding defined by the presence of 10 μM 5-HT represented 50–60% of total binding at a [3H]-SB-269970 concentration of 1 nM. The KD of 1.7±0.3 nM was similar to that determined at the human cloned 5-HT7(a) receptor (Table 1). In the presence of WAY 100635 and GR 127935, [3H]-5-CT also bound saturably and apparently monophasically to guinea-pig cortex, with a KD of 0.67±0.12 nM (Table 1, data not shown). The Bmax for [3H]-SB-269970 binding to guinea-pig cortex membranes was similar to that determined for [3H]-5-CT (125±8.2 and 143±19 fmoles mg protein−1 respectively), consistent with both radioligands labelling the same receptor population and suggesting that, as seen for the human cloned 5-HT7(a) receptor, most sites labelled appeared to be in the agonist high affinity state. However, as seen for the h5-HT7(a)/293 binding studies, GTPγS (10 nM–100 μM), produced only a weak inhibition of (0.5 nM) [3H]-5-CT binding to guinea-pig cortex membranes (11±3.2% inhibition at 100 μM) and had no effect on (1 nM) [3H]-SB-269970 binding (Figure 5b). Similarly, GppNHp (0.1–100 μM) produced a marginal inhibition of [3H]-5-CT binding (8.1±4.8% inhibition at 100 μM) and did not affect [3H]-SB-269970 binding to guinea-pig cortex membranes (data not shown).
Figure 3.
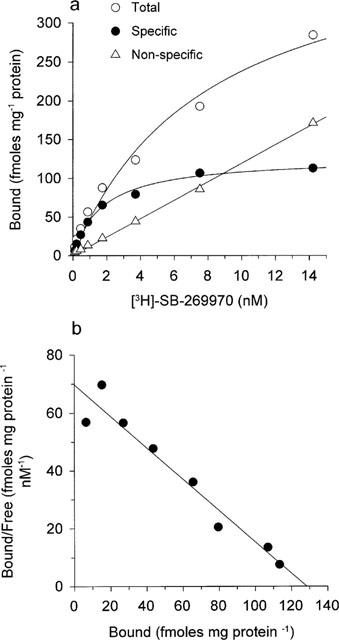
(a) Saturation analysis of [3H]-SB-269970 binding to guinea-pig cerebral cortex membranes. Data points show total binding, non-specific binding (defined in the presence of 10 μM 5-HT) and specific binding, calculated by subtracting non-specific binding from total binding. Data are from a typical experiment performed using triplicate determinations and repeated twice (n=3). (b) Scatchard plot (bound in pmoles mg protein−1 versus bound/free (pmoles mg protein−1 nM−1)) of the specific binding data shown in (a).
Pharmacological characterization of [3H]-SB-269970 and [3H]-5-CT binding to h5-HT7(a)/293 and guinea-pig cerebral cortex membranes
The pharmacological profile of [3H]-SB-269970 (1 nM) and [3H]-5-CT (0.5 nM) binding to both h5-HT7(a)/293 and guinea-pig cerebral cortex membranes was investigated using a range of 5-HT7 receptor agonists and antagonists. Figure 4a shows representative inhibition curves for displacement of [3H]-SB-269970 binding to h5-HT7(a)/293 membranes. The profile of inhibition of [3H]-SB-269970 binding to h5-HT7(a)/293 membranes correlated well (correlation coefficient 0.98) with that for [3H]-5-CT binding (Table 2, Figure 6) and was consistent with that reported previously for the h5-HT7(a) receptor (Bard et al., 1993; Thomas et al., 1998). SB-269970-A potently inhibited [3H]-SB-269970 binding to h5-HT7(a)/293 membranes, displaying a pKi of 8.61±0.10. For the majority of compounds, Hill slopes for inhibition of both [3H]-SB-269970 and [3H]-5-CT binding to 5-HT7(a)/293 membranes were close to 1. The Hill slope for 5-CT inhibition of [3H]-SB-269970 binding was slightly less than 1 (0.82±0.04). However, Hill slopes for inhibition of [3H]-SB-269970 and [3H]-5-CT binding by all other agonists tested were close to 1 and for each radioligand, the overall inhibitory profile appeared consistent with binding to a single population of receptors.
Figure 4.
