

Abstract
Ischaemia-reperfusion injury causes cell death by both necrosis and apoptosis. Caspase activation is a major event in apoptosis. We therefore examined the effect of caspase inhibitors during reperfusion upon myocardial infarction. Rat isolated hearts were subjected to 35 min coronary occlusion and 120 min reperfusion. Treatment groups were perfused with caspase inhibitors during early reperfusion. We assessed a non-selective caspase inhibitor (Z-VAD·fmk, 0.1 μM), a caspase-8 inhibitor (Z-IETD·fmk, 0.07 μM), a caspase-9 inhibitor (Z-LEHD·fmk, 0.07 μM) and a caspase-3 inhibitor (Ac-DEVD·cmk, 0.07 μM). All caspase inhibitors limited infarct size (infarct-risk ratio per cent: control 38.5±2.6; Z-VAD·fmk 24.6±3.4; Z-LEHD·fmk 19.3±2.4; Z-IETD·fmk 23.0±5.4; Ac-DEVD·cmk 27.8±3.3; P<0.05 when compared with control value, 1-way ANOVA). We conclude that caspase inhibition during early reperfusion protects myocardium against lethal reperfusion injury.
Keywords: Caspases, caspase inhibitors, infarct size, myocardium, reperfusion
Introduction
Myocardial ischaemia, with or without reperfusion, results in irreversible cell injury leading to myocardial infarction. Although, traditionally, necrosis is regarded as the pathological hallmark of myocardial infarction, there is accumulating evidence that apoptosis also contributes to myocardial cell death during infarction (Anversa et al., 1998). It has been suggested that the apoptotic program is initiated shortly after the onset of ischaemia but there is evidence that the process is amplified during reperfusion (Gottlieb et al., 1994; Fliss & Gattinger, 1996; Scarabelli et al., 1999). This enhancement may be due to the replenishment of high-energy phosphates which is necessary to sustain apoptosis, together with cytosolic and intra-mitochondrial calcium overload, and reactive oxygen species generation (Fliss, 1998).
Apoptosis is crucially dependent on the activation of certain intracellular proteases, called caspases, typically activated in a cascade fashion. There are currently about 14 known caspases. The ‘initiator' caspases, caspase-8 and caspase-9, and the ‘effector' caspase-3 seem to be play pivotal roles in apoptosis. We hypothesized that inhibition of apoptosis during reperfusion, by administration of synthetic peptide inhibitors of caspase activation, would reduce the extent of myocardial infarction. To test this hypothesis, we examined the effects of a non-selective (‘broad spectrum') caspase inhibitor (Z-VAD·fmk), an inhibitor of caspase-8 (Z-IETD·fmk), an inhibitor of caspase-9 (Z-LEHD·fmk) and an inhibitor of caspase-3 (Ac-DEVD·cmk). All agents are cell permeable and known to induce irreversible inhibition of caspases and, with the exception of Z-VAD·fmk, are reported to be selective inhibitors (Rudel, 1999). Our results are the first to indicate the ability of caspase inhibitors to limit myocardial infarction when administered only during early reperfusion.
Methods
Sources of materials
Caspase inhibitors were obtained from Calbiochem (Nottingham, U.K.). All other materials were of analytical grade from BDH. We used a non-selective caspase inhibitor, Z-VAD·fmk (Z-Val-Ala-Asp(OMe)-CH2F), a caspase-8 inhibitor, Z-IETD·fmk (Z-Ile-Glu-Thr-Asp(OMe)-CH2F), a caspase-9 inhibitor, Z-LEHD·fmk (Z-Leu-Glu-OMe)-His-Asp(OMe)-CH2F) and a caspase-3 inhibitor, Ac-DEVD·cmk (Ac-Asp-Glu-Val-Asp-CH2Cl). All these inhibitors are cell permeable and irreversible inhibitors of the respective caspases. They were dissolved in dimethylsulphoxide and aliquots were frozen. The aliquots were diluted in Krebs-Henseleit buffer (see below) immediately before use. The final concentrations of the dimethylsulphoxide in the buffer solution did not exceed 0.02%, a concentration which has previously been shown to have no effect on cardiac function or infarct size in this model.
