

Abstract
Block of hKv1.5 channels by R-bupivacaine has been attributed to the interaction of the charged form of the drug with an intracellular receptor. However, bupivacaine is present as a mixture of neutral and charged forms both extra- and intracellularly.
We have studied the effects produced by the R(+) enantiomer of a quaternary bupivacaine derivative, N-methyl-bupivacaine, (RB+1C) on hKv1.5 channels stably expressed in Ltk− cells using the whole-cell configuration of the patch-clamp technique.
When applied from the intracellular side of the membrane, RB+1C induced a time- and voltage-dependent block similar to that induced by R-bupivacaine. External application of 50 μM RB+1C reduced the current at +60 mV by 24±2% (n=10), but this block displayed neither time- nor voltage-dependence.
External RB+1C partially relieved block induced by R-bupivacaine (61±2% vs 56±3%, n=4, P<0.05), but it did not relieve block induced by internal RB+1C. In addition, it did not induce use-dependent block, but when applied in combination with internal RB+1C a use-dependent block that increased with pulse duration was observed.
These results indicate that RB+1C induces different effects on hKv1.5 channels when applied from the intra or the extracellular side of the membrane, suggesting that the actions of bupivacaine are the resulting of those induced on the external and the internal side of hKv1.5 channels.
Keywords: Bupivacaine, hKv1.5, K+ channels, local anaesthetics
Introduction
Bupivacaine, an amide type local anaesthetic that blocks native cardiac Na+ and K+ channels at the same range of concentrations (Clarkson & Hondeghem, 1985; Sanchez-Chapula, 1988; Castle, 1990; Valenzuela et al., 1995a,1995b), is a weak base (pKa=8.1) that is predominantly present in its protonated form at the physiological pH. Moreover, the two enantiomers of bupivacaine block cardiac Na+ and hKv1.5 channels in a stereoselective manner, the R- enantiomer being 1.6 fold and 7 fold more potent than S-bupivacaine, respectively (Valenzuela et al., 1995a,1995b). The blocking properties of tertiary amine local anaesthetics (bupivacaine, ropivacaine, mepivacaine) and antiarrhythmic drugs (quinidine, zatebradine) on hKv1.5 channels are very similar to those previously described for tetraethylammonium (TEA) and its alkyl derivatives (QA) on Shaker K+ channels when they are applied from the inside of the membrane (Armstrong, 1971; Yellen et al., 1991; Snyders et al., 1992; Choi et al., 1993; Valenzuela et al., 1995a; 1996; 1997; Longobardo et al., 1998a). Block induced by these drugs is voltage-dependent, consistent with a fractional electrical distance (δ) ranging between 0.15 and 0.19, suggesting that the cationic form of these drugs have to penetrate ∼17% of the transmembrane electrical field until they can bind to the channel and block the ion pore. This common binding site has been located at the S6 transmembrane segment; particularly at positions T505 and V512 of the channel protein (Choi et al., 1993; Yeola et al., 1996). Moreover, both amino acids, together with L508, are the molecular determinants for the stereoselective block of hKv1.5 channels produced by bupivacaine (Franqueza et al., 1997). Together, these results support the role of these amino acids as molecular determinants for binding of local anaesthetics, antiarrhythmic drugs and hydrophobic TEA derivatives.
In addition, preliminary results obtained at different extracellular pH, to modify the extracellular and the intracellular concentrations of the charged and uncharged forms of R-bupivacaine, suggested the existence of an external binding site for the drug in hKv1.5 channels (Longobardo et al., 1998b). In order to test this hypothesis without the added complication of concomitant effects of pH changes, in the present study we have analysed the effects of the R(+) enantiomer of a membrane-impermeant permanently charged N-methyl-bupivacaine (RB+1C) derivative on hKv1.5 channels stably expressed in Ltk− cells. Preliminary results of the present study have been published in abstract form (Longobardo et al., 1999).
Methods
Electrophysiological recording
Use of the stable Ltk− cell lines expressing wild type hKv1.5, Kv2.1 and various mutants has been described previously in detail (Snyders et al., 1993; Yeola et al., 1996; Franqueza et al., 1997). The intracellular pipette filling solution contained (in mM): K-aspartate 80, KCl 50, phosphocreatine 3, KH2PO4 10, MgATP 3, HEPES-K 10, EGTA 5 and was adjusted to pH 7.25 with KOH. The bath solution contained (in mM): NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1, HEPES-Na 10, and glucose 10, and was adjusted to pH 7.40 with NaOH.
Experiments were performed in a small volume (0.5 ml) bath mounted on the stage of an inverted microscope (Nikon model TMS, Garden City, NY, U.S.A.) perfused continuously at a flow rate of 0.5–1.0 ml min−1. hKv1.5 currents were recorded at room temperature (20–22°C) using the whole-cell voltage-clamp configuration of the patch-clamp technique (Hamill et al., 1981) with an Axopatch 1C patch-clamp amplifier (Axon Instruments, Foster City, CA, U.S.A.). Currents were filtered at 2 kHz (four-pole Bessel filter), sampled at 4 kHz. Data acquisition and command potentials were controlled by the PCLAMP 6.0.1 software (Axon Instruments). Micropipettes were pulled from borosilicate glass capillary tubes (Narishige, GD-1, Tokyo, Japan) on a programmable horizontal puller (Sutter Instrument Co., San Rafael, CA, U.S.A.) and heat-polished with a microforge (Narishige). When filled with the intracellular solution and immersed into the bath (external solution), the pipette tip resistance ranged between 1 and 3 MΩ. The micropipettes were gently lowered onto the cells to obtain a gigaohm seal (16±6 GΩ) after applying suction. After seal formation, cells were lifted from the bottom of the perfusion bath and the membrane patch was ruptured with brief additional suction. The capacitive transients elicited by symmetrical 10-mV steps from −80 mV were recorded at 50 kHz (filtered at 10 kHz) for subsequent calculation of capacitative surface area, access resistance, and input impedance. Thereafter, capacitance and series resistence compensation were optimized, and 80% compensation of the effective access resistance was usually obtained.
Drugs
Permanently charged R-bupivacaine [R-(+)-1-butyl-1-methyl-2′,6′-pipecoloxylidide] (Astra, Södertälje, Sweden) was dissolved in ethanol (50%) to yield a stock solution of 10 mM from which further dilutions were made to obtain the desired final concentration. R-bupivacaine (Astra, Södertälje, Sweden) was dissolved in distilled deionized water to yield stock solutions of 10 mM from which further dilutions were made to obtain the desired final concentration.
