

Abstract
We investigated the rate of penetration into and the intra-relationship between the serum, cerebrospinal fluid (CSF) and regional brain extracellular fluid (bECF) compartments following systemic administration of lamotrigine in rat.
The serum pharmacokinetics were biphasic with an initial distribution phase, (half-life approximately 3 h), and then a prolonged elimination phase of over 30 h. The serum pharmacokinetics were linear over the range 10–40 mg kg−1.
Using direct sampling of CSF with concomitant serum sampling, the calculated penetration half-time into CSF was 0.42±0.15 h. At equilibrium, the CSF to total serum concentration ratio (0.61±0.02) was greater than the free to total serum concentration (0.39±0.01).
Using in vivo recovery corrected microdialysis sampling in frontal cortex and hippocampus with concomitant serum sampling, the calculated penetration half-time of lamotrigine into bECF, 0.51±0.11 h, was similar to that for CSF and was not area or dose dependent. At equilibrium, the bECF to total serum concentration ratio (0.40±0.04) was similar to the free to total serum concentration (0.39±0.01), and did not differ between hippocampus and frontal cortex.
The species specific serum kinetics can explain the prolonged action of lamotrigine in rat seizure models. Lamotrigine has a relatively slow penetration into both CSF and bECF compartments compared with antiepileptic drugs used in acute seizures. Furthermore, the free serum drug concentration is not the sole contributor to the CSF compartment, and the CSF concentration is an overestimate of the bECF concentration of lamotrigine.
Keywords: Lamotrigine, cerebrospinal fluid, pharmacokinetics, brain extracellular fluid, microdialysis, antiepileptic drug
Introduction
Lamotrigine (3,5-diamino-6-(2,3-dichlorophenyl)-1,2,4-triazine) is a phenyltriazine derivative. In clinical trials, lamotrigine has been shown to be an effective antiepileptic drug (AED) with a broad spectrum of activity (Fitton & Goa, 1995). It protects against lesions produced by kainate (McGeer & Zhu, 1990); at high doses it can reduce cortical infarct volume, and protect against global cerebral ischaemia (Rataud et al., 1994; Smith & Meldrum, 1995; Crumrine et al., 1997; Wiard et al., 1995). It thus has potential to be used acutely as a neuroprotectant. Its mechanism of action is, in part, mediated through inhibition of voltage-activated sodium channels, and also an action on pre-synaptic N-type and P-type calcium channels (Walker & Sander, 1999). It has a favourable pharmacokinetic profile in humans with good absorption, linear pharmacokinetics and minimal effect on the pharmacokinetics of other drugs (Fitton & Goa, 1995; Walker & Sander, 1999).
Although serum concentration monitoring of AEDs is widely and effectively used in the management of patients, its primary purpose is as an index of brain concentrations (i.e. the concentration at the site of action). Serum pharmacokinetics may, however, be a very poor index of brain pharmacokinetics following acute administration of AEDs such as are used in the treatment of status epilepticus (Sechi et al., 1989; Wilder et al., 1977). An understanding of the neuropharmacokinetics of such drugs is critical for: optimization of therapy, determining the value of serum concentration monitoring, and ascertaining drug modes and mechanisms of action. Additionally it is not enough to picture the brain as a single compartment. The brain consists of extracellular, intracellular, and cerebrospinal fluid (CSF) compartments and depending on where a drug acts depends on which compartment's kinetics is of most relevance. The constitution of and access to these compartments is different (Davson & Segal, 1995). The pharmacokinetics of AEDs in whole brain are determined by non-specific binding to brain lipids and proteins; they are thus unlikely to represent the pharmacodynamically relevant compartment. Receptors on neurons, and ion channels on axons are surrounded by brain extracellular fluid (bECF), and it is likely that the pharmacodynamics of drugs that act on these receptors and ion channels are determined by the unbound concentration of drugs in the bECF (Sechi et al., 1989). Although, the bECF and CSF are produced independently, they are in direct communication with one another so that changes in the composition of one are often reflected in changes in the composition of the other (Davson & Segal, 1995). CSF drug concentrations could thus be an indirect index of bECF concentrations. Measurements of CSF penetration, however, do not necessarily give an accurate indication of blood brain permeability, and there are circumstances when CSF concentrations are a poor indicator of brain tissue and bECF concentrations (Thomas & Segal, 1998).
In order to study CSF pharmacokinetics, direct sampling of CSF with simultaneous serum sampling has been successfully used (Patsalos et al., 1992; Semba et al., 1993; Lolin et al., 1994; Walker et al., 1998; Doheny et al., 1999; Nagaki et al., 1999). The limited accessibility of CSF and the impracticability of repeated sampling in humans have meant that most of these drug studies have been carried out in animal models.
Various methods have been used to study drug pharmacokinetics in bECF; bECF phenytoin has been measured in the left temperoparietal region using implanted polypropylene balls in dogs (Sechi et al., 1989). The technique of microdialysis has enabled the temporal measurement of bECF drug concentrations in discrete brain areas following peripheral administration. This technique has a temporal and spatial resolution suitable for the study of drug neuropharmacokinetics, without significant perturbation to the system measured (the method does not rely upon the extraction of fluid from a compartment of interest). Importantly the blood-brain barrier is intact shortly following slow implantation of microdialysis probes (de-Lange et al., 1997). Microdialysis is, thus, highly suited for studies of drug neuropharmacokinetics (Patsalos et al., 1995; de-Lange et al., 1997). The further advantage of the microdialysis technique is that it enables simultaneous measurement in two or more brain areas (de-Lange et al., 1995; Walker et al., 1996)
Using well-characterized freely behaving rat models that permit simultaneous sampling of CSF and blood or bECF and blood, we set out to characterize the temporal pharmacokinetic and neuropharmacokinetic inter-relationship of lamotrigine after acute administration. The contribution of dose and protein binding were also determined.
