

Abstract
The contribution of endothelium-derived hyperpolarizing factor (EDHF), nitric oxide (NO) and a prostanoid (PG) to endothelium-dependent hyperpolarization and relaxation were assessed in coronary and mammary arteries of guinea-pigs by integration of the responses evoked during discrete applications of acetylcholine (ACh). The results of this integration approach were compared with those using traditional peak analysis methods.
Nω-nitro-L-arginine methyl ester (L-NAME, 100 μM) and indomethacin (1 μM), alone or in combination, were without effect on peak hyperpolarizations or relaxations while they markedly reduced the integrated responses in both arteries.
Integrated responses attributed to NO and PG were larger than those attributed to EDHF in the coronary artery (at 2 μM ACh, hyperpolarization (mV s): NO, 4200±91; PG, 5046±157; EDHF, 1532±94; relaxation (mN s mm−1): NO, 2488±122; PG, 2234±96; EDHF, 802±54). Integrated responses attributed to NO, PG and EDHF were similar in the mammary artery (at 2 μM ACh, hyperpolarization: NO, 347±69; PG, 217±49; EDHF, 310±63; relaxation: NO, 462±94; PG, 456±144; EDHF, 458±40).
Gilbenclamide (1 μM) all but abolished the hyperpolarization attributable to NO and PG but not EDHF in both arteries allowing assessment of the role of the hyperpolarization in relaxation. Gilbenclamide was without effect on the integrated relaxation due to NO but significantly reduced the relaxation associated with PG in the two arteries.
In conclusion, integration of the responses enabled a more complete assessment of the contribution of EDHF, NO and PG to endothelium-dependent responses, which were strikingly different in the two arteries. There is commonality in the role of hyperpolarization in relaxation in both arteries: EDHF-dependent relaxation is strongly dependent on hyperpolarization; hyperpolarization plays an important role in PG relaxation, whereas it has a small facilitatory role in NO-dependent relaxation.
Keywords: Endothelium-derived hyperpolarizing factor, nitric oxide, prostacyclin, coronary artery, mammary artery, hyperpolarization, relaxation, vascular smooth muscle, membrane potential
Introduction
The endothelium is now recognised as a vital organ in haemodynamic regulation through the release of potent vasodilators. The endothelium can produce at least three vasodilators: nitric oxide (NO), a prostanoid (PG, mainly prostacyclin) and endothelium-derived hyperpolarizing factor (EDHF). The contribution of each of these factors to endothelium-dependent vasodilatation varies across vascular beds and also with the agent used to stimulate the endothelium. In general, the endothelium releases predominantly NO in large arteries (Nagao et al., 1992) such as rabbit carotid artery (Cohen et al., 1997), while the contribution of EDHF assumes importance in smaller resistance arteries (Nagao et al., 1992; Garland et al., 1995) such as the rat mesenteric artery (Shimokawa et al., 1996), and arterioles. The involvement of PG in the regulation of vascular tone has not been as widely reported, often overlooked due to experimental design in which cyclo-oxygenase blockers were present from the onset of experimentation. Therefore the role of PG in regulating vessels of different size awaits elucidation. In some vessels such as the coronary (Parkington et al., 1993) and mammary arteries (Hammarström et al., 1999) of guinea-pigs, hepatic artery of rats (Zygmunt et al., 1998) and mesenteric artery of rabbits (Murphy & Brayden, 1995) all three factors contribute to the vasodilator response evoked by stimulation of the endothelium. However, a comparative evaluation of the relative role of the three vasorelaxants in the same study is rare.