Pulse protocol and analysis
The holding potential was maintained at −80 mV unless indicated otherwise. After control data were obtained, bath perfusion was switched to drug-containing solution. The effects of drug infusion was monitored with test pulses to +60 mV, applied every 10 s until steady-state was obtained. Steady-state current-voltage relationships (IV) were obtained by averaging the current over a small window (2–5 ms) at the end of 250 ms depolarizing pulses. Between −80 and −40 mV only passive linear leak was observed and least squares fits to these data were used for passive leak correction. Deactivating ‘tail' currents were recorded at −40 mV. The activation curve was obtained from the tail current amplitude immediately after the capacitive transient. Measurements were done using the CLAMPFIT program of PCLAMP 6.0.1 and by a custom-made analysis program.
The activation kinetics of hKv1.5 channels display a sigmoidal time-course which can be quantitated with the dominant time constant derived from fitting a single exponential to the current after the initial delay (Snyders et al., 1993; Delpon et al., 1996; Valenzuela et al., 1996). Other kinetic processes were generally fitted using a sum of exponentials of the form:
where τ1, τ2 … τn are the system time constants, A1, A2 … An are the amplitudes of each component of the exponential, and C is the baseline value. The voltage dependence of activation curves were fitted with a Boltzmann equation: y=1/[1+exp (−(E−Eh)/s)], in which s represents the slope factor, E the membrane potential and Eh the voltage at which 50% of the channels are open. The curve-fitting procedure used a non-linear least-squares (Gauss-Newton) algorithm; results were displayed in linear and semilogarithmic format, together with the residuals of the fit. Goodness of fit was judged by the χ2 criterion and by inspection for systematic non-random trends in the residuals.
Voltage dependence of block was determined as follows: leak-corrected current in the presence of drug was normalized to matching control to yield the fractional block at each voltage (f=1−Idrug/Icontrol). The voltage dependence of block was fitted to:
where z, F, R and T have their usual meaning, δ represents the fractional electrical distance, i.e., the fraction of the transmembrane electrical field sensed by a single charge at the receptor site and KD* represents the apparent dissociation constant at the reference potential (0 mV).
Statistical methods
Results are expressed as mean±s.e.mean. Direct comparisons between mean values in control conditions and in the presence of drug for a single variable were performed by paired Student's t-test. Student's two tail t-test was also used to compare two regression lines. Differences were considered significant if P<0.05.
Results
Effects of externally applied RB+1C on hKv1.5 channels
External perfusion of hKv1.5 expressing Ltk− cells with RB+1C (RB+1Cout) readily induced a blockade that reached steady state after 3–4 min and was reversible in approximately 4 min upon perfusion with drug-free solution (92±1%; n=10), consistent with the expectation that this quaternary drug does not penetrate into the cells. Figure 1a shows original current traces obtained in the absence and in the presence of 50 and 500 μM RB+1C in the external solution. At 50 and 500 μM the quaternary bupivacaine derivative inhibited hKv1.5 current by 24±2% (n=10) and 49±8% (n=3), respectively, when measured at the end of 250 ms depolarizing pulses from −80 mV to +60 mV. Figure 1b shows the concentration-dependent induced block by RB+1Cout. Figure 2a shows hKv1.5 current records obtained in the absence, in the presence of 50 μM RB+1Cout and after 5 min of perfusion with drug-free external solution. RB+1Cout induced a decrease of the current that was time-independent, i.e. it scaled down the activated current during the depolarizing steps without inducing the time-dependent open channel block as previously described with R-bupivacaine (Valenzuela et al., 1995a). Figure 2b shows the IV relationship of hKv1.5 current obtained in the absence and in the presence of 50 μM RB+1Cout. At this concentration, RB+1Cout decreased the ionic current at all membrane potentials tested. When representing the relative current (IDrug/IControl) against membrane potential we observed an increase of block coinciding with the range of activation of the hKv1.5 current which reached a plateau at around 0 mV (Figure 2c). Thus, RB+1Cout induced block was not voltage dependent at membrane potentials at which the activation curve reached saturation (positive to 0 mV), in contrast to the voltage dependent block exhibited by R-bupivacaine (Valenzuela et al., 1995a).
Figure 1.
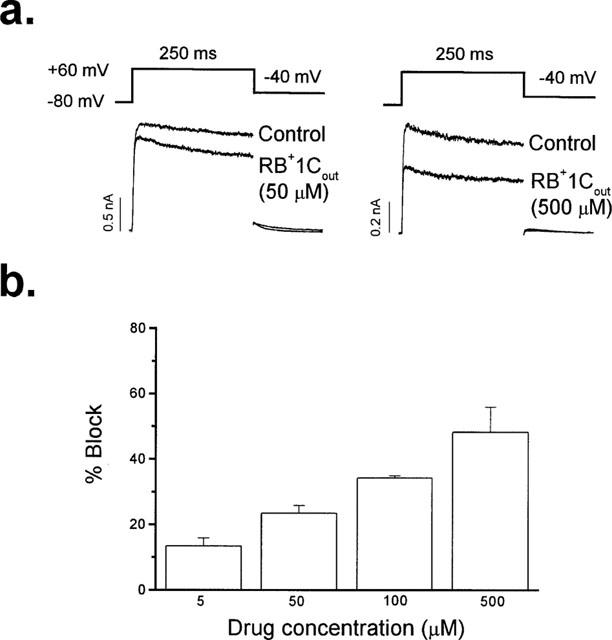
Concentration-dependent effects of externally applied RB+1C (RB+1Cout) in hKv1.5 channels. (a) Original hKv1.5 current traces obtained in the absence and in the presence of RB+1Cout (50 and 500 μM) recorded after depolarizing the membrane potential from −80 mV to +60 mV during 250 ms. Deactivating tail currents were recorded after repolarizing to −40 mV. (b) Histogram representing the concentration dependent block induced by RB+1Cout. Columns represent the mean±s.e.mean of 3–10 experiments.
Figure 2.