Ischaemia-reperfusion protocols
A previously characterized model of myocardial infarction in the rat isolated heart was used (Mocanu et al., 1999). Male Sprague-Dawley rats (300–350 g body weight) were anaesthetized with sodium pentobarbitone (55 mg kg−1 i.p.) and heparin (300 IU) was administered concomitantly. Under deep anaesthesia, the hearts were excised, placed in ice-cold buffer and mounted on a Langendorff perfusion system (80 mmHg constant pressure). Hearts were perfused retrogradely with a modified Krebs-Henseleit bicarbonate buffer, containing (in mM): NaCl 118.5, NaHCO3 25, KCl 4.8, MgSO4 1.2, KH2PO4 1.2, CaCl2 1.7, glucose 12. All solutions were filtered through a Whatman microfibre filter (2.0 μm pore), gassed with 95% O2/5% CO2, pH 7.4 (±0.4). Heart temperature was maintained at 37°C throughout. A latex isovolumic balloon was introduced in the left ventricle and inflated to give a preload of 8–10 mmHg. Left ventricular developed pressure, heart rate and coronary flow were recorded at intervals. For occlusion of the left main coronary artery, a 3/0 surgical silk suture mounted on a curved needle was placed under the artery close to its origin and the ends of the suture were passed through a small plastic snare. Regional ischaemia was induced by clamping the snare taut onto the epicardial surface with a haemostat forceps. Reperfusion was induced by releasing the snare and was confirmed by an increase in coronary flow rate above the preceding ischaemic value. Reperfusion-induced tachyarrhythmias were of low incidence and severity due to the relatively high K+ concentration of the perfusion buffer (Curtis & Hearse, 1989).
Drug treatment protocols
Hearts were randomized to the following treatment groups: (1) Controls (C, n=12): hearts were allowed to stabilize then underwent 35 min regional ischaemia followed by 120 min reperfusion. (2) Treated hearts received a caspase inhibitor added in the perfusate for 15 min, starting 5 min before reperfusion: 0.1 μM Z-VAD·fmk (n=9), 0.07 μM Z-LEHD·fmk (n=9), 0.07 μM Z-IETD·fmk (n=7), or 0.07 μM Ac-DEVD·cmk (n=8). The experimental protocol is presented in Figure 1.
Figure 1.
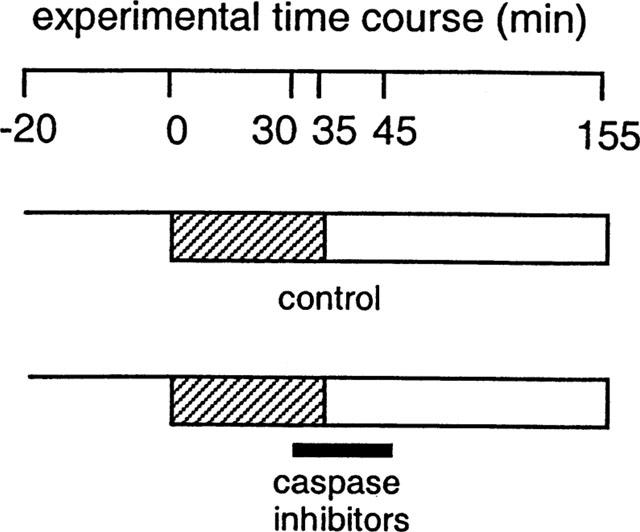
Experimental protocols. Following stabilization for 20 min, hearts were subjected to 35 min coronary artery occlusion followed by 120 min reperfusion after which infarct size was assessed. Hearts treated with caspase inhibitors were randomly assigned to receive one of four inhibitors, administered in the Krebs-Henseleit buffer. Perfusion with the inhibitors was commenced 5 min before reperfusion and stopped at 10 min reperfusion, indicated by the black bar. Thereafter hearts were perfused with normal Krebs-Henseleit buffer.