Each of the endothelial factors may rely on different mechanisms to produce relaxation. NO and prostacyclin are known to stimulate the production of guanosine 3′ : 5′ cyclic monophosphate (cyclic GMP) and adenosine 3′ : 5′ cyclic monophosphate (cyclic AMP) and associated intracellular pathways (Holzmann et al., 1980; Rapoport & Murad, 1983; Ignarro et al., 1984) which may facilitate relaxation by reducing cytoplasmic Ca2+ and decreasing the sensitivity of the contractile apparatus to Ca2+. These vasodilators may also open K+ channels either directly or through their respective second messengers (Archer et al., 1994; Bolotina et al., 1994; Kubo et al., 1994; Quayle et al., 1994). The K+ channels involved in the responses to NO and prostacyclin reportedly vary according to the artery and species. K+ channels implicated in NO-mediated responses include large conductance Ca2+-activated K+ channels (BKCa) (Archer et al., 1994; Bolotina et al., 1994) and ATP-sensitive K+ channels (Garland & McPherson, 1992; Murphy & Brayden, 1995; Parkington et al., 1995). BKCa and KATP and delayed rectifier K+ channels (Kv) are involved in the PG response to varying extents in a variety of arteries (Kubo et al., 1994; Parkington et al., 1995; Schubert et al., 1997; Nishiyama et al., 1998; Dong et al., 1998). Thus NO and prostacyclin can elicit relaxation via mechanisms that are both dependent and independent of membrane potential.
There is considerable controversy regarding the identity of EDHF, and whether it is a diffusible factor or reflects electrotonic spread of hyperpolarization from the endothelium to the underlying smooth muscle via myoendothelial gap junctions (Garland et al., 1995; Bény, 1997; Edwards & Weston, 1998; Edwards et al., 1998; Fisslthaler et al., 1999). The hyperpolarization evoked by EDHF, unlike that produced by NO and PG, is believed to play a key role in its relaxation. Inhibition of the EDHF hyperpolarization with a combination of charybdotoxin and apamin, inhibitors of BKCa and intermediate (IKCa) and small conductance Ca2+-activated K+ channels (SKCa), results in abolition of the EDHF relaxation (Zygmunt et al., 1998).
The contribution of membrane potential-dependent and -independent mechanisms to smooth muscle relaxation is likely to be complex. Membrane potential dependent mechanisms are likely to have more immediate effects on relaxation whereas the second messenger-mediated pathways will tend to prolong the response. Thus an optimal assessment of the involvement of each of the endothelium-derived factors to the total response must take into consideration both the amplitude and duration of the hyperpolarization and relaxation. This can be achieved by integrating the responses, which requires separate applications of different concentrations of ACh rather than cumulative application. In this study membrane potential and tension were recorded simultaneously, allowing the determination of the role of the hyperpolarization in the relaxation produced by each of the endothelial factors. The aims of the present study were: (1) to compare an integration method of analysis with conventional peak measurement methods. We then went on to use the integration method, (2) to investigate the relative contribution of EDHF, NO and PG to hyperpolarization and relaxation in two different arteries where all three factors are released. Finally, glibenclamide blocks hyperpolarization attributed to NO and/or PG in a variety of arteries (e.g. Garland & McPherson 1992; Murphy & Brayden, 1995; Parkington et al., 1995; Corriu et al., 1996), including guinea-pig coronary and mammary arteries, but is without effect on the hyperpolarization produced by EDHF. Thus, using glibenclamide, (3) the role of the hyperpolarization in relaxation produced by endogenous NO and PG and EDHF was investigated.
Methods
Preparations
Guinea-pigs of either gender (300–800 g) were killed by decapitation. Ring segments (1–2 mm in length) of mammary and main descending coronary arteries were mounted on a Mulvany-style myograph as previously described (Parkington et al., 1993). The resting mean internal diameters of coronary and mammary arteries were 382±12 μm (n=14) and 331±9 μm (n=10), respectively. The tissues were continuously superfused at 3.6 ml min−1 with Krebs' solution (at 35°C) containing (mM): NaCl 120; KCl 5; CaCl2 2.5; MgSO4 1.2; KH2PO4 1; NaHCO3 25; glucose 11; bubbled with O2 95% and CO2 5%. The segments of artery were stretched in increments to a tension equivalent to about 60 mmHg, the mean arterial blood pressure in guinea-pigs. Membrane potential and isometric tension were recorded simultaneously as described previously (Parkington et al., 1993). Conventional intracellular microelectrodes were filled with 1 M KCl and had resistances of around 100 MΩ. The data were recorded on a chart recorder (Graphtec Linearcorder, Japan) and stored on video cassette using a digital data recorder (Instrutech Corporation, U.S.A.) for later analysis.