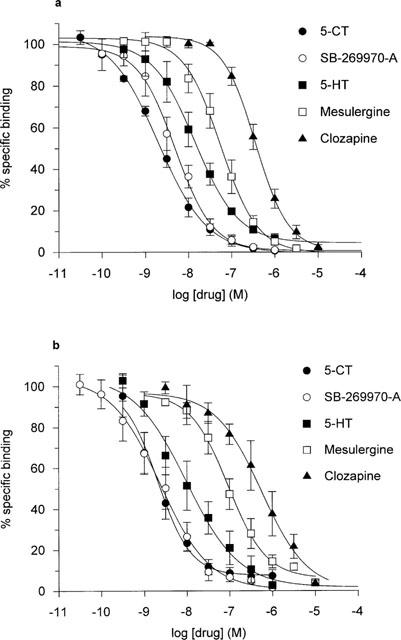
Inhibition of 1 nM [3H]-SB-269970 binding to (a) h5-HT7(a)/293 membranes and (b) guinea-pig cortex membranes, by 5-CT, SB-269970-A, 5-HT, mesulergine and clozapine. Data points represent the mean±s.e.mean of at least three separate experiments each performed using duplicate determinations. Results are expressed as per cent of specific binding where non-specific binding was defined using 10 μM 5-HT.
Table 2.
Potency of compounds to inhibit [3H]-5-CT and [3H]-SB-269970 binding to human 5-HT7(a)/293 and guinea-pig cortex membranes
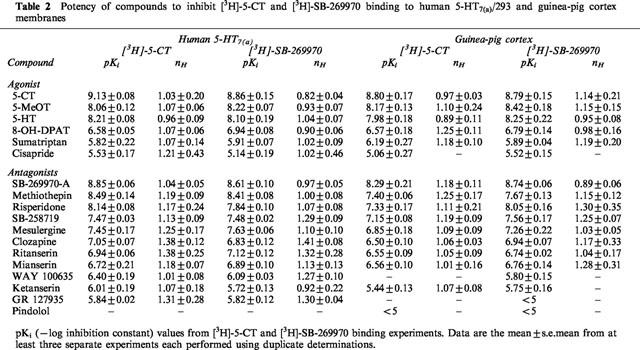
Figure 6.
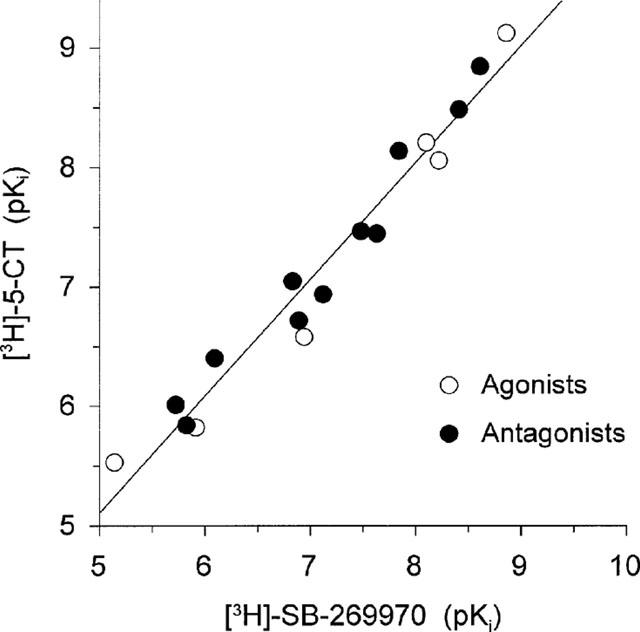
Correlation plot of drug potencies (expressed as pKi; values as shown in Table 1) to inhibit [3H]-5-CT and [3H]-SB-269970 binding to h5-HT7/293 membranes. Data points shown are for agonists and antagonists. The correlation plot gave a slope of 0.99 and correlation coefficient of 0.98.
As seen in the human 5-HT7(a)/293 recombinant system, the profile of inhibition of [3H]-SB-269970 (1 nM) binding to guinea-pig cerebral cortex membranes correlated well with that for [3H]-5-CT (0.5 nM) binding (correlation coefficient 0.98) (Table 2 and Figure 7). Figure 4b shows representative inhibition curves for displacement of [3H]-SB-269970 binding to guinea-pig cortex membranes. The profile seen for both [3H]-SB-269970 and [3H]-5-CT binding was consistent with that reported previously for the guinea-pig brain 5-HT7 receptor (e.g. To et al., 1995; Thomas et al., 1999a). In addition, for both agonists and antagonists Hill slopes for inhibition of both [3H]-SB-269970 and [3H]-5-CT binding were close to 1 (Table 2), consistent with binding to a single population of receptors. The pharmacological profile of binding of [3H]-SB-269970 and [3H]-5-CT to 5-HT7(a)/HEK293 membranes was similar to the corresponding profile seen for guinea-pig cortex membranes (Table 2), suggesting that both human recombinant and guinea-pig native 5-HT7 receptors display a similar pharmacological profile using either [3H]-SB-269970 or [3H]-5-CT.