Methods
Drugs
Lamotrigine (Glaxo-Wellcome, Cheshire) was dissolved in 50% propylene glycol (20 mg ml−1) for intraperitoneal administration or at low concentrations in artificial CSF (composition mM: NaCl 125, KCl 2.5, MgCl2 1.18 and CaCl2 1.26) for recovery experiments.
Animals
Male Sprague-Dawley rats (Charles River, Margate, Kent U.K.) weighing 260–360 g were used. Rats were individually housed under a 12 h light-dark cycle (lights on 0800 h), an ambient temperature of 25°C and with free access to water and to a normal laboratory diet (SDS R and M number 1 expanded, Scientific Dietary Services, Witham, Essex, U.K.).
CSF and blood sampling
Rats were anaesthetized with halothane (2%), and catheters were implanted in the cisterna magna for CSF sampling and the right jugular vein for blood sampling, using a previously described technique except that the position of the intersliding polythene tubing in the cisterna magna catheter was fixed with epoxy resin after implantation (Patsalos et al., 1992).
Two days later the CSF and blood catheters were checked for patency, and blood (200 μl) and CSF (20 μl) samples were collected at 30 min intervals for 1 h. Thirty minutes later the animals were given lamotrigine in propylene glycol by intraperitoneal injection (10 or 20 mg kg−1). Venous blood samples (200 μl) were withdrawn at 10, 20, 30, 40 and 60 min, then hourly to 12 h and then 3 hourly to 30 h after lamotrigine administration. After each sampling, the catheter was flushed with 100 μl of 5 u ml−1 heparinized saline to maintain patency and prevent hypovolaemia. CSF samples were taken at 20–30 min intervals for the first hour, then hourly to 12 h and then 3 hourly to 30 h after lamotrigine administration. Sera were separated by centrifugation, and sera and CSF were stored at −70°C until analysis.
bECF and blood sampling
Concentric dialysis probes with Filtral 12 (Hospal, Rugby, U.K.) dialysis membrane 4 mm long, 200 μm diameter were prepared as previously described (Hutson et al., 1985). On the day of surgery the in vitro recovery for each probe was calculated by placing the probes in a 8 μM solution of lamotrigine dissolved in artificial CSF at 37°C, and then perfusing the probes with artificial CSF at 2 μl min−1. Samples (40 μl) were collected every 20 min for 80 min, and stored at −70°C until analysis.
Rats were anaesthetized with halothane (2%), and microdialysis probes were implanted, and the jugular vein was catheterized using previously described procedures. The probes were slowly implanted in the hippocampus (from bregma 5.6 mm posterior, 5 mm lateral, 8.2 mm ventral) and the frontal cortex (from bregma 2.5 mm anterior, 1.5 mm lateral, 5.5 mm ventral) according to the atlas of Paxinos & Watson (1986), and held in place with dental cement and anchor screws (De Trey, Surrey, U.K.).
Two days after surgery when the animals were fully recovered (Patsalos et al., 1992), the jugular vein catheter, and dialysis probes were checked for patency. Artificial CSF was perfused through the microdialysis probes at 2 μl min−1. Three baseline dialysate samples (40 μl) and concomitant blood samples (200 μl) were taken in the first hour. The rats were then injected intraperitoneally with 20 or 40 mg kg−1 lamotrigine in propylene glycol; 40 mg kg−1 was higher than that used in the CSF experiments, because following 10 mg kg−1 i.p. lamotrigine, the concentrations of lamotrigine in dialysate would have been below the detectable limit of the h.p.l.c. analysis. Venous blood samples (200 μl) were withdrawn every 20–120 min and thereafter hourly until 300 min after lamotrigine administration. Dialysate samples (20 μl) were collected every 10 min for 120 min and then every 20 min (40 μl) for a further 180 min. Sera were separated by centrifugation, and sera and dialysate were stored at −70°C until analysis.
In vivo microdialysis probe recovery
The no-net-flux method for calculating the difference between in vivo and in vitro recoveries was used (Lonnroth et al., 1987). Concentric dialysis probes with Filtral 12 (Hospal, Rugby, U.K.) dialysis membrane 4 mm long, 200 μm diameter were prepared as previously described.
On the day of surgery the in vitro recovery for each probe was calculated by placing the probes in a 40 μM solution of lamotrigine dissolved in artificial CSF at 37°C, and then perfusing the probes with artificial CSF at 2 μl min−1. Samples (40 μl) were collected every 20 min for 80 min, and stored at −70°C until analysis.
Rats were anaesthetized with halothane (2%), and microdialysis probes were implanted using previously described techniques. The probes were slowly implanted in the frontal cortex (from bregma 2.5 mm anterior, 1.5 mm lateral, 5.5 mm ventral) according to the atlas of Paxinos & Watson (1986), and held in place with dental cement and anchor screws (De Trey, Surrey, U.K.).