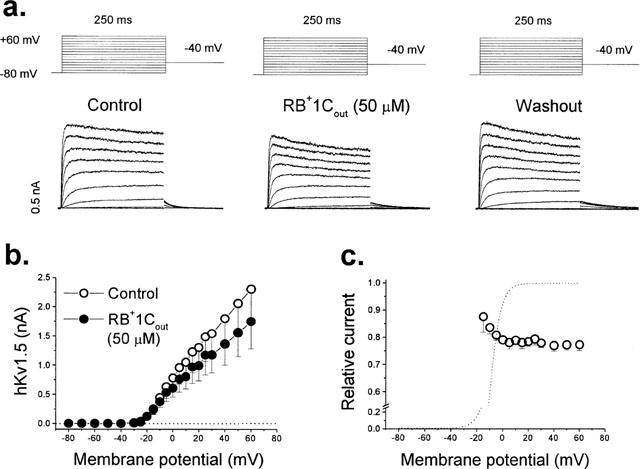
Effects of RB+1Cout on hKv1.5 channels. (a) Original records obtained in control conditions, in the presence of 50 μM RB+1Cout and after washout with drug-free external solution. Cells were held at −80 mV and 250 ms pulses every 10 s to membrane potentials between −80 and +60 mV in 10-mV steps, were applied. Depolarizing tail currents were recorded after repolarizing to −40 mV. (b) IV relationships obtained in the absence and in the presence of RB+1Cout. (c) Voltage dependence of block represented as the relative current at all membrane potentials studied. Individual data points represent the mean±s.e.mean of 10 experiments.
The effects induced by RB+1Cout could be explained if the drug binds either to an external domain in hKv1.5 channels or to the internal binding site, accessible from the external mouth of the channel. The latter explanation seems unlikely since RB+1Cout does not induce the typical time- and voltage-dependent open channel block observed in the presence of R-bupivacaine (Valenzuela et al., 1995a). To test this hypothesis further we studied the effects of RB+1Cout on hKv1.5 channels with pore mutations that alter the affinity of R-bupivacaine for its internal binding site (Franqueza et al., 1997). Figure 3 shows the effects of 50 μM RB+1Cout on the S6 mutants T505A and V512M, and the P-loop mutant T477S. Neither of these internal pore mutations altered the blocking effect of externally applied RB+1C. Thus, block was 26±2% (n=3, P>0.05), 19±1% (n=3, P>0.05), and 25±5% (n=3, P>0.05) in T505A, V512M and T477S channels, respectively (compared with 24% for WT hKv1.5 channels, see above).
Figure 3.
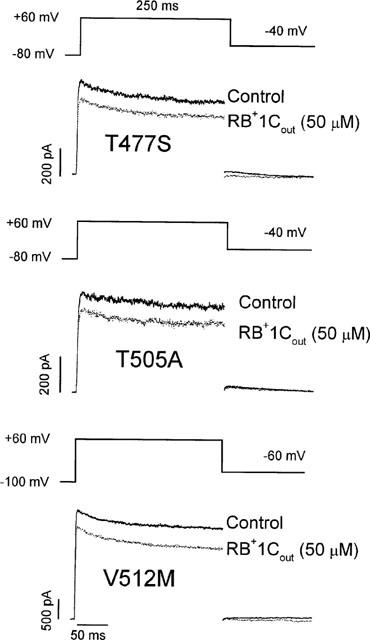
Effects of RB+1Cout (50 μM) in hKv1.5 pore mutant channels located at the P-loop (T477S) and the S6 segment (T505A and V512M). Current traces were elicited after applying 250 ms depolarizing pulses from a holding potential of −80 mV (T477S and T505A) or −100 mV (V512M) to +60 mV. Tail currents were recorded at −40 mV (T477S and T505A) or at −60 mV (V512M).
Whereas the internal pore sequence is very conservative among different Kv channel isoforms, K+ channels vary greatly in the external pore sequence and the amino acids at the extracellular end of the S4 helix (Deal et al., 1996). Therefore, in order to assess if the effects induced by RB+1Cout in hKv1.5 channels are due to an interaction with some domain of the external face of the channel or a non-specific effect on the cell membrane, we analysed the effects of externally applied RB+1C in another Kv channel, Kv2.1, stably expressed in the same cell line (Ltk−). Figure 4a shows original traces obtained in the absence and in the presence of 50 μM RB+1Cout. As it can be observed, RB+1Cout induced a significantly higher degree of block of Kv2.1 channels measured at +60 mV when compared to the blocking effects on hKv1.5 channels (39±2% vs 24±2%, P<0.01). Figure 4b shows the IV relationship of Kv2.1 current obtained in the absence and in the presence of 50 μM RB+1Cout. At this concentration, RB+1Cout decreased the ionic current at all membrane potentials tested. When representing the relative current against membrane potential we observed a steep voltage dependence block coinciding with the range of activation of the Kv2.1 current and a shallower one at membrane potentials positive to 0 mV consistent with a δ value of 0.21±0.02 (n=8) from the external side of the membrane (Figure 4c).
Figure 4.
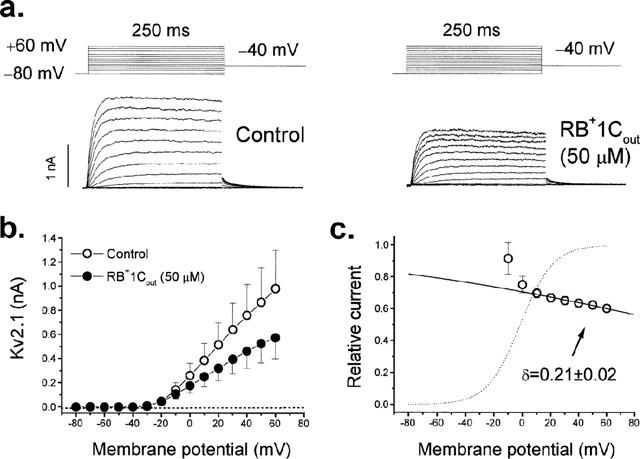
Effects of RB+1Cout on Kv2.1 channels. (a) Original records obtained in control conditions and in the presence of 50 μM RB+1Cout. (b) IV relationships obtained in the absence and in the presence of RB+1Cout. The dotted line represents the zero level. (c) Voltage dependence of block represented as the relative current at all membrane potentials studied. The dotted line represents the activation curve and the solid line is the Boltzmann fit to data points positive to 0 mV. Individual data points represent the mean±s.e.mean of eight experiments.