Figure 7.
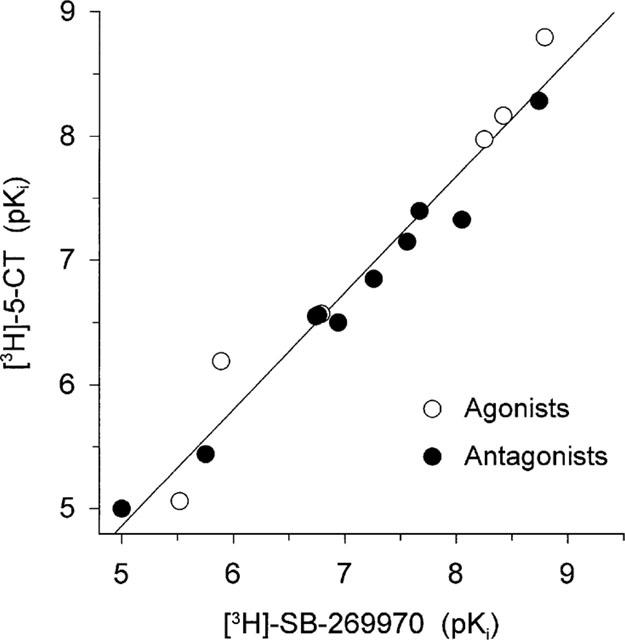
Correlation plot of drug potencies (expressed as pKi; values as shown in Table 1) to inhibit [3H]-5-CT and [3H]-SB-269970 binding to guinea-pig cerebral cortex membranes. Data points shown are for agonists and antagonists. The correlation plot gave a slope of 0.92 and correlation coefficient of 0.98.
Discussion
5-HT7 receptor binding studies in recombinant and native tissues have, to date, utilized a number of non-selective, agonist radioligands, including [3H]-5-HT and [3H]-5-CT. These studies have provided useful information on both the pharmacological profile and native tissue localization of 5-HT7 receptors. However, the use of non-selective radioligands has, in a number of studies, resulted in labelling of multiple binding sites and in all studies to date, has required the use of blocking drugs to mask binding to non-5-HT7 sites. There has therefore been a need for a selective 5-HT7 receptor radioligand to aid investigation of 5-HT7 receptors in native and recombinant tissues. In the present study we have characterized the binding of the selective 5-HT7 receptor antagonist radioligand, [3H]-SB-269970, to both human cloned and guinea-pig native 5-HT7 receptors.
Specific [3H]-SB-269970 binding to well-washed membranes prepared from HEK293 cells stably expressing the human cloned 5-HT7(a) receptor (h5-HT7(a)/293 membranes) was reversible, saturable, of high affinity and represented approximately 90% of total membrane binding (at 1 nM [3H]-SB-269970). Saturation analysis of the [3H]-SB-269970 binding data revealed an apparently monophasic binding profile with a KD of 1.25 nM, similar to the KD derived from analysis of [3H]-SB-269970 association and dissociation kinetics (1.0 nM). [3H]-5-CT also bound saturably and apparently monophasically to h5-HT7(a)/293 membranes, with a slightly higher binding affinity (KD 0.42 nM) compared to [3H]-SB-269970. The Bmax for [3H]-SB-269970 (5.8±0.4 pmoles mg protein−1) was similar to that for [3H]-5-CT (6.2±0.2 pmoles mg protein−1), suggesting that both radioligands appeared to be labelling the same receptor population in h5-HT7(a)/293 membranes. The similar Bmax also suggested that the majority of receptors were present in a high agonist affinity, presumably G-protein-coupled state. However, GTPγS (10 nM–100 μM), which would be predicted to induce receptor-G-protein uncoupling, with a consequent loss of the agonist high affinity state, did not alter the level of either [3H]-5-CT or [3H]- SB-269970 binding, producing only a marginal inhibition of [3H]-5-CT binding at the highest concentration tested (100 μM). Similarly, GppNHp did not markedly inhibit [3H]-5-CT binding to h5-HT7(a)/293 membranes. In contrast, a marked inhibition by both GTPγS (100 μM) and GppNHp (100 μM) of [3H]-5-CT binding to h5-HT1B/CHO cell membranes could be detected (data not shown), confirming that the assay conditions used in the present study were suitable for detecting guanine nucleotide-induced effects on agonist binding. Consistent with the lack of a marked effect of guanine nucleotides on [3H]-5-CT binding to h5-HT7(a)/293 membranes, it has previously been reported that the rate of [35S]-GTPγS binding to h5-HT7(a)/293 membranes is unaltered in response to 5-HT7 receptor stimulation (Thomas et al., 1998). However, 5-HT7 receptor agonist-mediated stimulation of adenylyl cyclase activity can be measured in h5-HT7(a)/293 membranes, suggesting that the 5-HT7 receptor couples to Gs in this system (Thomas et al., 1998). It is possible therefore, that the receptor is present in a high agonist affinity, G protein-coupled state, which is relatively resistant to guanine nucleotide-induced uncoupling under the assay conditions used. Studies are ongoing to further investigate these findings.