One day after surgery, the rats were injected with lamotrigine, 40 mg kg−1 i.p. 20 h later, artificial CSF was perfused through the microdialysis probes at 2 μl min−1. Three baseline dialysate samples (60 μl) were taken in the first 1.5 h. Then 5, 10, 15 and 20 μM lamotrigine in artificial CSF was perfused through the microdialysis probe 2 μl min−1 for 0.5 h after which a dialysate sample (60 μl) was collected over 0.5 h. After the lamotrigine perfusions, artificial CSF was perfused through the microdialysis probes at 2 μl min−1 for 0.5 h and a further three dialysate samples (60 μl) were collected. Dialysate was stored at −70°C until analysis.
Lamotrigine analysis
The concentrations of lamotrigine in sera, CSF and dialysate were determined by high performance liquid chromatography (h.p.l.c.) with ultraviolet detection as follows. Sera (50 μl) and acetonitrile (50 μl) containing 3.75 μg ml−1 10-methoxycarbamazepine as the internal standard were pipetted into a 1.5 ml polyethylene tube (Treff AG, Switzerland), vortex mixed and then centrifuged for 5 min at 9500×g (Abbott Micro-Centrifuge, Abbott, Maidenhead, U.K.). Fifty μl of the supernatant extract were mixed with 25 μl of mobile phase and 10 μl were injected into the h.p.l.c. system. The h.p.l.c. system comprised a Spectrasystem AS3000 autosampler, a Spectrasystem P4000 pump, a Spectrasystem UV2000 detector and an SP 4270 integrator/printer plotter with LABNET data system controller (Spectra-Physics, Maidenhead, U.K.). For CSF and dialysate analysis, 20 μl were directly injected into the h.p.l.c. system without acetonitrile extraction. Chromatograms were run at ambient temperature on a Merck LiChroCART column (125×4.0 mm) packed with Lichrosorb RP-8, 5 μm (BDH, Poole, U.K.). Mobile phases of 0.045 M phosphate buffer with 15% acetonitrile and 0.0275 M phosphate buffer with 37.5% acetonitrile were run on a gradient. The column effluent was monitored at 215 nm with a sensitivity range of 0.02 absorption units full scale. The procedures for determining the non-protein bound, serum lamotrigine concentration were exactly that for sera, except that the sera were first filtered through an Amicon Centrifree Micropartition System (Amicon, Stonehouse, U.K.) at a temperature of 25°C using a Sorvall RC-5B refrigerated centrifuge (Du Pont, Stevenage, U.K.), and 50 μl of the ultrafiltrate was used in the analysis.
Pharmacokinetic analysis and statistics
For determination of in vivo microdialysis probe recoveries, the no-net-flux method was used (Lonnroth et al., 1987). The difference between lamotrigine concentration out and lamotrigine concentration in for each perfusate concentration was plotted against lamotrigine concentration in. At steady-state concentrations, the slope of the line through the points represents the dialysis recovery for lamotrigine. A least mean squares linear regression analysis was used to calculate the slope. The in vivo recoveries were compared to the in vitro recoveries in order to calculate an in vivo to in vitro recovery ratio; this was used to correct all subsequent in vitro recoveries.
For serum pharmacokinetics: time to maximum concentration (Tmax) and maximum concentration (Cmax) were estimated from the graphs. The serum data for prolonged sampling (30 h) were modelled using Microcal Origin v5 assuming a biexponential decay, Ct=Ae−αt+Be−βt where Ct is the concentration at time t, β represents the elimination rate constant, and α represents a redistribution rate constant. In all cases β>>α, and indeed it was not possible to calculate β accurately despite 30 h of sampling. Thus for ease and accuracy of modelling the equation was approximated to Ct=Ae−αt+B.
The dialysate concentrations were corrected for probe recovery (see above). The ratios of CSF concentration to serum concentration and the ratios of corrected dialysate concentration to serum concentration were calculated for each individual animal at each time point for which there were concurrent data. The ratio versus time graphs were modelled using R=Rss(1-e−kt) where R is the ratio of CSF or dialysate concentration to serum concentration, Rss is the ratio at steady state and k is the rise constant (Loscher & Frey, 1984; Mayer et al., 1959). Where appropriate pharmacokinetic parameters were compared using analysis of variance (ANOVA).
Results
Serum pharmacokinetics
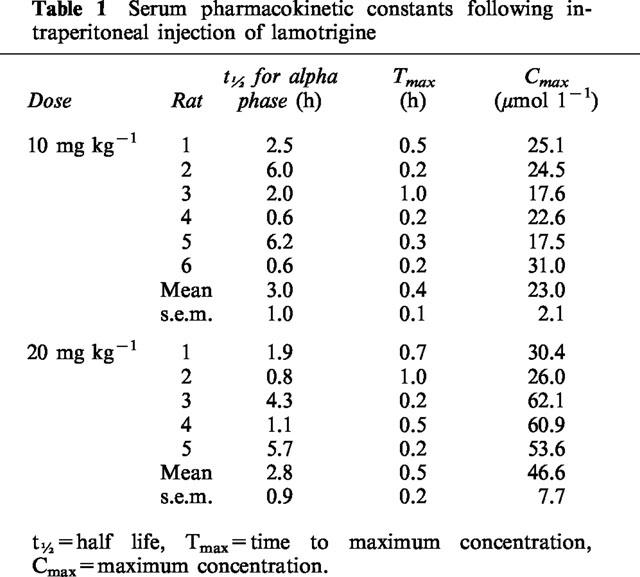
The serum concentration versus time profiles of lamotrigine during prolonged sampling (Figure 1a) demonstrate rapid absorption following intraperitoneal injection with peak concentrations achieved at 0.2–1.0 h for 20 mg kg−1 and 0.2–0.5 h for 10 mg kg−1 (Table 1). Thereafter there is a biphasic fall in sera concentrations. The initial alpha phase has a half-life of approximately 3 h at both doses (Table 1). The beta phase is prolonged and an accurate estimate of the decay constant was not possible despite 30 h of sampling. The serum lamotrigine concentration-time profile for the rats with CSF catheters at a dose of 20 mg kg−1 was similar to that for rats with microdialysis probes at the same dose (Figure 1b). Over 5 h of sampling, the serum lamotrigine concentration-time profiles were linearly related to dose (Figure 1b); the area under the curve for the concentration-time profiles over 5 h corrected for dose were not significantly different (P>0.1).