Experiments
The arteries were depolarized and constricted by including the stable thromboxane A2 analogue U 46619 in the superfusate. To stimulate the endothelium, acetylcholine (ACh) was injected into the perfusion line close to the tissue (1 s delay) for a duration of 10 s. The concentration of ACh reached equilibrium within 3–5 s (see Parkington et al., 1993). Each concentration of ACh (8 nM–20 μM) was applied discretely and the membrane potential and tension were allowed to recover completely before the next concentration was tested. Production of NO by the endothelium was blocked using Nω-nitro-L-arginine methyl ester (L-NAME) (50 or 100 μM). Synthesis of PGs was inhibited using indomethacin (1 μM). These blockers were added to the superfusate for at least 30 min prior to and during endothelial stimulation. These concentrations of the blockers were selected as higher concentrations were found to be without additional inhibitory effect.
Analysis of data
The peak amplitudes of the hyperpolarizations and relaxations were measured from the mean level of membrane potential (mV) and tension (mN) just prior to application of ACh to the maximum level of the response. Relaxations were corrected for the length of the tissue (mN mm−1) (see Mulvany & Halpern, 1977).
To take into consideration the duration as well as the amplitude of the responses, the hyperpolarizations and relaxations were integrated from chart records using SigmaScan software (Jandel Scientific, Inc., CA, U.S.A.). In Figure 3 the responses evoked by each endothelial vasodilator were deduced using the following method: the response attributable to the PG was calculated by subtracting the integrated response in the presence of L-NAME plus indomethacin from that obtained in L-NAME alone. Similarly, the component of the response ascribed to NO was determined by subtracting the responses in L-NAME plus indomethacin from the responses obtained in the presence of indomethacin alone. The responses that remained in the presence of both blockers were attributed to EDHF.
Figure 3.
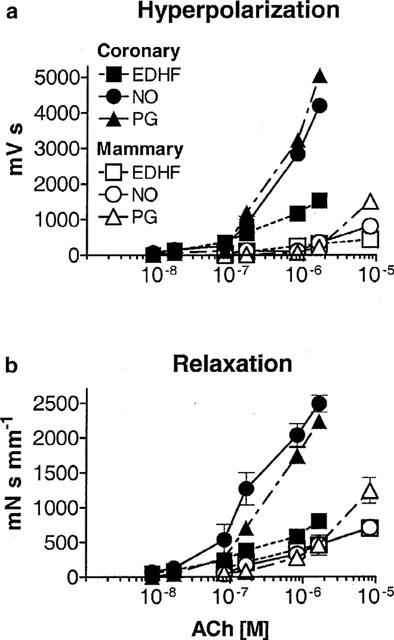
Absolute integrated hyperpolarization (a) and relaxation (b) responses. The components ascribed to NO (n=5) and a PG (n=5) were obtained by subtracting the integral of the responses in L-NAME plus indomethacin (that is, the response attributed to EDHF, n=10) from those obtained in indomethacin or L-NAME alone, respectively (see Methods).
When concentration-response curves were calculated, a sigmoid curve was fitted to the data using the least-squares method, with the aid of the computer software package Prism (GraphPad Software, Inc., CA, U.S.A.). The concentration of agonist which was effective in producing a response that was 50% of maximum (EC50) was determined for each curve. pD2 values (−log EC50) have been quoted throughout.
The standard error based on the number of animals used (n) is quoted with each value of the mean. Student's t-test was used to test for significance between means. To examine the relationship between hyperpolarization and relaxation two way analysis of variance (ANOVA) with repeated measures was used. A level of P<0.05 was accepted as significant throughout.