Pharmacological characterization of [3H]-SB-269970 binding to h5-HT7(a)/293 membranes revealed that drug potencies to inhibit [3H]-SB-269970 binding correlated well with the corresponding potencies to inhibit [3H]-5-CT binding. For example, 5-CT, SB-269970-A and risperidone potently inhibited binding, whereas 8-OH-DPAT and ketanserin were weaker inhibitors of binding. For the majority of test compounds, Hill slopes for inhibition of both [3H]-SB-269970 and [3H]-5-CT binding to 5-HT7(a)/293 membranes were close to 1. The Hill slope for 5-CT inhibition of [3H]-SB-269970 binding was slightly less than one (0.82±0.04), which could reflect the presence of multiple agonist affinity states of the h5-HT7(a) receptor. However, this possibility appears unlikely since Hill slopes for inhibition of [3H]-SB-269970 binding by all other agonists tested were close to 1. In addition, as already discussed, the similar Bmax values seen for [3H]-SB-269970 and [3H]-5-CT binding to h5-HT7(a)/293 membranes appears to suggest that the majority of receptors are present in a high agonist affinity, presumably G protein-coupled state. The overall pharmacological profile, for both [3H]-SB-269970 and [3H]-5-CT binding to h5-HT7(a)/293 membranes, was consistent with binding to 5-HT7 receptors, and consistent with the profile reported previously for the h5-HT7(a) receptor (Bard et al., 1993; Thomas et al., 1998).
[3H]-SB-269970 (0.1–10 nM) binding to guinea-pig cerebral cortex membranes displayed a similar profile to that seen for the human cloned receptor, being saturable and of high affinity. Saturation analysis of the [3H]-SB-269970 binding data also revealed an apparently monophasic binding profile with a Bmax of 125±8.2 fmoles mg protein−1 and KD of 1.7±0.3 nM, similar to that determined at the human cloned 5-HT7(a) receptor. Specific binding was somewhat lower than that at the human cloned receptor, representing 50–60% of total binding at 1 nM [3H]-SB-269970. For comparison, [3H]-5-CT binding to guinea-pig cortex membranes was carried out in the presence of WAY-100635 (1 μM) and GR-127935 (10 μM), to inhibit binding to 5-HT1A and 5-HT1B/1D receptors respectively. As seen for [3H]-SB-269970, [3H]-5-CT bound saturably and apparently monophasically to guinea-pig cortex membranes, with a KD of 0.67±0.12 nM. The Bmax for [3H]-5-CT binding (143±19 fmoles mg protein−1) was similar to that for [3H]-SB-269970. This suggested that, as seen for the human recombinant receptor, most sites labelled appeared to be present in an agonist high affinity state. However, also consistent with the human recombinant studies, neither GTPγS, nor GppNHp (at concentrations up to 100 μM) markedly altered the binding of [3H]-5-CT or [3H]-SB-269970 to guinea-pig cortex membranes. The lack of a clear effect of guanine nucleotides on agonist binding could be explained if, in both the recombinant and native tissue systems, the 5-HT7 receptor is present in a low agonist affinity, G-protein uncoupled state. However, this explanation appears unlikely, since pKi values for agonists in the present study are comparable or higher than their corresponding pEC50 values determined in 5-HT7 receptor functional studies (Thomas et al., 1998; 1999a), the latter presumably reflecting interaction with receptors in a high agonist affinity, G protein-coupled state.