Figure 1.
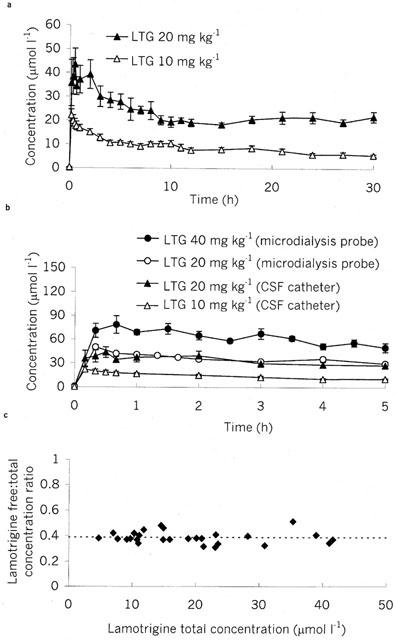
Serum pharmacokinetics following intraperitoneal injection of lamotrigine (LTG: 10, 20 or 40 mg kg−1) in (a) rats with CSF catheters over 30 h and (b) rats with microdialysis probes compared with serum concentrations from rats with CSF catheters over 5 h. Data are presented as mean±s.e.mean of 5–6 rats. (c) Serum lamotrigine free/total concentration ratio at differing total lamotrigine concentrations demonstrating no concentration effect on lamotrigine serum protein binding. Dotted line indicates mean value of ratio.
Table 1.
Serum pharmacokinetic constants following intraperitoneal injection of lamotrigine
Over the concentration range of 5–52 μmol l−1, the lamotrigine non-protein bound/total serum concentration ratio was 0.39±0.01 (n=27, Figure 1c).
Microdialysis probe recoveries
A comparison of in vivo and in vitro recoveries were made in three rats, using the finding that the serum lamotrigine concentrations from 15–30 h after administration do not significantly change. The rats were injected with lamotrigine 1 day after surgery, and the study was performed the next day (i.e. at the same time interval for the other microdialysis experiments). The in vitro recovery of these probes was 10.1±0.9%. The difference between dialysate concentration of lamotrigine (lamotrigine out) and perfusate concentration of lamotrigine (lamotrigine in) for five different perfusate lamotrigine concentrations was linearly related to perfusate concentration of lamotrigine (Figure 3a). The calculated in vivo recovery using the no-net-flux method was 4.8±0.3%. To confirm that the extracellular concentration of lamotrigine had not changed over the study period (a necessary prerequisite of the no-net-flux method), we compared the dialysate lamotrigine concentration whilst perfusing with artificial CSF containing no lamotrigine at the beginning of the study and at the end (0.80±0.07 and 0.90±0.03 μM, respectively; P>0.4). The ratio of in vitro to in vivo recoveries was 2.11±0.16, and this figure was used subsequently to correct the in vitro probe recoveries.
Figure 3.
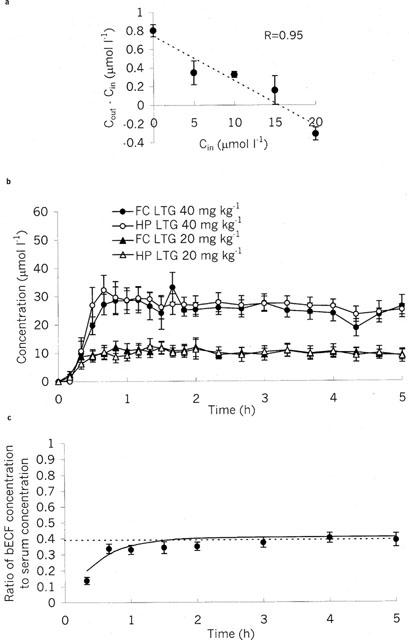
Extracellular fluid pharmacokinetics of lamotrigine. (a) Calculation of in vivo microdialysis probe recoveries. Cout is the concentration of lamotrigine in dialysate. Cin is concentration of lamotrigine in perfusate. Slope of line through points gives recovery (−0.048±0.003), and intercept gives extracellular concentration (−15.6±1.3 μmol l−1). Data are presented as mean±s.e.mean for three rats. (b) Microdialysis concentrations of lamotrigine corrected for in vivo recovery in hippocampus (HP) and frontal cortex (FC) following intraperitoneal injection of lamotrigine (LTG: 20 or 40 mg kg−1). Data are presented as mean±s.e.mean of six rats. (c) Lamotrigine bECF:serum concentration ratio-time profile. Dotted line indicates free serum concentration ratio. Continuous line indicates modelled values (see Methods). Data are presented as mean±s.e.mean of eight rats.