Effects of internally applied RB+1C in hKv1.5 channels
The effects of RB+1C on the intracellular side of the cell membrane were studied by adding the drug at the desired concentration to the pipette solution (RB+1Cin). In these experiments the tip of the pipette was filled with drug-free internal solution by dipping the pipette tip for ∼2 s into the normal internal solution, in order to obtain ‘control' records. Therefore, the first traces obtained after breaking the membrane seal were considered as the ‘control' for each experiment. Under these conditions, RB+1Cin induced a decrease of the current which reached steady state 15 min after breaking the seal (Figure 5a). Moreover, and contrary to extracellular application, RB+1Cin induced a time-dependent decay of the current at the beginning of the depolarizing pulse which became faster as the drug concentration was increased (Figure 5a, b). The time constants of this initial fast decline can be taken as a good approximation of the time constant of block (τB) (Snyders et al., 1992; Valenzuela et al., 1995a) and averaged 25.6±3.8 ms (n=12), 15.3±1.6 ms (n=6) and 5.1±0.4 ms (n=5) in the presence of 20, 100 and 500 μM RB+1Cin, respectively. Under these experimental conditions 100 μM and 500 μM RB+1Cin induced an inhibition of the current of 18±3% (n=8) and 66±4% (n=5), when measured at +60 mV against the ‘control' records. The concentration-dependence of block by RB+1Cin is shown in Figure 5c.
Figure 5.
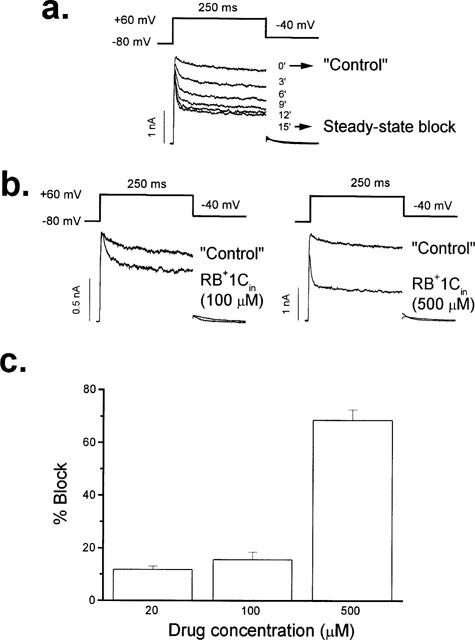
Effects of internally applied RB+1C (RB+1Cin) in hKv1.5 channels. In these experiments, the pipette was filled with an internal solution containing RB+1C and the tip of the pipette was filled with drug-free internal solution. (a) Development of block induced by RB+1Cin (500 μM) during the first 15 min after rupture of the seal membrane, which was taken as time 0′. (b) Original hKv1.5 current traces obtained in the absence and in the presence of RB+1Cin at 100 μM and 500 μM. (c) Histogram representing the concentration dependent block induced by RB+1Cin. Columns represent the mean±s.e.mean of 5–12 experiments.
Figure 6 summarizes the effects of 500 μM RB+1Cin. Figure 6a shows that the time dependent decline of the current is more marked at more positive potentials (when channel activation is much faster than the time course of drug-induced block). Figure 6b shows the IV relationship obtained just after breaking the seal (‘control') and after 15 min of internal perfusion with RB+1Cin. It can be observed that RB+1Cin decreased hKv1.5 currents at all membrane potentials tested. When measured at +60 mV, RB+1Cin induced block averaged 66±4% (n=5). In contrast to the results for externally applied RB+1C (see Figure 2c), block induced by RB+1Cin was voltage-dependent over the whole range of membrane potentials between −20 and +60 mV. This voltage dependence was consistent with a δ value of 0.34±0.04 (n=8), measured from the inside of the membrane. Thus, the characteristics of RB+1C induced block of hKv1.5 channels differed depending whether the drug was externally or internally applied, and only the blockade induced by RB+1Cin was voltage- and time-dependent similarly to that previously described for R-bupivacaine (Valenzuela et al., 1995a).
Figure 6.
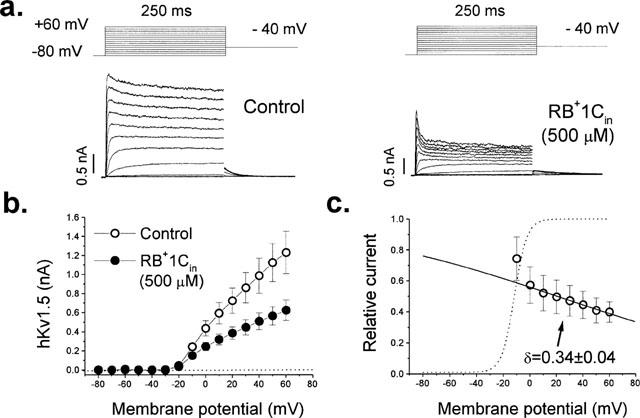
Effects of RB+1Cin on hKv1.5 channels. (a) Original records obtained just after breaking the seal (‘control') and when the steady state block induced by 500 μM RB+1Cin was achieved (∼15′). (b) IV relationships under ‘control' conditions and after 15′ of dialysis with RB+1C-containing internal solution. (c) Voltage dependence of block, plotted as the relative current at all membrane potentials studied. This voltage dependence was consistent with a fractional electrical distance (δ) of 0.34±0.04. Individual data points represent the mean±s.e.mean of six experiments.
Competition between R-bupivacaine and RB+1C
In the extracellular solution, R-bupivacaine is present as a mixture of charged and uncharged forms, although at physiological pH it predominates in its cationic form. Since any effects of R-bupivacaine binding at the putative external binding site are confounded by its open channel block properties, we analysed its interaction with RB+1C at the external binding site. Thus, we performed competition experiments between both agents. In these experiments we first applied R-bupivacaine at a concentration (5 μM) close to its apparent KD (Valenzuela et al., 1995a). After block reached steady-state, we perfused the cells with an external solution containing both R-bupivacaine and RB+1C. Figure 7 (left top panel) shows original records obtained in the presence of 5 μM R-bupivacaine and in the presence of R-bupivacaine plus RB+1Cout (50 μM). If RB+1Cout acts independently from R-bupivacaine, the total amount of block would be expected to increase by approximately 25% (see Figure 1b) when RB+1Cout is added. Contrary to this expectation, block induced by R-bupivacaine was slightly relieved after addition of RB+1Cout (from 61±2% to 56±3%, n=4; P<0.05).
Figure 7.