The profile of inhibition of [3H]-SB-269970 binding to guinea-pig cerebral cortex membranes by standard agonists and antagonists correlated well with that seen for [3H]-5-CT binding and was consistent with the receptor binding profile reported previously for 5-HT7 receptors in guinea-pig cortex (To et al., 1995; Boyland et al., 1996). Hill slopes for drug inhibition of both [3H]-SB-269970 and [3H]-5-CT binding were close to 1, consistent with binding to a single population of receptors in this tissue. However, pKi values for antagonists, determined using [3H]-5-CT, were slightly lower than those determined using [3H]-5-CT at the human cloned receptor. To et al., (1995), who also reported differences in drug potencies to inhibit [3H]-5-CT binding between recombinant and native tissue, suggested that this may result from the use of an agonist radioligand, reflecting differences in coupling efficiency between cloned and native receptors. The data in the present study support this explanation, since absolute drug potencies to inhibit binding of the antagonist radioligand [3H]-SB-269970 to h5-HT7(a)/293 and guinea-pig cortex membranes were similar. Furthermore, the similar pharmacological profile for [3H]-SB-269970 binding to h5-HT7(a)/293 and guinea-pig cortex membranes is consistent with previous reports that human recombinant and guinea-pig native 5-HT7 receptors display a similar pharmacological profile (Boyland et al., 1996; Tsou et al., 1994).
The density of 5-HT7 receptors in guinea-pig cortex, determined in the present study using either [3H]-SB-269970 or [3H]-5-CT, is somewhat higher than that reported previously by To et al. (1995) using [3H]-5-CT, who reported a Bmax of 69 fmoles mg protein−1 in this tissue. This difference could be explained by the different combination of blocking drugs used by To et al. who used 1 μM (−)-cyanopindolol and 1 μM sumatriptan to inhibit [3H]-5-CT binding to 5-HT1A/1B/1D receptors. As discussed by To et al. (1995) this combination of blocking drugs could produce a partial blockade of 5-HT7 sites, resulting in an underestimation of the maximal binding density. In contrast, it is likely that the Bmax of 125 fmoles mg protein−1, determined in guinea-pig cortex using [3H]-SB-269970, represents an accurate estimate of the density of 5-HT7 receptors in this tissue. In human clonal cell lines, SB-269970 displays at least 100 fold selectivity for 5-HT7 receptors versus other 5-HT receptor subtypes, apart from the 5-HT5A receptor (50 fold selective) and in addition, in a commercial screening package (Cerep), displayed at least 100 fold selectivity for the human 5-HT7(a) receptor versus a total of 50 other receptors, enzymes or ion channels (Lovell et al., 2000). Furthermore, although the density of 5-HT5A receptors in guinea-pig brain has not been reported, previous studies in other species suggest that, if the 5-HT5A receptor is present in guinea-pig brain, the density is likely to be low (Rees et al., 1994; Grailhe et al., 1999). It is therefore unlikely that [3H]-SB-269970 would label the 5-HT5A receptor (or other non-5-HT7 receptor sites) under the assay conditions used in the present study. This conclusion is supported by the finding that SB-258719, which displays a low affinity for the human 5-HT5A receptor (pKi 5.0, unpublished observation), in the present study, completely inhibited [3H]-SB-269970 binding to guinea-pig cortex membranes and with a potency similar to that reported previously at 5-HT7 receptors in guinea-pig hippocampus (Thomas et al., 1999a). Furthermore, since [3H]-5-CT displayed a Bmax comparable to that for [3H]-SB-269970 in guinea-pig cortex and like [3H]-SB-269970, displayed an apparently monophasic binding profile, it appears that the blocking drugs used inhibited [3H]-5-CT binding to 5-HT1A/1B/1D receptors without significantly inhibiting the 5-HT7 binding component. Consistent with this, WAY-100635 has previously been shown to display low affinity for 5-HT7 receptors in guinea-pig brain (pKi<5.5, Thomas et al., 1999a) and in the present study, GR-127935 did not inhibit [3H]-SB-269970 binding to guinea-pig cortex (pKi<5).
In conclusion [3H]-SB-269970 has been shown to radiolabel, with high affinity, both the human cloned and guinea pig native 5-HT7 receptor. [3H]-SB-269970 is the first selective antagonist radioligand for 5-HT7 receptors which represents a valuable tool with which to further characterize 5-HT7 receptors in recombinant and native tissues and which should aid elucidation of the tissue distribution of the 5-HT7 receptor and its role in brain function.