CSF and bECF neuropharmacokinetics
Lamotrigine was detectable in the CSF at the first time point, 10 min post-dose (Figure 2a): At both doses the concentration then rose over a period of 0.5–1 h up to a maximum, and then slowly declined. Similarly, lamotrigine appears rapidly in brain bECF (Figure 3b). The neuropharmacokinetics of lamotrigine in hippocampus and frontal cortex were indistinguishable (Figure 3b). The CSF to serum concentration ratio increased to a steady state value of 0.61 with a half-time of 0.42±0.15 h (Figure 2b), and the corrected dialysate to serum concentration ratio increased to a steady state value of 0.40 with a half-time of 0.51±0.11 h (Figure 3c). Using multifactorial ANOVA, there was no significant effect of dose, brain area (hippocampus vs frontal cortex) or fluid compartment (CSF or bECF) on the rise constant. The steady state ratio was not dose dependent, but was dependent on compartment with a significant difference between CSF and corrected bECF dialysate to serum concentration ratios (P<0.001).
Figure 2.
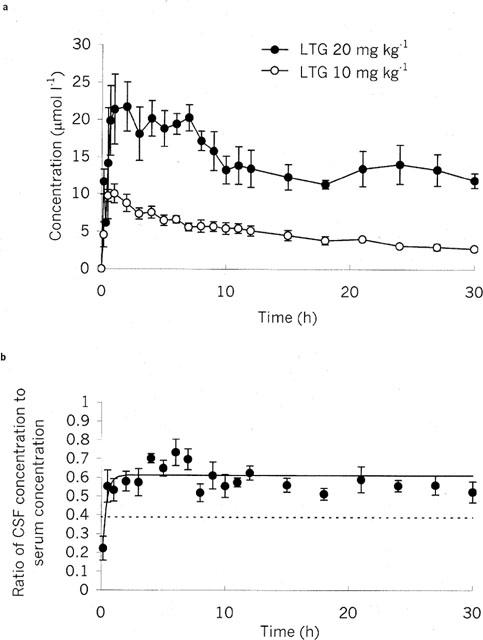
(a) Cerebrospinal fluid pharmacokinetics of lamotrigine following intraperitoneal injection of lamotrigine (LTG: 10 or 20 mg kg−1). Data are presented as mean±s.e.mean of 5–6 rats. (b) Lamotrigine cerebrospinal fluid:serum concentration ratio-time profile. Dotted line indicates free serum concentration ratio. Continuous line indicates modelled values (see Methods). Data are presented as mean±s.e.mean of eight rats.
Discussion
Although as part of its preclinical development many studies were undertaken to determine the blood (serum) pharmacokinetics of lamotrigine in a variety of species, there is a sparcity of published data (Parsons et al., 1995). Furthermore, as far as we are aware, there are no published data describing acute neuropharmacokinetics of lamotrigine and this is the first report on the temporal inter-relationship of lamotrigine serum pharmacokinetics, CSF and bECF neuropharmacokinetics. The rat models used have numerous advantages: (1) as each rat serves as its own control, interindividual variability (as occurs when composite values of individual rats killed at different time points after drug administration are used) is minimized and thus the number of experimental animals needed to obtain detailed kinetic data is reduced; (2) long sampling protocols can be used (up to 30 h in the present study) in determining drug kinetics; and (3) microdialysis permits monitoring of pharmacodynamically relevant bECF compartment and simultaneous sampling from more than one brain region. Microdialysis techniques result in minimal pertubation to the system measured; CSF sampling, however, does involve the withdrawal of fluid. The volume of fluid collected represents less than 12% of the total CSF volume, and since the rate of secretion is 2 μl min−1, then the volume would be replaced in 10 min (Baudrie et al., 1990; Davson & Segal, 1995). The maximum rate of sampling of 20 μl every 20 min is significantly less than the CSF replacement rate, and would thus not be expected and has not been observed to perturb significantly the concentration profile of other antiepileptic drugs (Patsalos et al., 1992; Lolin et al., 1994; Nagaki et al., 1999).
After intraperitoneal administration, lamotrigine rapidly appeared in serum (mean Tmax 0.5 h) suggesting ready penetration from the peritoneal cavity. The serum pharmacokinetics are biphasic with the first phase probably representing distribution from the blood compartment, and the second phase representing mainly elimination. The prolonged elimination phase in rat is due to rat's inability to efficiently conjugate lamotrigine, and indeed, the method of elimination differs significantly between rat and man (Dickins et al., 1995). In addition, lamotrigine accumulates in the kidney of male rats such that the kidney to serum concentration ratio can be up to 300 : 1 (Parsons et al., 1995). This effect is both gender and species specific, and could also partly explain the maintenance of serum concentrations in our study. These kinetic effects would predict a prolonged action of lamotrigine in rat models of epilepsy, and are important in the determination of lamotrigine's effects in chronic animal models (e.g. kindling), and in comparing lamotrigine with other AEDs in rat models. Indeed, in a comparison of AEDs in the maximal electroshock model, oral lamotrigine had the longest duration of action of a number of AEDs; it had an almost constant ED50 from 1–8 h post dose, and at 25 h post dose the ED50 was only 3–4 times the peak ED50 (Miller et al., 1986).
The mean±s.e.mean serum free fraction of lamotrigine as determined by ultracentrifugation was 0.39±0.01% and was not concentration dependent over the concentration range of 5–52 μmol l−1. This is similar to a previously reported value of 0.46 for rat serum determined by equilibrium dialysis (Parsons et al., 1995).