Infarct size assessment
At the end of 120 min reperfusion the snare was tightened to re-occlude the coronary artery and Evans' blue solution (0.12%) was infused slowly into the aorta. After freezing at −20°C for 1–4 h, hearts were sliced into 1 mm-thick transverse sections and incubated in triphenyltetrazolium chloride solution (1% in phosphate buffer, pH 7.4) at 37°C for 10–12 min. The stained sections were then fixed in 10% formalin, overnight. At the end of this procedure, the viable tissue was stained red and the infarcted tissue remained pale. Using computerized planimetry (Summa Sketch II, Summagraphics, Connecticut, U.S.A.) the percentage of infarcted tissue within the myocardium at risk was calculated.
Statistical analysis
All data are expressed as mean±s.e. mean. Infarct size data were analysed using 1-way analysis of variance with post hoc comparisons using the Fisher's protected least significant difference test. Differences in coronary flow and rate-pressure product were analysed by repeated measures ANOVA. Differences between groups were considered significant when P<0.05.
Results
A total of 52 hearts were used for this study. Five hearts were excluded prior to coronary artery occlusion due to low coronary flow rate, left ventricular developed pressure less than 80 mmHg or heart rate less than 300 beats min−1. A further two hearts were excluded due to failure to reperfuse. Fast ventricular tachycardia or ventricular fibrillation occurred in several hearts during reperfusion (incidence 11–42% among the different experimental groups) but converted to sinus rhythm within 2 min. No hearts were excluded as a result of tachyarrhythmias. Thus we report infarct data from 45 successfully completed experiments.
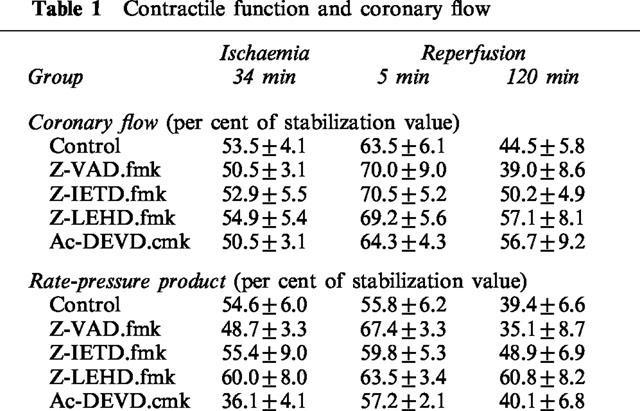
Table 1 summarizes myocardial contractile function (as rate-pressure product) and coronary flow at the end of ischaemia, during early reperfusion and at the end of reperfusion. During prolonged Langendorff perfusion without coronary occlusion, coronary flow rate and contractile function decline steadily during the experimental time course. Coronary occlusion resulted in immediate decline in coronary flow rate which increased upon reperfusion and thereafter declined steadily during the reperfusion period. At the end of the 120 min reperfusion period there were no significant variations between groups with regard to coronary flow rate or contractile performance which is consistent with previous experience with this model (Mocanu et al., 1999). Although contractile function and coronary flow were not primarily endpoints assessed in this study, a tendency towards better recovery of both flow and contractility was noticeable in the caspase-9 inhibitor group.
Table 1.
Contractile function and coronary flow
The ischaemic risk zone volume (i.e. the volume of myocardium at risk of infarction) was similar in all the experimental groups at around 0.5 cm3 (Figure 2a). In control hearts, the percentage of infarction within the risk zone was 38.5±2.6%, a value consistent with previous reports using this experimental model of ischaemia-reperfusion (Mocanu et al., 1999). Within control hearts 38.5±2.6% of the risk zone was infarcted. The percentage of infarction was markedly reduced in all the caspase inhibitor-treated groups compared with the control group (Figure 2b). It is of interest to note that treatment with the non-selective caspase inhibitor (Z-VAD·fmk) did not confer greater protection against infarction than any of the selective inhibitors when these were administered alone (Z-VAD·fmk 24.6±3.4%; Z-LEHD·fmk 19.3±2.4%; Z-IETD·fmk 23.0±5.4%; Ac-DEVD·cmk 27.8±3.3%, differences not significant).