Drugs
ACh, L-NAME and indomethacin were obtained from Sigma (St. Louis, U.S.A.), U 46619 was from Cayman Chemical Co. (Ann Arbor, U.S.A.), and glibenclamide was a kind gift from Hoechst.
Indomethacin was dissolved in 0.1 M Na2CO3 (stock 10 mM). U 46619 was diluted using 12% ethylacetate and 88% deionized water (stock 100 μM). Glibenclamide was dissolved in dimethyl sulphoxide (DMSO) (stock 10 mM). All stock solutions were further diluted with Krebs' solution. The final dilution of DMSO in Krebs' solution was 1 in 10,000. The final concentration of both DMSO and ethylacetate in the Krebs' solution were without effect on the membrane potential or tension of either artery.
Results
Depolarization and constriction of the arteries with U 46619
Exposure of the coronary and mammary arteries to either L-NAME or indomethacin alone was associated with membrane depolarization (membrane potentials: coronary: −53±2 mV, n=6, P<0.05, and −57±1 mV, n=6, P<0.05; mammary: −58±1 mV, n=5, P<0.05, and −60±1 mV, n=5, P>0.05, in L-NAME or indomethacin, respectively; refer to Table 1 for resting membrane potentials in the absence of blockers). In the presence of either L-NAME or indomethacin, U 46619 (30–70 nM) evoked sustained depolarization to −38±1 mV (n=13) and contraction of 6.0±0.3 mN mm−1 (56% of maximal contraction, n=13) in coronary arteries. A similar level of tension was achieved using 20–30 nM U 46619 in the mammary artery, which evoked depolarization to −38±1 mV (n=10) and contraction of 6.5±0.4 mN mm−1 (58% of maximal, n=10) (Figure 1). Oscillations of approximately 5 mV in amplitude occurred in the membrane potential, and these were often topped with spike action potentials, and were associated with phasic oscillations in tension (Figure 1). Thus, in the presence of U 46619 and one blocker (either L-NAME or indomethacin), the level of the membrane potential and tension in both arteries were similar at the commencement of each experiment.
Table 1.
Level of membrane potential and tension in coronary and mammary artery

Figure 1.
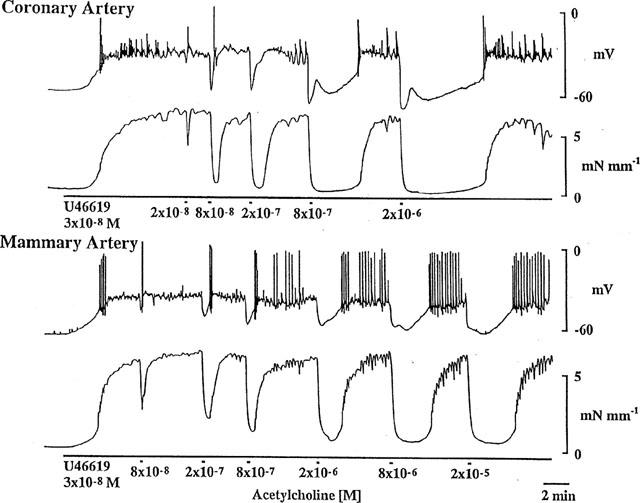
In the presence of indomethacin (1 μM), U 46619 (30 nM) depolarized (upper panel) and constricted (lower panel) the smooth muscle of ring segments of coronary and mammary arteries. ACh evoked concentration-dependent hyperpolarization and relaxation in both arteries. Continuous impalement in each tissue.
Characteristics of the responses evoked by discrete applications of ACh
In the presence of U 46619, ACh caused cessation of oscillations and action potential activity and concentration-dependent membrane hyperpolarization in both arteries (Figure 1). At lower concentrations of ACh the hyperpolarizations were typically monophasic, that is, consisting of only one discernible component. At higher concentrations the initial rapid hyperpolarization was followed by a slower component of longer duration, and thus the responses were biphasic in appearance. Both the monophasic and biphasic hyperpolarizations were associated with relaxations that were essentially monophasic and which were sustained for the duration of the hyperpolarization (Figures 1 and 4).