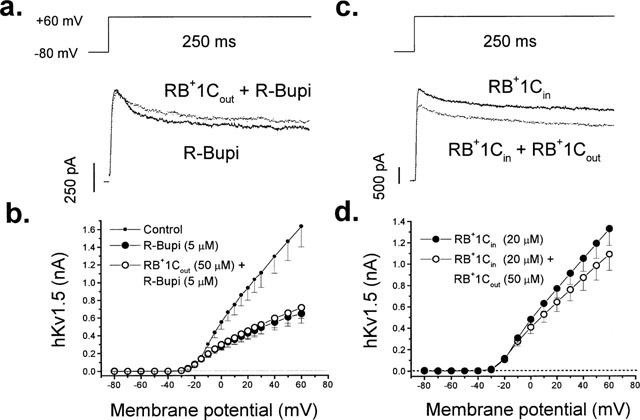
Competition between R-bupivacaine and RB+1Cout (left panels) and effects of RB+1Cout in cells dialyzed with RB+1Cin (right panels). (a) Original current records elicited by the application of a depolarizing pulse from −80 to +60 mV in the presence of R-bupivacaine (5 μM) alone and in the presence of the combination of R-bupivacaine plus RB+1Cout (50 μM). (b) The IV relationship obtained under control conditions, in the presence of R-bupivacaine and in the presence of the combination of R-bupivacaine plus RB+1Cout. Individual data points represent the mean±s.e.mean of four experiments. (c) Original records obtained in the presence of 20 μM RB+1Cin alone and in the presence of RB+1Cin plus RB+1Cout. (d) Shows the IV relationship obtained in cells internally perfused with RB+1Cin in the absence and in the presence of RB+1Cout. Individual data points represent the mean±s.e.mean of 10 experiments.
These results could be due to the competition between RB+1Cout and R-bupivacaine at an hypothetical bupivacaine binding external site. Alternatively, binding of RB+1Cout at some external domain of hKv1.5 could (allosterically) reduce the affinity of R-bupivacaine for its internal binding site. To address this possibility we analysed the effects of RB+1Cout (50 μM) in cells dialyzed with 20 μM RB+1Cin. If this hypothesis is correct, we would expect an increase of the current upon perfusion with RB+1Cout. As shown in Figure 7 (right top panel), RB+1Cout induced-block of hKv1.5 channels in cells dialyzed with 20 μM RB+1Cin was similar to that induced in cells internally perfused with drug-free internal solution (21±3% vs 24±2% with and without RB+1C in the internal solution; n=8, respectively, P>0.05) (compare Figures 1 and 7). Similar results were obtained in cells dialyzed with 500 μM RB+1Cin [29±6% (n=4) vs 24±2% (n=10) with and without RB+1Cin; P>0.05]. These results indicate that RB+1Cout does not noticeably alter the affinity of R-bupivacaine for its internal binding site.
Lack of use-dependent effects induced by RB+1Cout on hKv1.5 channels
Many local anaesthetics and antiarrhythmic drugs induce use-dependent accumulation of block during repetitive pulse trains. To test whether this also occurs in the presence of RB+1Cout, we studied its use-dependent effects when applying trains of 15 pulses of different pulse duration (250 and 1000 ms) with a fixed interpulse duration of 1000 ms. RB+1Cout (50 μM) only induced a significant use-dependent inhibition of the hKv1.5 current after applying a 250 ms pulse duration train (11.3±1.5% vs 16.1±2.4%; n=6; P<0.05). With trains of longer pulse duration (1000 ms) it only slightly decreased the current (18.3±0.6 vs 20.3±1.6%; n=5; P>0.05) (Figure 8a). These differences are better shown in Figure 8b that shows the relative current elicited during the application of the train protocols as a function of the number of pulses in the train. The small reduction of current in control conditions presumably reflects an accumulation of channels in the C-type inactivated state. Since the recovery interval was identical, the reduced use-dependence with longer steps (which promote C-type inactivation) suggests that external RB+1C block and C-type inactivation are mutually exclusive.
Figure 8.
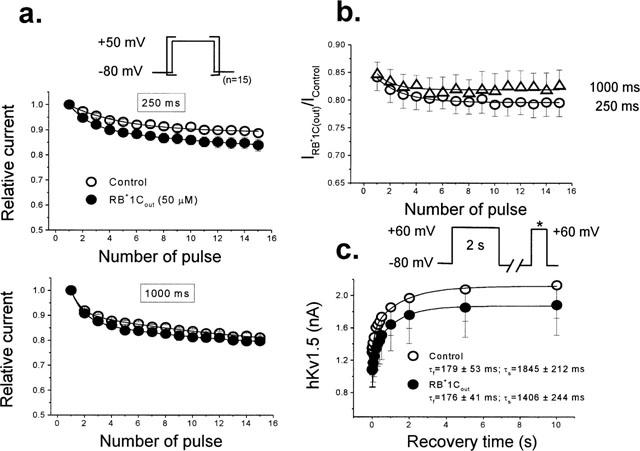
Lack of use-dependent effects of RB+1Cout in hKv1.5 channels. (a) Normalized outward hKv1.5 peak current amplitude in control conditions and in the presence of RB+1Cout (50 μM) during trains of 15 pulses of 250 ms and 1000 ms in duration applied from −80 to +50 mV in which the depolarizing steps were separated from each other by a fixed interstimulus interval of 1000 ms. (b) Relative current (IDrug/IControl) plotted as a function of the number of pulses in the train. (c) Recovery process of hKv1.5 current under control conditions and in the presence of RB+1Cout. The maximum peak amplitude of outward hKv1.5 currents elicited by a test pulse was plotted as a function of the interstimulus interval in the absence and presence of RB+1Cout. Continuous lines represent the best biexponential fit of the increase of hKv1.5 currents as a function of the interstimulus interval from which the recovery time constants were obtained. Individual data points represent the mean±s.e.mean of five experiments.
The kinetics of the recovery process of RB+1Cout blocked channels were also compared to those observed under control conditions. The kinetics of the recovery process using the following experimental protocol: a conditioning prepulse of 2000 ms in duration from −80 mV to +60 mV was followed by a fixed test pulse of 250 ms in duration to +60 mV after an interstimulus interval of variable duration (between 1 and 10,000 ms) at −80 mV. Figure 8c shows the mean results obtained in five cells. hKv1.5 channels recovered following a biexponential kinetics, with a τfast of 179±53 ms and a τslow of 1845±212 ms (n=5). After perfusion of the cells with RB+1Cout (50 μM), neither τfast (176±41 ms; n=5; P>0.05) nor τslow (1406±244 ms; n=5; P>0.05) were modified. Moreover, the contribution of the fast component to the total recovery process was similar in the absence and in the presence of RB+1Cout (41±3 vs 36±4%; n=5; P>0.05).