Figure 2.
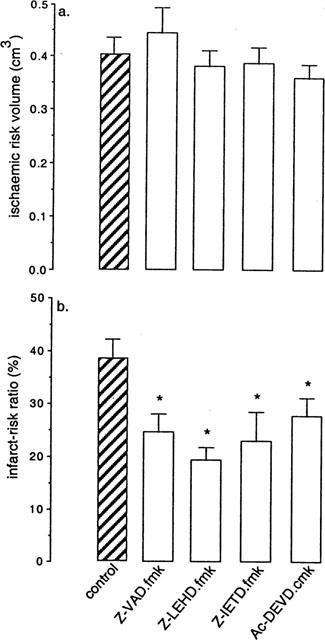
(a) Myocardial ischaemic risk volume. (b) Infarct size expressed as a percentage of myocardial risk volume. Z-VAD·fmk, non-selective caspase inhibitor; Z-LEHD·fmk, a caspase-9 inhibitor; Z-IETD·fmk, a caspase-8 inhibitor; Ac-DEVD·cmk, a caspase-3 inhibitor. Treatment with each caspase inhibitor resulted in significant limitation of infarct size. *P<0.05 vs control (1-way ANOVA).
Discussion
Infarct size is a critical determinant of acute and long-term prognosis in patients following coronary thrombosis. At present, prompt reperfusion is regarded as the primary means of salvaging ischaemic myocardium but, paradoxically, reperfusion may be associated with further exacerbation of the infarct process (‘lethal reperfusion injury') (Yellon & Baxter, 2000). The identification of drugs which influence the rate and extent of tissue death during both ischaemia and reperfusion is a major therapeutic goal. In particular, agents which could be administered as adjuncts to thrombolysis to attenuate lethal reperfusion injury, and would thereby provide further benefit than reperfusion therapy alone, might significantly influence clinical outcome from myocardial infarction. In the present study, we report that a range of caspase inhibitors when administered as adjuncts to reperfusion in an experimental model of myocardial infarction, provide substantial protection. There are several lines of evidence that myocardial cell death during ischaemia-reperfusion can take place via apoptosis and necrosis but the individual contributions of these two phenomena and at what point they contribute to tissue death is unclear (Bromme & Holtz, 1996; Buja & Entman, 1998; Gottlieb & Engler, 1999). Since caspase activation via death ligands or mitochondrial damage is a crucial event in apoptosis and since apoptosis appears to be accelerated during the process of reperfusion, we were interested to study the effect upon reperfusion injury of caspase inhibitors given at the onset of reperfusion. Our primary hypothesis was that apoptosis contributes substantially to myocardial infarction and that its role is most relevant during reperfusion. Although some studies provide evidence that apoptosis occurs during experimental myocardial ischaemia (Anversa et al., 1998), work from our own and other laboratories suggests that subsequent reperfusion increases the extent of cardiomyocyte and vascular endothelial cell apoptosis quite substantially (Gottlieb et al., 1994; Fliss & Gattinger, 1996; Scarabelli et al., 1999). Previous studies have shown that application of caspase inhibitors prior to ischaemia confers protection against ischaemia-reperfusion injury in myocardium. For example, Z-VAD·fmk administered before, and continuously during and after an ischaemic insult resulted in better recovery and a smaller infarct size in rat heart in vivo (Yaoita et al., 1998). Holly et al. (1999) have reported that caspase inhibition with the non-selective inhibitor Y-VAD·cmk prior to coronary artery occlusion in the rabbit heart in vivo led to limitation of infarct size. Since these compounds inhibit caspases irreversibly it is likely that caspases were also inhibited during reperfusion. In contrast, in the present study the caspase inhibitors were given specifically at early reperfusion. We demonstrate that under these circumstances they also reduce the extent of infarction significantly, providing evidence that the key signaling pathways controlling apoptosis may mediate reperfusion injury.