In order to determine the rate of entry into the CSF and bECF compartment, we used a mathematical model that assumes that the rise in serum concentration is instantaneous and that the serum concentration then does not fall. It remains a useful estimate in situations in which there is a rapid rise in serum concentration and then a slow decay, as with our data (Loscher & Frey, 1984; Mayer et al., 1959). The effect of using intraperitoneal injection and so a slower rise time for the serum concentration is to underestimate the half-time for rise of the CSF or bECF to concentration ratio. Thus the estimated penetration half-times of 0.42 h for CSF and 0.51 h for bECF are likely to be underestimates. Lamotrigine thus has a slow rate of CNS penetration, which is slower than values derived for phenytoin, phenobarbitone, and the benzodiazepines (Loscher & Frey, 1984). Indeed, this slow penetrance could militate against lamotrigine's use in acute seizures.
The CSF to serum concentration ratio rises to a steady-state value (0.62) that is greater than the unbound to total serum concentration ratio. This can be explained by the fact that the hypothesis that the free serum drug concentration is the tissue exchangeable drug concentration is incorrect; indeed, a number of studies have demonstrated that serum protein bound drug is often available for transport into the brain (Urien et al., 1987; Pardridge et al., 1983; Cornford et al., 1992; Lolin et al., 1994). The CSF to serum concentration ratio at steady state is substantially less than the total brain to serum concentration ratio at steady state, which in rats is approximately two (Walton et al., 1996). This suggests that there is significant binding and/or intracellular accumulation in the brain that could act as a potential reservoir.
Due to the peculiar serum pharmacokinetics of lamotrigine – the serum concentration decays only slowly after approximately 8 h – it was possible for us to calculate the in vivo recovery of the microdialysis probes using the no-net-flux method (Lonnroth et al., 1987; de-Lange et al., 1997). This is dependent on an unvarying bECF concentration of the drug during the study – a fact that we confirmed by comparing the dialysate concentration before and after the no-net-flux study. The in vitro microdialysis probe recovery was approximately double the calculated in vivo microdialysis probe recovery. There are a number of factors that may contribute to this (de-Lange et al., 1997). The in vitro recovery is performed in a solution of the substrate, which is very different from the in vivo situation in which the microdialysis probe is surrounded by brain tissue, which may inhibit free diffusion. Also the microdialysis probes are in place for 2 days before the experiment takes place; this is to allow recovery of the animals from surgery (Patsalos et al., 1992). During this time, however, proteins bind to the dialysis membrane and are likely to decrease substantially the permeability of the membrane; in vivo microdialysis probe recoveries after a similar period of time after implantation have been shown to be up to 2 fold lower than the in vitro recoveries (de-Lange et al., 1994). The marked difference between in vivo and in vitro recoveries emphasizes the importance of calculating in vivo recoveries in order to get accurate measures of the bECF concentration. Correcting for estimated in vivo recovery, resulted in bECF concentration measurements that at steady-state approach the free serum concentration. These values are significantly smaller than the measured CSF concentrations at steady-state, emphasizing the difference between these two compartments; thus lamotrigine concentrations within the CSF compartment are not always an accurate indicator of concentrations in the bECF compartment. Furthermore the higher concentrations in the CSF suggest that the choroid plexus is playing a permissive role, perhaps actively transporting the drug.
The concentrations in hippocampus and frontal cortex were identical; this contrasts with studies in which we found higher concentrations of phenytoin in the hippocampus than in the frontal cortex (Walker et al., 1996), and higher concentrations of vigabatrin in the frontal cortex than in the hippocampus (Patsalos et al., 1999). The brain distribution of AEDs may partly explain spectrum of antiepileptic activity, and it is interesting that lamotrigine and phenytoin, two drugs that have similar mechanisms of action, should have different spectrums of antiepileptic activity – lamotrigine, but not phenytoin is effective in absences.
This study emphasizes the importance of determining both the peripheral (serum) and central (CSF and bECF) kinetics of a drug using techniques that permit concurrent sampling over an extensive period. Such data are essential if appropriate dosing strategies are to be employed to study the pharmacodynamics of a drug and also to aid appropriate interpretation of experimental data.
Acknowledgments
We wish to thank the Wellcome Trust for supporting the work of M.C. Walker and H. Perry, Mr A.A. Elyas for his invaluable assistance in the h.p.l.c. analysis and Glaxo-Wellcome for the supply of lamotrigine.
Abbreviations
- AED
antiepileptic drug
- bECF
brain extracellular fluid
- Cmax
maximum concentration
- CSF
cerebrospinal fluid
- Ct
concentration at time t
- FC
frontal cortex
- HC
hippocampus
- h.p.l.c.