Use dependent effects of RB+1Cout in cells internally dialyzed with RB+1C
Figure 9a shows that the use-dependent inhibition of hKv1.5 current by RB+1Cin (20 μM) was increased when trains of longer pulses were applied (12±1% and 30±2% after applying trains of pulses of 250 and 1000 ms in duration; n=5). After addition of RB+1Cout (50 μM), the degree of inhibition of the current observed during the application of trains of pulses of 250 ms in duration was similar to that obtained in cells not internally dialyzed with RB+1C (e.g. Figure 8a). Since the open channel block proceeded with a τB of 25 ms (Figure 4), we would expect similar steady-state block with both 250 and 1000 ms pulses. However, the inhibition of the current induced by RB+1Cout observed in cells with RB+1Cin after applying trains of 1000 ms pulses was significantly higher than that obtained in non drug-dialyzed cells (18±1 vs 30±2%, in the absence and in the presence of RB+1Cin, respectively, n=5, P<0.01). The marked enhancement of block with the longer pulse duration suggests an interaction between internal RB+1C block and C-type inactivation, e.g. drug-induced block enhances C-type inactivation (Baukrowitz & Yellen, 1996a). Perfusion of these cells with RB+1Cout (50 μM) induced a further inhibition of the current that was also higher after applying trains of longer depolarizing pulses (19±2 and 43±4% after applying trains of 250 and 1000 ms duration pulses, respectively, P<0.01). These results are in contrast to those obtained in the absence of RB+1Cin (see Figure 8b) and suggest that RB+1Cin might induce a conformational change of the ion channel that modifies the RB+1Cout effects.
Figure 9.
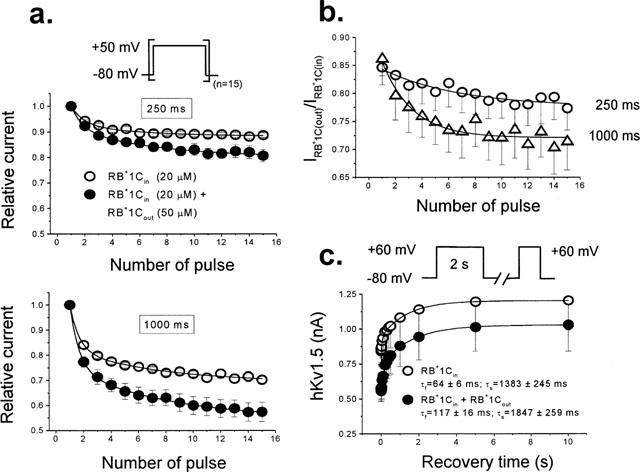
Use-dependent effects of RB+1Cout in hKv1.5 channels in cells dialyzed with RB+1Cin. (a) Outward hKv1.5 peak current amplitude in the presence of RB+1Cin (20 μM) alone and the combination of RB+1Cin plus RB+1Cout (50 μM) when applying trains of 15 pulses of 250 and 1000 ms in duration applied from −80 to +50 mV in which depolarizing steps were separated each other by a fixed interstimulus interval of 1000 ms. (b) Relative current plotted as a function of the number of pulses in the train. (c) Recovery process of hKv1.5 current in the presence of RB+1Cin and of RB+1Cin plus RB+1Cout. The maximum peak amplitude of outward hKv1.5 currents elicited by a test pulse was plotted as a function of the interstimulus interval in both experimental conditions. Continuous lines represent the best biexponential fit of the increase of hKv1.5 currents as a function of the interstimulus interval from which the time constants of recovery were obtained. Individual data points represent the mean±s.e.mean of five experiments.
Use-dependent block suggests that the recovery process of blocked channels is slower than the return to the rested state of drug-free channels. To explore this possibility, we analysed the recovery kinetics of the hKv1.5 current using the experimental protocol as described above. Figure 9c shows the mean results obtained in seven experiments. The hKv1.5 current recovered following a biexponential kinetics in cells dialyzed with RB+1Cin (20 μM), with a τfast of 64±6 ms and a τslow of 1383±245 ms (n=7), which were faster than those calculated under control conditions (179±53 ms, P<0.05, and 1845±212 ms, P>0.05, for the τfast and τslow, respectively). In the presence of the combination of RB+1Cin plusRB+1Cout (50 μM), the τfast became slower (117±16 ms, n=7; P<0.05), whereas the τslow remained unchanged (1847±259 ms, n=7; P>0.05). In both cases, the contribution of the fast component to the total recovery process was similar (30±2 vs 39±5%; n=7; P>0.05). This slowing of the τfast might explain the use dependent block induced by RB+1Cout in this series of experiments.
Discussion
The main findings of the present study are: (1) RB+1Cout induced-block of hKv1.5 channels was not time- or voltage-dependent, (2) RB+1Cin blocks hKv1.5 channels with higher potency than RB+1Cout, in a time- and voltage-dependent manner consistent with an open channel block mechanism and a fractional electrical distance of 0.33 measured from the inside of the cell. These contrasting effects of RB+1C observed when applied from the external and the internal side of the membrane, together with those obtained from the competition experiments between R-bupivacaine and RB+1C, suggest that some of the electrophysiological effects induced by R-bupivacaine are due to its charged form acting on the external face of hKv1.5 channels. The increase in use-dependent block produced by RB+1Cout in cells dialyzed with RB+1Cin suggests that drug binding to the internal bupivacaine binding site may induce a conformational change of the ion channel that modifies the effects of RB+1Cout.
Effects of RB+1C on the external and the internal face of hKv1.5 channels
The effects observed on hKv1.5 channels after perfusion with RB+1Cout developed very fast and were readily reversible after perfusion of the cells with drug-free external solution, consistent with the idea that this quaternary compound cannot cross the cell membrane. RB+1Cout inhibited hKv1.5 current in a time independent manner upon applying depolarizing pulses, so that current amplitude was simply scaled down. On the contrary, block induced by RB+1Cin exhibited a time dependent decay at the beginning of the depolarizing pulses that was faster as the concentration of RB+1Cin was increased and similar to that produced by R-bupivacaine (Valenzuela et al., 1995a). These different effects observed when RB+1C was added to the external or to the internal side of the membrane may suggest that it binds to two different binding sites (external and internal) which mediate different types of channel blockade.
Permanently charged and membrane impermeant drugs have been widely used to study block of voltage-gated Na+ channels by local anaesthetics. The lidocaine analogue, QX-314, binds to a single receptor site in brain rH1 Na+ channels that is accessible both from the external and from the internal side of the ion pore (Qu et al., 1995). If a similar situation occurs for block of hKv1.5 channels by RB+1C, we should observe the same type of block both when RB+1C was applied from the extracellular or the intracellular side of the ion pore. However, when RB+1C was applied from the external side we did not observe the time-dependent block observed when the drug was applied from the inside of the membrane (see Figure 6). Moreover, mutations in the domain S6 (T505A and V512M), which reduce the affinity for R-bupivacaine (Franqueza et al., 1997), did not modify the effects of RB+1Cout. Taken together, these data suggest that externally applied RB+1C does not have access to the internal bupivacaine binding site and that its effects are most likely due to its interaction with an external binding site (Franqueza et al., 1997). Another possible explanation would be that RB+1Cout will penetrate into the lipid bilayer inducing some non-specific effect on hKv1.5 channels. However, the finding that RB+1Cout produced a 2 fold increase in the degree of block of Kv2.1 channels as compared to that induced in hKv1.5 channels expressed in the same cell line seems to support the hypothesis of an interaction between the drug and some external domain of the ion channel.