Abbreviations
- AC
adenylyl cyclase
- 5-CT
5-carboxamidotryptamine
- GppNHp
5′-guanylylimidodiphosphate
- GTPγS
guanosine-5′-O-(3-thiotriphosphate)
- 8-OH-DPAT
8-hydroxy-dipropylaminotetralin
- SB-258719
((R)-3,N-Dimethyl-N-[1-methyl-3-(4-methylpiperidin-1-yl)propyl]benzene-sulphonamide)
- SB-269970
(R)-3-(2-(2-(4-Methyl-piperidin-1-yl)ethyl)-pyrrolidine-1-sulphonyl)-phenol
References
- ATKINSON P.A., THOMAS D.R., HO M., BROMIDGE S.M., HAGAN J.J., MIDDLEMISS D.N., PRICE G.W. [3H]-SB-269970 radiolabels 5-HT7 receptors in guinea pig cerebral cortex membranes. Br. J. Pharmacol. 1999;128:286P. [Google Scholar]
- BARD J.A., ZGOMBICK J., ADHAM N., VAYSSE P., BRANCHEK T.A., WEINSHANK R.L. Cloning of a novel human serotonin receptor (5-HT7) positively linked to adenylate cyclase. J. Biol. Chem. 1993;268:23422–23426. [PubMed] [Google Scholar]
- BOWEN W.P., JERMAN J.C. Nonlinear regression using spreadsheets. TiPS. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- BOYLAND P.S., EASTWOOD S., ELLIS C., BERGSMA D., JONES B.J., GLOGER I.S., UPTON N., MIDDLEMISS D.N. High specific activity [3H]-5-CT binding: correlation of guinea-pig cortex with human cloned 5-HT7 receptors. Br. J. Pharmacol. 1996;117:132P. [Google Scholar]
- BRANCHEK T.A., GUSTAFSON E.L., DURKIN M.M., BARD J.A., WEINSHANK R.L. Autoradiographic localisation of 5-HT7 and its mRNA in rat CNS by radioligand binding and in situ hybridization histochemistry. Br. J. Pharmacol. 1994;112 Suppl.:100P. [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLEMETT D.A., KENDALL D.A., COCKETT M.I., MARSDEN C.A., FONE K.C.F. Pindolol-insensitive [3H]-5-hydroxytryptamine binding in the rat hypothalamus; identity with 5-hydroxytryptamine7 receptors. Br. J. Pharmacol. 1999;127:236–242. doi: 10.1038/sj.bjp.0702503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNCAN M.J., SHORT J., WHEELER D.L. Comparison of the effect of aging on 5-HT7 and 5-HT1A receptors in discrete regions of the circadian timing system of hamsters. Brain Res. 1999;829:39–45. doi: 10.1016/s0006-8993(99)01311-6. [DOI] [PubMed] [Google Scholar]
- FLETCHER A., FORSTER E.A., BILL D.J., BROWN G., CLIFFE I.A., HARTLEY J.E., JONES D.E., MCLENACHAN A., STANHOPE K.J., CRITCHLEY D.J.P., CHILDS K.J., MIDDLEFELL V.C., LANFUMEY L., CORRADETTI R., LAPORTE A.-M., GOZLAN H., HAMON M., DOURISH C.T. Electrophysiological, biochemical, neurohormonal and behavioural studies with WAY-100635, a potent, selective and silent 5-HT1A receptor antagonist. Behav. Brain Res. 1996;73:337–353. doi: 10.1016/0166-4328(96)00118-0. [DOI] [PubMed] [Google Scholar]
- FORBES I.T., DABBS S., DUCKWORTH D.M., JENNINGS A.J., KING F.D., LOVELL P.J., COLLIN L., BROWN A.M., HAGAN J.J., MIDDLEMISS D.N., RILEY G.J., THOMAS D.R., UPTON N. (R)-3,N-Dimethyl-N-[1-methyl-3-(4-methylpiperidin-1-yl)propyl]benzene sulfonamide: The First Selective 5-HT7 Receptor Antagonist. J. Med. Chem. 1998;41:655–657. doi: 10.1021/jm970519e. [DOI] [PubMed] [Google Scholar]
- GOBBI M., PAROTTI L., MENINI T. Are 5-hydroxytryptamine7 receptors involved in [3H]5-hydroxytryptamine binding to 5-hydroxytryptamine1nonA-nonB receptors in rat hypothalamus. Mol. Pharmacol. 1996;49:556–559. [PubMed] [Google Scholar]
- GRAILHE R., WAEBER C., DULAWA S.C., HORNUNG J.P., ZHUANG X., BRUNNER D., GEYER M.A., HEN R. Increased exploratory activity and altered response to LSD in mice lacking the 5-HT5A receptor. Neuron. 1999;22:581–591. doi: 10.1016/s0896-6273(00)80712-6. [DOI] [PubMed] [Google Scholar]