It is not clear to what extent apoptosis and necrosis individually contribute to tissue infarction. Relatively recent information suggests that necrosis and apoptosis are governed by similar mechanisms, and contrary to earlier belief they may share common molecular pathways (Shimizu et al., 1996; Gottlieb & Engler, 1999). Moreover, if the high energy phosphate reserves are exhausted, cells undergoing apoptosis can switch to secondary necrosis (Leist & Nicotera, 1997; Daemen et al., 1999). Thus, the precise mechanism(s) by which caspase inhibitors lead to limitation of infarction is not certain. In spite of the fact that the inhibitors we used are reported to exert specific and selective anti-caspase effects at the concentration used, their specificity should be accepted with some caution. Indeed, the fact that all the selective inhibitors were effective to approximately the same degree as the non-selective inhibitor was surprising to us. Therefore, we cannot exclude the possibility that caspase inhibitors may exert non-specific actions and might inhibit other proteases, such as calpains for example, which have been previously implicated in ischaemia-reperfusion injury (Iwamoto et al., 1999). There is evidence that calpains, which are structurally related to caspases, are involved not only in necrotic processes but also in apoptosis (McGinnis et al., 1999). Conversely, there is accumulating evidence that apoptosis and necrosis are linked phenomena sharing common pathways, and in the pathology of ischemic reperfused myocardium it is difficult to distinguish between these two cell death pathways (Shimizu et al., 1996; Gottlieb & Engler, 1999). Indeed, recent evidence suggests that in addition to their well-established role in apoptosis, caspases may also mediate necrotic injury (Edelstein et al., 1999).
In conclusion, we present the first evidence that caspase inhibitors administered as adjuncts to reperfusion limit infarct size. Although the precise mechanism underlying the protection remains to be clarified, these observations indicate that inhibition of caspases may be a promising route for development of therapies to attenuate reperfusion-induced injury in the heart.
Acknowledgments
This work was supported by the British Heart Foundation. The authors are grateful for the continued support of the Hatter Foundation.
Abbreviations
- Ac-DEVD·cmk
Ac-Asp-Glu-Val-Asp-CH2Cl
- Z-IETD·fmk
Z-Ile-Glu-Thr-Asp(OMe)-CH2F
- Z-LEHD·fmk
Z-Leu-Glu-(OMe)-His-Asp(OMe)-CH2F
- Z-VAD·fmk
Z-Val-Ala-Asp(OMe)-CH2F
References
- ANVERSA P., CHENG W., LIU Y., LERI A., REDAELLI G., KAJSTURA J. Apoptosis and myocardial infarction. Basic Res. Cardiol. 1998;93 Suppl:8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- BROMME H.J., HOLTZ J. Apoptosis in the heart: when and why. J. Mol. Cell Biochem. 1996;163–164:261–275. doi: 10.1007/BF00408667. [DOI] [PubMed] [Google Scholar]
- BUJA L.M., ENTMAN M.L. Modes of myocardial cell injury and cell death in ischemic heart disease. Circulation. 1998;98:1355–1357. doi: 10.1161/01.cir.98.14.1355. [DOI] [PubMed] [Google Scholar]
- CURTIS M.J., HEARSE D.J. Ischaemia-induced and reperfusion-induced arrhythmias differ in their sensitivity to potassium: implications for mechanisms of initiation and maintenance of ventricular fibrillation. J. Mol. Cell Cardiol. 1989;21:21–40. doi: 10.1016/0022-2828(89)91490-9. [DOI] [PubMed] [Google Scholar]