Furthermore, in contrast to the results obtained in hKv1.5 channels, the blockade induced by RB+1Cout in Kv2.1 channels was voltage dependent at potentials positive to 0 mV. These differential effects could be due to the different amino acid sequence at the external side between these two isoforms of K+ channels. The equivalent position to threonine 449 (T449) in hKv1.5 and Kv2.1 channels is an arginine (R485) and a tyrosine (Y369), respectively. In fact, when the T449 of Shaker K+ channels is replaced by a tyrosine, the affinity for external TEA is increased by nearly 50 fold and the voltage dependence of block is decreased from a δ value of 0.19 to 0.04 (Heginbotham & MacKinnon, 1992). These authors explained the decrease in δ values by the different amino acid present in the outer mouth of the ion channel, that represents the external TEA binding site in Shaker channels. Under this proposed framework, we could explain the different voltage dependence induced by RB+1Cout in hKv1.5 and Kv2.1 channels, by their distinct external amino acid sequence, in such a way that RB+1Cout could enter more deeply into the pore and therefore induce a more voltage dependent block.
The results obtained in the competition experiments between R-bupivacaine and RB+1Cout suggest that the cationic moiety of R-bupivacaine interacts similarly to RB+1Cout with some external domain of hKv1.5 channels. Moreover, the results from Figure 7 suggest that the internal pore block per se (with permanently charged RB+1Cin) does not modify the external blocking effect of RB+1C. However, if both the cationic form of R-bupivacaine and RB+1Cout act as external channel blockers, then adding both drugs should result in an increase of block rather than the small relief of block observed in the present experiments (Figure 6). These results could be explained if both, the charged form of R-bupivacaine and RB+1C inhibit the C-type inactivation of hKv1.5 channels as previously reported for quaternary ammonium derivatives in Shaker channels (Baukrowitz & Yellen, 1996b). Then, if RB+1C is more potent than the charged form of R-bupivacaine to induce this effect, in the presence of both agents they would compete for binding to an external domain. Alternatively, if there exists an external bupivacaine receptor, it could exhibit two binding sites for which RB+1C will exhibit negative cooperativity.
In addition to its blocking effects, RB+1Cin induced an acceleration of the fast component of the recovery process (Figure 9), but at the same time there was accumulation of use-dependent block during trains with long depolarizing steps. However, this kinetic change would lead, theoretically, to less accumulation of use dependent inhibition of the current. Therefore, the observed acceleration of the fast time of the recovery process likely involves a drug-induced change of the channel kinetics independent of its blocking properties, which could derive from an interaction with some structural domain of the channel involved in the channel gating.
Effects of RB+1Cout in cells internally perfused with RB+1C
Block of hKv1.5 channels induced by RB+1Cout in cells internally perfused with RB+1C exhibited several differences from that induced in control cells. The steady state block observed in the IV relationships was similar in both situations. However, in cells internally perfused with RB+1C, we observed a use dependent block which increased at longer depolarizing train pulses, in contrast to the lack of use dependent block of hKv1.5 channels observed in cells not internally dialyzed with RB+1C (compare Figures 8 and 9). These results suggest that binding of RB+1Cin to the internal bupivacaine receptor site induces a conformational change of the channel protein which modifies the effects of RB+1Cout on channel gating as well as its blocking effects. One possible explanation would be that binding of RB+1Cin to its internal binding site would enhance the affinity of RB+1Cout for the inactivated state of the channel. Alternatively, the present results could also be explained by drug-induced changes in channel kinetics which would favour the transition to the inactivated state. In both cases, binding of RB+1Cin is assumed to promote changes in hKv1.5 channel gating. It has been demonstrated that K+ and β subunits induce changes in potassium channel gating that primarily affect the inactivation process (Uebele et al., 1996). In fact, K+ modifies Shaker channel gating after binding to its external and internal binding sites (Choi et al., 1991; Lopez-Barneo et al., 1993; Baukrowitz & Yellen, 1995; 1996b; Chen et al., 1997; Harris et al., 1998; Kiss & Korn, 1998). Moreover, modifications in the external or internal K+ concentrations abolish the ‘agonist' effect of low concentrations of benzocaine and influence its blocking properties (Delpon et al., 1999). Therefore, a possible mechanism by which RB+1C alters hKv1.5 channel gating could also involve an interaction with K+ external and/or internal binding sites, which themselves are linked to inactivation (Baukrowitz & Yellen, 1996b).
Conclusions
The present study demonstrates that RB+1Cin causes open channel block of hKv1.5 channels similar to that caused by R-bupivacaine, presumably by binding to the same internal binding site. RB+1Cout induced a blockade that was not time- or voltage-dependent, but which may compete with the C-type inactivation of hKv1.5 channels. The contrasting effects observed with RB+1C when added from the external and the internal side of the membrane, together with the results obtained in hKv1.5 S6 mutant and Kv2.1 channels, suggest the existence of an external binding site for bupivacaine in hKv1.5 channels. The fact that the blocking effects of application of RB+1Cout in cells dialyzed with RB+1Cin are not purely additive suggests that binding of RB+1C to the internal binding site induces a conformational change of the ion channel that might modify the RB+1Cout effects.
Acknowledgments
The authors want to express their thanks to Guadalupe Pablo for her excellent technical assistance. We also thank Astra Pain Control for supplying us with R-bupivacaine and R-(+)-1-Butyl-1-Methyl-2′,6′-pipecoloxylidide (Dr R. Sandberg, Södertälje, Sweden). This work was supported by CICYT SAF98-0058 (C. Valenzuela), CICYT SAF99-0069 (J. Tamargo), CAM 08.4/0016198 (E. Delpón), NIH HL49330 (M.M. Tamkun) and U.S.-Spain Science and Technology Program 98131 (M.M. Tamkun and C. Valenzuela) Grants.