- GUSTAFSON E.L., DURKIN M.M., BARD J.A., ZGOMBICK J., BRANCHEK T.A. A receptor autoradiographic and in situ hybridization analysis of the distribution of the 5-ht7 receptor in rat brain. Br. J. Pharmacol. 1996;117:657–666. doi: 10.1111/j.1476-5381.1996.tb15241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGAN J.J., PRICE G.W., JEFFREY P., DEEKS N.J., STEAN T., PIPER D., SMITH M.I., UPTON N., MEDHURST A.D., MIDDLEMISS D.N., RILEY G., LOVELL P.J., BROMIDGE S., THOMAS D.R.Characterisation of SB-269970-A, a selective 5-HT7 receptor antagonist Br. J. Pharmacol. 2000. in press [DOI] [PMC free article] [PubMed]
- HEIDMAN D.E.A., METCALF M.A., KOHEN R., HAMBLIN M.W. Four 5-hydroxytryptamine7 (5-HT7) receptor isoforms in human and rat produced by alternative splicing: Species differences due to altered intron-exon organisation. J. Neurochem. 1997;68:1372–1381. doi: 10.1046/j.1471-4159.1997.68041372.x. [DOI] [PubMed] [Google Scholar]
- HEMEDAH M., COUPAR I.M., MITCHELSON F.J. [3H]-Mesulergine labels 5-HT7 sites in rat brain and guinea-pig ileum but not rat jejunum. Br. J. Pharmacol. 1999;126:179–188. doi: 10.1038/sj.bjp.0702293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOYER D., CLARKE D.E., FOZARD J.R., HARTIG P.R., MARTIN G.R., MYLECHARANE E.J., SAXENA F.R., HUMPHREY P.P.A. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol. Rev. 1994;46:157–243. [PubMed] [Google Scholar]
- JASPER J.R., TO Z.P., KOSAKA A., EGLEN R.M., CHANG D.J. Cloning, expression and pharmacology of a truncated splice variant of the human 5-HT7 receptor (h5-HT7(b)) Br. J. Pharmacol. 1997;122:126–132. doi: 10.1038/sj.bjp.0701336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVELL P.J., BROMIDGE S.M., DABBS S., DUCKWORTH D.M., FORBES I.T., JENNINGS A.J., KING F.D., MIDDLEMISS D.N., RAHMAN S.K., SAUNDERS D.V., COLLIN L.L., HAGAN J.J., RILEY G.J., THOMAS D.R. A novel, potent and selective 5-HT7 antagonist: (R)-3-(2-(2-(4-methyl-piperidin-1-yl)ethyl)-pyrrolidine-1-sulfonyl)-phenol (SB-269970) J. Med. Chem. 2000;43:342–345. doi: 10.1021/jm991151j. [DOI] [PubMed] [Google Scholar]
- LOVENBERG T.W., BARON B., DE LECEA L., MILLER J.D., PROSSER R.A., REA M.A., FOYE P.E., RACKE M., SLONE A.L., SIEGEL B.W., DANIELSON P.E., SUTCLIFFE J.G., ERLANDER M.G. A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythyms. Neuron. 1993;11:449–458. doi: 10.1016/0896-6273(93)90149-l. [DOI] [PubMed] [Google Scholar]
- PLASSAT J., AMLAIKY N., HEN R. Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase. Mol. Pharmacol. 1993;44:229–236. [PubMed] [Google Scholar]
- PRICE G.W., ATKINSON P.A., HO M., BROMIDGE S.M., HAGAN J.J., MIDDLEMISS D.N., THOMAS D.R. [3H]-SB-269970 – a selective antagonist radioligand for 5-HT7 receptors. Br. J. Pharmacol. 1999;128:233P. doi: 10.1038/sj.bjp.0703318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES S., DEN DAAS I., FOORD S., GOODSON S., BULL D., KILPATRICK G., LEE M. Cloning and characterisation of the human 5-HT5A receptor. FEBS Lett. 1994;355:242–246. doi: 10.1016/0014-5793(94)01209-1. [DOI] [PubMed] [Google Scholar]
- ROTH B.L., CRAIGO S.C., CHOUDHARY M.S., ULUER A., MONSMA F.J., JR, SHEN Y., MELTZER H.Y., SIBLEY D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharm. Exp. Ther. 1994;268:1403–1410. [PubMed] [Google Scholar]