Figure 4.
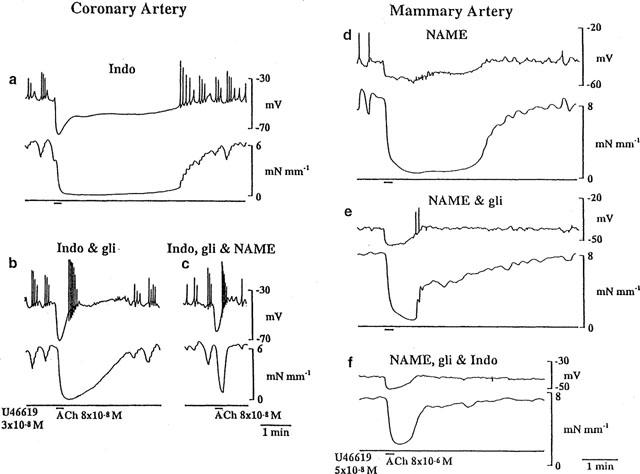
One example of the effects of each sequence of blockers used is shown in the coronary (a,b,c) and mammary (d,e,f) arteries. The arteries were depolarized and constricted with U 46619 in the presence of either indomethacin (1 μM) (a) or L-NAME (100 μM) (d). The endothelium was stimulated with ACh to evoke hyperpolarization and relaxation. This was repeated in the presence of glibenclamide (b,e) and then once more in the presence of all three inhibitors: +L-NAME (c) or +indomethacin (f).
As described previously for coronary and mammary artery, the slow component of the hyperpolarization and associated relaxation were attributed to the combined actions of NO and a prostanoid (PG) whereas the initial hyperpolarization and accompanying relaxation were attributed to EDHF (Parkington et al., 1995; Hammarström et al., 1999). When only a single inhibitor was applied, either L-NAME or indomethacin alone, the contribution of EDHF and a PG or NO could be distinguished only in the membrane potential response (Figures 1 and 4). In contrast, the relaxation always appeared monophasic, as a consequence of the merging of the individual contributions from EDHF and the other factor (NO or PG).
Actions of L-NAME and indomethacin: peak amplitude versus integrated responses
Peak responses
Since endothelium-dependent responses have previously been analysed in terms of peak amplitude, we used this method in the first instance. The amplitude of the initial component of the hyperpolarization, attributed to the actions of EDHF, was not influenced by L-NAME or indomethacin, or the combination of both blockers (Table 2) in either coronary (Figure 2a) or mammary arteries (Figure 2b). In contrast, the slow component of the hyperpolarization was virtually abolished by a combination of both L-NAME and indomethacin in both of these arteries (Figure 2a,b; Tables 1 and 2). The peak amplitudes of the relaxations in the coronary and mammary arteries were not influenced by any combination of the blockers used (Figure 2c,d; Table 1) and the pD2s are listed in Table 2.
Table 2.
pD2's for peak amplitude of hyperpolarization and relaxation evoked by ACh
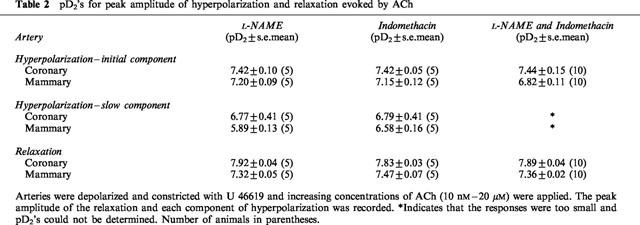
Figure 2.