high performance liquid chromatography
- LTG
lamotrigine
- Tmax
time to maximum concentration
References
- BAUDRIE V., ROULLET J.B., GOUREAU Y., CHAOULOFF F., ELGHOZI J.L. Determination of cerebrospinal fluid production rate using a push-pull perfusion procedure in the conscious rat. Fundam. Clin. Pharmacol. 1990;4:269–274. doi: 10.1111/j.1472-8206.1990.tb00494.x. [DOI] [PubMed] [Google Scholar]
- CORNFORD E.M., YOUNG D., PAXTON J.W., SOFIA R.D. Blood-brain barrier penetration of felbamate. Epilepsia. 1992;33:944–954. doi: 10.1111/j.1528-1157.1992.tb02205.x. [DOI] [PubMed] [Google Scholar]
- CRUMRINE R.C., BERGSTRAND K., COOPER A.T., FAISON W.L., COOPER B.R. Lamotrigine protects hippocampal CA1 neurons from ischemic damage after cardiac arrest. Stroke. 1997;28:2230–2236. doi: 10.1161/01.str.28.11.2230. [DOI] [PubMed] [Google Scholar]
- DAVSON H., SEGAL M.B. Physiology of the CSF and blood brain barriers. Florida: CRC Press; 1995. [Google Scholar]
- DE-LANGE E.C., BOUW M.R., MANDEMA J.W., DANHOF M., DE-BOER A.G., BREIMER D.D. Application of intracerebral microdialysis to study regional distribution kinetics of drugs in rat brain. Br. J. Pharmacol. 1995;116:2538–2544. doi: 10.1111/j.1476-5381.1995.tb15107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE-LANGE E.C., DANHOF M., DE-BOER A.G., BREIMER D.D. Critical factors of intracerebral microdialysis as a technique to determine the pharmacokinetics of drugs in rat brain. Brain Res. 1994;666:1–8. doi: 10.1016/0006-8993(94)90276-3. [DOI] [PubMed] [Google Scholar]
- DE-LANGE E.C., DANHOF M., DE-BOER A.G., BREIMER D.D. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Brain Res. Rev. 1997;25:27–49. doi: 10.1016/s0165-0173(97)00014-3. [DOI] [PubMed] [Google Scholar]
- DICKINS M., SAWYER D.A., MORLEY T.J., PARSONS D.N.Lamotrigine: Chemistry and biotransformation Antiepileptic drugs 1995New York: Raven Press; 871–875.ed. Levy, R.H., Mattson, R.H. & Meldrum, B.S. pp [Google Scholar]
- DOHENY H.C., RATNARAJ N., WHITTINGTON M.A., JEFFERYS J.G., PATSALOS P.N. Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Res. 1999;34:161–168. doi: 10.1016/s0920-1211(98)00104-1. [DOI] [PubMed] [Google Scholar]
- FITTON A., GOA K.L. Lamotrigine. An update of its pharmacology and therapeutic use in epilepsy. Drugs. 1995;50:691–713. doi: 10.2165/00003495-199550040-00008. [DOI] [PubMed] [Google Scholar]
- HUTSON P.H., SARNA G.S., KANTAMANENI B.D., CURZON G. Monitoring the effect of a tryptophan load on brain indole metabolism in freely moving rats by simultaneous cerebrospinal fluid monitoring and brain dialysis. J. Neurochem. 1985;44:1266–1274. doi: 10.1111/j.1471-4159.1985.tb08753.x. [DOI] [PubMed] [Google Scholar]
- LOLIN Y.I., RATNARAJ N., HJELM M., PATSALOS P.N. Antiepileptic drug pharmacokinetics and neuropharmacokinetics in individual rats by repetitive withdrawal of blood and cerebrospinal fluid: phenytoin. Epilepsy Res. 1994;19:99–110. doi: 10.1016/0920-1211(94)90020-5. [DOI] [PubMed] [Google Scholar]
- LONNROTH P., JANSSON P.A., SMITH U. A microdialysis method allowing characterization of intercellular water space in humans. Am. J. Physiol. 1987;253:E228–E231. doi: 10.1152/ajpendo.1987.253.2.E228. [DOI] [PubMed] [Google Scholar]
- LOSCHER W., FREY H.H. Kinetics of penetration of common antiepileptic drugs into cerebrospinal fluid. Epilepsia. 1984;25:346–352. doi: 10.1111/j.1528-1157.1984.tb04199.x. [DOI] [PubMed] [Google Scholar]
- MAYER S., MAICKEL R.P., BRODIE B.B. Kinetics of penetration of drugs and other foreign compounds into the cerebrospinal fluid and brain. J. Pharmacol. Exp. Ther. 1959;127:205–211. [Google Scholar]
- MCGEER E.G., ZHU S.G. Lamotrigine protects against kainate but not ibotenate lesions in rat striatum. Neurosci. Lett. 1990;112:348–351. doi: 10.1016/0304-3940(90)90229-3. [DOI] [PubMed] [Google Scholar]
- MILLER A.A., WHEATLEY P., SAWYER D.A., BAXTER M.G., ROTH B. Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: I. Anticonvulsant profile in mice and rats. Epilepsia. 1986;27:483–489. doi: 10.1111/j.1528-1157.1986.tb03572.x. [DOI] [PubMed] [Google Scholar]
- NAGAKI S., RATNARAJ N., PATSALOS P.N. Blood and cerebrospinal fluid pharmacokinetics of primidone and its primary pharmacologically active metabolites, phenobarbital and phenylethylmalonamide in the rat. Eur. J. Drug Metab. Ph. 1999;24:255–264. doi: 10.1007/BF03190029. [DOI] [PubMed] [Google Scholar]