- DAEMEN M.A., VAN'T VEER C., DENECKER G., HEEMSKERK V.H., WOLFS T.G., CLAUSS M., VANDENABEELE P., BUURMAN W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Invest. 1999;104:541–549. doi: 10.1172/JCI6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDELSTEIN C.L., SHI Y., SCHRIER R.W. Role of caspases in hypoxia-induced necrosis of rat renal proximal tubules. J. Am. Soc. Nephrol. 1999;10:1940–1949. doi: 10.1681/ASN.V1091940. [DOI] [PubMed] [Google Scholar]
- FLISS H. Accelerated apoptosis in reperfused myocardium: friend or foe. Basic. Res. Cardiol. 1998;93:90–93. doi: 10.1007/s003950050067. [DOI] [PubMed] [Google Scholar]
- FLISS H., GATTINGER D. Apoptosis in ischemic and reperfused rat myocardium. Circ. Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- GOTTLIEB R.A., BURLESON K.O., KLONER R.A., BABIOR B.M., ENGLER R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. Clin. Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOTTLIEB R.A., ENGLER R.L. Apoptosis in myocardial ischemia-reperfusion. Ann. N.Y. Acad. Sci. 1999;874:412–426. doi: 10.1111/j.1749-6632.1999.tb09255.x. [DOI] [PubMed] [Google Scholar]
- HOLLY T.A., DRINCIC A., BYUN Y., NAKAMURA S., HARRIS K., KLOCKE F.J., CRYNS V.L. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J. Mol. Cell Cardiol. 1999;31:1709–1715. doi: 10.1006/jmcc.1999.1006. [DOI] [PubMed] [Google Scholar]
- IWAMOTO H., MIURA T., OKAMURA T., SHIRAKAWA K., IWATATE M., KAWAMURA S., TATSUNO H., IKEDA Y., MATSUZAKI M. Calpain inhibitor-1 reduces infarct size and DNA fragmentation of myocardium in ischemic/reperfused rat heart. J. Cardiovasc. Pharmacol. 1999;33:580–586. doi: 10.1097/00005344-199904000-00010. [DOI] [PubMed] [Google Scholar]
- LEIST M., NICOTERA P. The shape of cell death. Biochem. Biophys. Res. Commun. 1997;236:1–13. doi: 10.1006/bbrc.1997.6890. [DOI] [PubMed] [Google Scholar]
- MCGINNIS K.M., GNEGY M.E., PARK Y.H., MUKERJEE N., WANG K.K. Procaspase-3 and poly(ADP)ribose polymerase (PARP) are calpain substrates. Biochem. Biophys. Res. Commun. 1999;263:94–99. doi: 10.1006/bbrc.1999.1315. [DOI] [PubMed] [Google Scholar]
- MOCANU M.M., GADGIL S., YELLON D.M., BAXTER G.F. Mibefradil, a T-type and L-type calcium channel blocker, limits infarct size through a glibenclamide-sensitive mechanism. Cardiovasc. Drugs Ther. 1999;13:115–122. doi: 10.1023/a:1007732025184. [DOI] [PubMed] [Google Scholar]
- RUDEL T. Caspase inhibitors in prevention of apoptosis. Herz. 1999;24:236–241. doi: 10.1007/BF03044967. [DOI] [PubMed] [Google Scholar]
- SCARABELLI T.M., KNIGHT R.A., RAYMENT N.B., COOPER T.J., STEPHANOU A., BRAR B.K., LAWRENCE K.M., SANTILLI G., LATCHMAN D.S., BAXTER G.F., YELLON D.M. Quantitative assessment of cardiac myocyte apoptosis in tissue sections using the fluorescence-based tunel technique enhanced with counterstains. J. Immunol. Methods. 1999;228:23–28. doi: 10.1016/s0022-1759(99)00090-3. [DOI] [PubMed] [Google Scholar]
- SHIMIZU S., EGUCHI Y., KAMIIKE W., WAGURI S., UCHIYAMA Y., MATSUDA H., TSUJIMOTO Y. Retardation of chemical hypoxia-induced necrotic cell death by Bcl-2 and ICE inhibitors: possible involvement of common mediators in apoptotic and necrotic signal transductions. Oncogene. 1996;12:2045–2050. [PubMed] [Google Scholar]
- YAOITA H., OGAWA K., MAEHARA K., MARUYAMA Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- YELLON D.M., BAXTER G.F. Protecting the ischaemic and reperfused myocardium in acute myocardial infarction: distant dream or near reality. Heart. 2000;83:381–387. doi: 10.1136/heart.83.4.381. [DOI] [PMC free article] [PubMed] [Google Scholar]