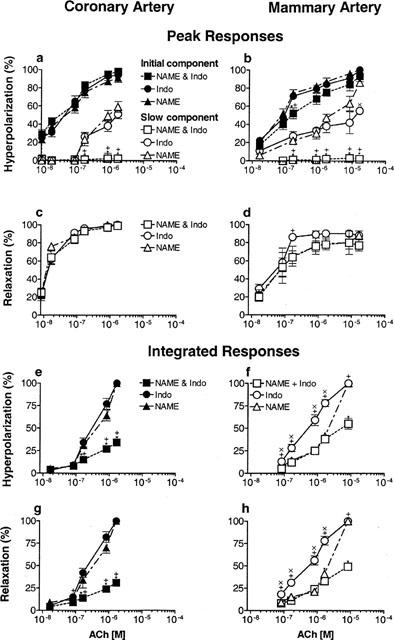
Arteries were depolarized and constricted with U 46619 and increasing concentrations of ACh between 8 nM and 1.7 μM in the coronary (a,c,e,g) and between 17 nM and 17 μM in the mammary artery (b,d,f,h), were applied. The effect of L-NAME (50 μM, n=5), indomethacin (1 μM, n=5) and a combination of both blockers (n=10) was examined on the peak level of the initial and slow components of the hyperpolarization (a,b) and the relaxation (c,d). The peaks were expressed as a percentage of the maximum peak hyperpolarization or relaxation. Results of integrating the hyperpolarization (e,f) and relaxation (g,h) responses from which the peak measurements had been made. The integrated results are expressed as a percentage of the largest integrated hyperpolarization or relaxation. *Significant difference between L-NAME and L-NAME plus indomethacin (Indo); +significant difference between indomethacin and L-NAME plus indomethacin; ×significant difference between L-NAME and indomethacin (P<0.05).
Integrated responses
The contribution of both the duration and amplitude to the hyperpolarizations and relaxations were analysed by integrating the same records as used for the peak amplitude analysis. Since the entire response was integrated, results for the hyperpolarization were not separated into initial and slow components. In the coronary artery, when one blocker was present the total integrated hyperpolarization was the same magnitude for every concentration of ACh regardless of whether NO+EDHF (in indomethacin alone) or PG+EDHF (in L-NAME alone) contributed to the response (Figure 2e). The same was observed for the integrated relaxation (Figure 2g). When both L-NAME and indomethacin were present, the integrated hyperpolarization (Figure 2e) and relaxation (Figure 2g) were significantly reduced. In the mammary artery the total integrated hyperpolarization and relaxation were conspicuously larger over the majority of the ACh concentration range when indomethacin alone was present (Figure 2f,h). The responses in L-NAME alone were only significantly different from those in the combination of L-NAME and indomethacin for the highest concentration of ACh (Figure 2f,h).
Contribution of NO, PG and EDHF to endothelium-dependent hyperpolarization and relaxation
The individual contributions of EDHF, NO and PG to the hyperpolarization and relaxation evoked by ACh were determined by subtraction of the integrated data (see Methods), and the results are presented in Figure 3. In the coronary artery, the integrated hyperpolarization and relaxation attributable to EDHF increased steadily as the concentration of ACh was raised. At the lower concentrations of ACh, NO and EDHF made approximately similar contributions to the integrated hyperpolarization. Above 100 nM ACh, the actions of a PG became evident and thereafter it and NO contributed approximately equally to the hyperpolarization and relaxation and to a much greater extent than EDHF (Figure 3a). Similar trends were also seen in the relaxation (Figure 3b). The proportionally smaller contribution of EDHF to the overall integrated responses is attributed to the short duration of the response because, as seen above, EDHF alone was capable of achieving full (99%) relaxation (Table 1). The integrated relaxation in the coronary artery attributable to NO and a PG were parallel, with the PG-evoked relaxation requiring a significant 2 fold higher concentration of ACh for its release (Figure 3b), although the hyperpolarizations attributable to these relaxants did not require different concentrations of ACh.
In preparations of mammary artery the integrated hyperpolarization attributable to NO and EDHF were similar up to 2 μM ACh, while the PG hyperpolarization was reduced. The relaxations produced by all factors seemed to be superimposed over this range. At the highest concentrations of ACh, the contribution of PG to hyperpolarization and relaxation was greater than the other two factors (Figure 3).