- PARDRIDGE W.M., SAKIYAMA R., FIERER G. Transport of propranolol and lidocaine through the rat blood-brain barrier. Primary role of globulin-bound drug. J. Clin. Invest. 1983;71:900–908. doi: 10.1172/JCI110844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARSONS D.N., DICKINS M., MORLEY T.J.Lamotrigine: absorption, distribution and excretion Antiepileptic drugs 1995New York: Raven Press; 877–881.ed. Levy, R.H., Mattson, R.H. & Meldrum, B.S. pp [Google Scholar]
- PATSALOS P.N., ABED W.T., ALAVIJEH M.S., O'CONNELL M.T. The use of microdialysis for the study of drug kinetics: some methodological considerations illustrated with antipyrine in rat frontal cortex. Br. J. Pharmacol. 1995;115:503–509. doi: 10.1111/j.1476-5381.1995.tb16362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATSALOS P.N., ALAVIJEH M.S., SEMBA J., LOLIN Y.I. A freely moving and behaving rat model for the chronic and simultaneous study of drug pharmacokinetics (blood) and neuropharmacokinetics (cerebrospinal fluid): hematological and biochemical characterization and kinetic evaluation using carbamazepine. J. Pharmacol. Toxicol. Methods. 1992;28:21–28. doi: 10.1016/1056-8719(92)90061-5. [DOI] [PubMed] [Google Scholar]
- PATSALOS P.N., TONG X., RATNARAJ N. Intracerebral microdialysis shows that vigabatrin does not exhibit ubiquitous brain extracellular distribution. Seizure. 1999;8:372. [Google Scholar]
- PAXINOS G., WATSON C. The rat brain in stereotaxic co-ordinates. 2nd edition. San Diego: Academic Press; 1986. [Google Scholar]
- RATAUD J., DEBARNOT F., MARY V., PRATT J., STUTZMANN J.M. Comparative study of voltage-sensitive sodium channel blockers in focal ischaemia and electric convulsions in rodents. Neurosci. Lett. 1994;172:19–23. doi: 10.1016/0304-3940(94)90652-1. [DOI] [PubMed] [Google Scholar]
- SECHI G.P., PETRUZZI V., ROSATI G., TANCA S., MONACO F., FORMATO M., RUBATTU L., DE-RIU P. Brain interstitial fluid and intracellular distribution of phenytoin. Epilepsia. 1989;30:235–239. doi: 10.1111/j.1528-1157.1989.tb05460.x. [DOI] [PubMed] [Google Scholar]
- SEMBA J., CURZON G., PATSALOS P.N. Antiepileptic drug pharmacokinetics and neuropharmacokinetics in individual rats by repetitive withdrawal of blood and cerebrospinal fluid: milacemide. Br. J. Pharmacol. 1993;108:1117–1124. doi: 10.1111/j.1476-5381.1993.tb13514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH S.E., MELDRUM B.S. Cerebroprotective effect of lamotrigine after focal ischemia in rats. Stroke. 1995;26:117–121. doi: 10.1161/01.str.26.1.117. [DOI] [PubMed] [Google Scholar]
- THOMAS S.A., SEGAL M.B. The transport of the anti-HIV drug, 2′,3′-didehydro-3′-deoxythymidine (D4T), across the blood-brain and blood-cerebrospinal fluid barriers. Br. J. Pharmacol. 1998;125:49–54. doi: 10.1038/sj.bjp.0702044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- URIEN S., PINQUIER J.L., PAQUETTE B., CHAUMET R.P., KIECHEL J.R., TILLEMENT J.P. Effect of the binding of isradipine and darodipine to different plasma proteins on their transfer through the rat blood-brain barrier. Drug binding to lipoproteins does not limit the transfer of drug. J. Pharmacol. Exp. Ther. 1987;242:349–353. [PubMed] [Google Scholar]
- WALKER M.C., ALAVIJEH M.S., SHORVON S.D., PATSALOS P.N. Microdialysis study of the neuropharmacokinetics of phenytoin in rat hippocampus and frontal cortex. Epilepsia. 1996;37:421–427. doi: 10.1111/j.1528-1157.1996.tb00586.x. [DOI] [PubMed] [Google Scholar]
- WALKER M.C., SANDER J.W.Lamotrigine Antiepileptic Drugs: pharmacology and therapeutics 1999Berlin: Springer-Verlag; 331–338.ed. M.J. Eadie, M.E. & Vajda, F. pp [Google Scholar]
- WALKER M.C., TONG X., BROWN S., SHORVON S.D., PATSALOS P.N. Comparison of single- and repeated-dose pharmacokinetics of diazepam. Epilepsia. 1998;39:283–289. doi: 10.1111/j.1528-1157.1998.tb01374.x. [DOI] [PubMed] [Google Scholar]
- WALTON N.Y., JAING Q., HYUN B., TREIMAN D.M. Lamotrigine vs phenytoin for treatment of status epilepticus: comparison in an experimental model. Epilepsy Res. 1996;24:19–28. doi: 10.1016/0920-1211(96)00007-1. [DOI] [PubMed] [Google Scholar]
- WIARD R.P., DICKERSON M.C., BEEK O., NORTON R., COOPER B.R. Neuroprotective properties of the novel antiepileptic lamotrigine in a gerbil model of global cerebral ischemia. Stroke. 1995;26:466–472. doi: 10.1161/01.str.26.3.466. [DOI] [PubMed] [Google Scholar]
- WILDER B.J., RAMSAY R.E., WILLMORE L.J., FEUSSNER G.F., PERCHALSKI R.J., SHUMATE J.B., JR Efficacy of intravenous phenytoin in the treatment of status epilepticus, kinetics of central nervous system penetration. Ann. Neurol. 1977;1:511–518. doi: 10.1002/ana.410010602. [DOI] [PubMed] [Google Scholar]