Abbreviations
- RB+1C
R-(+)-1-butyl-1-methyl-2′,6′-pipecoloxylidide
- QA
Quaternary ammonium derivatives
- TEA
Tetraethylammonium
- δ
Fractional electrical distance
- τB
Time constant of block
References
- ARMSTRONG C.M. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUKROWITZ T., YELLEN G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- BAUKROWITZ T., YELLEN G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996a;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- BAUKROWITZ T., YELLEN G. Two functionally distinct subsites for the binding of internal blockers to the pore of voltage-activated K+ channels. Proc. Natl. Acad. Sci. U.S.A. 1996b;93:13357–13361. doi: 10.1073/pnas.93.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASTLE N.A. Bupivacaine inhibits the transient outward K+ current but not the inward rectifier in rat ventricular myocytes. J. Pharmacol. Exp. Ther. 1990;255:1038–1046. [PubMed] [Google Scholar]
- CHEN F.S., STEELE D., FEDIDA D. Allosteric effects of permeating cations on gating currents during K+ channel deactivation. J. Gen. Physiol. 1997;110:87–100. doi: 10.1085/jgp.110.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K.L., ALDRICH R.W., YELLEN G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K.L., MOSSMAN C., AUBE J., YELLEN G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- CLARKSON C.W., HONDEGHEM L.M. Mechanism for bupivacaine depression of cardiac conduction: fast block of sodium channels during the action potential with slow recovery from block during diastole. Anesthesiology. 1985;62:396–405. [PubMed] [Google Scholar]
- DEAL K.K., ENGLAND S.K., TAMKUN M.M. Molecular physiology of cardiac potassium channels. Physiol. Rev. 1996;76:49–76. doi: 10.1152/physrev.1996.76.1.49. [DOI] [PubMed] [Google Scholar]
- DELPON E., CABALLERO R., VALENZUELA C., LONGOBARDO M., SNYDERS D., TAMARGO J. Benzocaine enhances and inhibits the K+ current through a human cardiac cloned channel (Kv1.5) Cardiovasc. Res. 1999;42:510–520. doi: 10.1016/s0008-6363(99)00043-7. [DOI] [PubMed] [Google Scholar]
- DELPON E., VALENZUELA C., PEREZ O., FRANQUEZA L., GAY P., SNYDERS D.J., TAMARGO J. Mechanisms of block of a human cloned potassium channel by the enantiomers of a new bradycardic agent: S-16257-2 and S-16260-2. Br. J. Pharmacol. 1996;117:1293–1301. doi: 10.1111/j.1476-5381.1996.tb16728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANQUEZA L., LONGOBARDO M., VICENTE J., DELPON E., TAMKUN M.M., TAMARGO J., SNYDERS D.J., VALENZUELA C. Molecular determinants of stereoselective bupivacaine block of hKv1.5 channels. Circ. Res. 1997;81:1053–1064. doi: 10.1161/01.res.81.6.1053. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HARRIS R.E., LARSSON H.P., ISACOFF E.Y. A permanent ion binding site located between two gates of the Shaker K+ channel. Biophys. J. 1998;74:1808–1820. doi: 10.1016/s0006-3495(98)77891-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEGINBOTHAM L., MACKINNON R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- KISS L., KORN S.J. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONGOBARDO M., DELPON E., CABALLERO R., TAMARGO J., VALENZUELA C. Structural determinants of potency and stereoselective block of hKv1.5 channels induced by local anesthetics. Mol. Pharmacol. 1998a;54:162–169. doi: 10.1124/mol.54.1.162. [DOI] [PubMed] [Google Scholar]
- LONGOBARDO M., DELPON E., CABALLERO R., TAMARGO J., VALENZUELA C. Effects of changes in extracellular pH on R(+)-bupivacaine-induced block of hKv1.5 channels. Br J Pharmacol. 1998b;124:110P. [Google Scholar]
- LONGOBARDO M., NAVARRO-POLANCO R., CABALLERO R., DELPON E., TAMARGO J., SNYDERS D.J., TAMKUN M.M., VALENZUELA C. Evidences of an external bupivacaine receptor in hKv1.5 channels. Biophys J. 1999;76:A189. [Google Scholar]
- LOPEZ-BARNEO J., HOSHI T., HEINEMANN S.H., ALDRICH R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors & Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- QU Y.S., ROGERS J., TANADA T., SCHEUER T., CATTERALL W.A. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11839–11843. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-CHAPULA J. Effects of bupivacaine on membrane currents of guinea-pig ventricular myocytes. Eur. J. Pharmacol. 1988;156:303–308. doi: 10.1016/0014-2999(88)90274-9. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., KNOTH K.M., ROBERDS S.L., TAMKUN M.M. Time-, voltage-, and state-dependent block by quinidine of a cloned human cardiac potassium channel. Mol. Pharmacol. 1992;41:322–330. [PubMed] [Google Scholar]
- SNYDERS D.J., TAMKUN M.M., BENNETT P.B. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J. Gen. Physiol. 1993;101:513–543. doi: 10.1085/jgp.101.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UEBELE V.N., ENGLAND S.K., CHAUDHARY A.C., TAMKUN M.M., SNYDERS D.J. Functional differences in Kv1.5 currents expressed in mammalian cell lines are due to the presence of endogenous Kvβ2.1 subunits. J. Biol. Chem. 1996;271:2406–2412. doi: 10.1074/jbc.271.5.2406. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., DELPON E., TAMKUN M.M., TAMARGO J., SNYDERS D.J. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys. J. 1995a;69:418–427. doi: 10.1016/S0006-3495(95)79914-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALENZUELA C., DELPON E., FRANQUEZA L., GAY P., PEREZ O., TAMARGO J., SNYDERS D.J. Class III antiarrhythmic effects of zatebradine. Time-, state-, use-, and voltage-dependent block of hKv1.5 channels. Circulation. 1996;94:562–570. doi: 10.1161/01.cir.94.3.562. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., DELPON E., FRANQUEZA L., GAY P., SNYDERS D.J., TAMARGO J. Effects of ropivacaine on a potassium channel (hKv1.5) cloned from human ventricle. Anesthesiology. 1997;86:718–728. doi: 10.1097/00000542-199703000-00025. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., SNYDERS D.J., BENNETT P.B., TAMARGO J., HONDEGHEM L.M. Stereoselective block of cardiac sodium channels by bupivacaine in guinea pig ventricular myocytes. Circulation. 1995b;92:3014–3024. doi: 10.1161/01.cir.92.10.3014. [DOI] [PubMed] [Google Scholar]
- YELLEN G., JURMAN M.E., ABRAMSON T., MACKINNON R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- YEOLA S.W., RICH T.C., UEBELE V.N., TAMKUN M.M., SNYDERS D.J. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ. Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]