- RUAT M., TRAIFFORT E., LEURS R., TARDIVEL-LACOMBE J., DIAZ J., ARRANG J.-M., SCWARTZ J.-C. Molecular cloning, characterisation, and localisation of a high-affinity serotonin receptor (5-ht7) activating cAMP formation. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8547–8551. doi: 10.1073/pnas.90.18.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHOEFFTER P., ULLMER C., BOBIRNAC I., GABBIANI G., LUBBERT H. Functional, endogenously expressed 5-hydroxytryptamine 5-ht7 receptors in human vascular smooth muscle cells. Br. J. Pharmacol. 1996;117:993–994. doi: 10.1111/j.1476-5381.1996.tb16687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENKER A., MAAYANI S., WEINSTEIN H., GREEN J.P. Pharmacological characterisation of two 5-hydroxytryptamine receptors coupled to adenylate cyclase in guinea pig hippocampal membranes. Mol. Pharmacol. 1987;31:357–367. [PubMed] [Google Scholar]
- SKINGLE M., SLEIGHT A.J., FENIUK Effects of the 5-HT1D receptor antagonist GR127935 on extracellular levels of 5-HT in the guinea-pig frontal cortex as measured by microdialysis. Neuropharmacology. 1995;34:377. doi: 10.1016/0028-3908(94)00167-q. [DOI] [PubMed] [Google Scholar]
- SLEIGHT A.J., CAROLO C., PETIT N., ZWINGELSTEIN C., BOURSON A. Identification of 5-Hydroxytryptamine7 receptor binding sites in rat hypothalamus: sensitivity to chronic antidepressant treatment. Mol. Pharmacol. 1995;47:99–103. [PubMed] [Google Scholar]
- STOWE R.L., BARNES N.M. Selective labelling of 5-HT7 recognition sites in rat brain using [3H]-5-carboxamidotryptamine. Neuropharm. 1998;37:1611–1619. doi: 10.1016/s0028-3908(98)00117-8. [DOI] [PubMed] [Google Scholar]
- THOMAS D.R., GITTINS S.A., COLLIN L.L., MIDDLEMISS D.N., RILEY G., HAGAN J., GLOGER I., ELLIS C.E., FORBES I.T., BROWN A.M. Functional characterisation of the human cloned 5-HT7 receptor (long form); antagonist profile of SB-258719. Br. J. Pharmacol. 1998;124:1300–1306. doi: 10.1038/sj.bjp.0701946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D.R., MIDDLEMISS D.N., TAYLOR S.G., NELSON P., BROWN A.M. 5-CT stimulation of adenylyl cyclase activity in guinea pig hippocampus: evidence for involvement of 5-HT7 and 5-HT1A receptors. Br. J. Pharmacol. 1999a;128:158–164. doi: 10.1038/sj.bjp.0702759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D.R., BROMIDGE S.M., HAGAN J.J., MIDDLEMISS D.N., PRICE G.W. SB-269970-A is a potent and selective antagonist at human cloned and guinea pig brain 5-HT7 receptors. Br. J. Pharmacol. 1999b;128:232P. [Google Scholar]
- TO Z.P., BONHAUS D.W., EGLEN R.M., JAKEMAN L.B. Characterisation and distribution of putative 5-ht7 receptors in guinea-pig brain. Br. J. Pharmacol. 1995;115:107–116. doi: 10.1111/j.1476-5381.1995.tb16327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSOU A.P., KOSAKA A., BACH C., ZUPPAN P., YEE C., TOM L., ALVAREZ R., RAMSEY S., BONHAUS D.W., STEFANICH E., JAKEMAN L., EGLEN R.M., CHAN H.W. Cloning and expression of a 5-hydroxytryptamine7 receptor positively coupled to adenylyl cyclase. J. Neurochem. 1994;63:456–464. doi: 10.1046/j.1471-4159.1994.63020456.x. [DOI] [PubMed] [Google Scholar]
- WAEBER C., MOSKOWITZ M.A. Autoradiographic visualisation of [3H]-5-carboxamidotryptamine binding sites in guinea pig and rat brain. Eur. J. Pharmacol. 1995;283:31–46. doi: 10.1016/0014-2999(95)00275-p. [DOI] [PubMed] [Google Scholar]
- WATSON J.M., BURTON M.J., PRICE G.W., JONES B.J., MIDDLEMISS D.N. GR 127935 acts as a partial agonist at recombinant human 5-HT1Dα and 5-HT1Dβ receptors. Eur. J. Pharmacol. 1996;314:365–372. doi: 10.1016/s0014-2999(96)00579-1. [DOI] [PubMed] [Google Scholar]
- YING S.W., RUSAK B. 5-HT7 receptors mediate serotonergic effects on light-sensitive suprachiasmatic nucleus neurons. Brain Res. 1997;755:246–254. doi: 10.1016/s0006-8993(97)00102-9. [DOI] [PubMed] [Google Scholar]