The integrated responses to ACh in the mammary artery were conspicuously smaller than those evoked in the coronary artery (Figure 3). This is attributed to the overall shorter duration of the responses in the mammary artery, particularly those parts of the response evoked by NO and PG and the smaller amplitude of the responses evoked by EDHF (Figure 1; Table 1).
Contribution of the NO- and PG-induced hyperpolarization to relaxation
In a separate series of experiments, glibenclamide (1 μM) was used to selectively inhibit the hyperpolarization produced by NO/PG (Figure 4b,e). The effects of blocking the hyperpolarization on the relaxation were investigated. The actions of glibenclamide were assessed using both peak amplitude and integration methods of analysis.
Peak responses
The ‘control' response was obtained in the presence of one blocker, either L-NAME or indomethacin, so that the actions of glibenclamide on the PG and NO response could be assessed directly. Therefore two separate series of experiments were completed: one where the blocker present throughout the entire experiment was L-NAME (Figures 5a,c,e,g; 6a,c), and the other series where the blocker was indomethacin (Figures 5b,d,f,h; 6b,d). In the presence of L-NAME or indomethacin, glibenclamide was without effect on the pD2 for the initial component of the hyperpolarization attributed to EDHF in either coronary (Table 3; Figure 5ab) or mammary (Table 3; Figure 5e,f) arteries. Subsequent addition of the second blocker, in the continued presence of glibenclamide was without effect on pD2 in the coronary or mammary arteries (Table 3, Figure 5a,b,e,f). There was a small but significant reduction in the maxima of the curves in the mammary artery (2-way ANOVA, L-NAME versus L-NAME+glibenclamide+indomethacin, P=0.047, Figure 5e; indomethacin or indomethacin+glibenclamide versus indomethacin+glibenclamide+L-NAME, P=0.02 and P=0.01, respectively, Figure 5f). In the mammary artery there is greater overlap between the initial and slow components of the hyperpolarization at the highest concentrations of ACh and this accounts for the apparent effect of glibenclamide on the initial component. Essentially, glibenclamide was without effect on the initial component of the hyperpolarization. In contrast, glibenclamide markedly depressed the peak amplitude of the slow component of the hyperpolarization due to PG (Figure 5c,g) and NO (Figure 5d,h) in both arteries.
Figure 5.
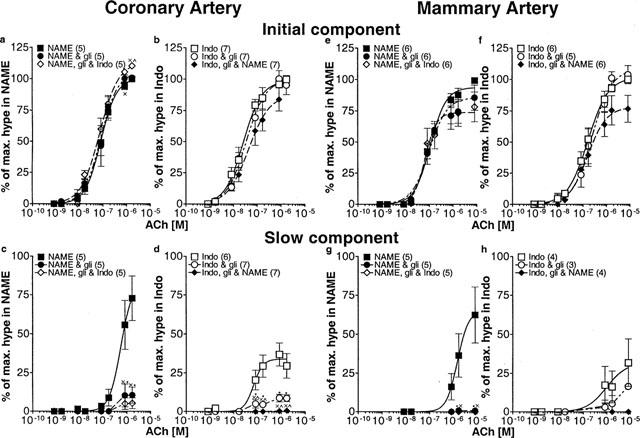
Effects of glibenclamide on the peak amplitudes of the initial and slow components of the ACh-induced hyperpolarization. Coronary (a–d) and mammary arteries (e–h) were depolarized and constricted with U 46619 and increasing concentrations of ACh were applied. Arteries were initially exposed to either L-NAME (100 μM) (a,c,e,g) or indomethacin (1 μM) (b,d,f,h) and then glibenclamide (1 μM) was applied before the addition of the other blocker (indomethacin or L-NAME). The initial component (a,b,e,f) and the slow components (c,d,g,h) of the hyperpolarization were expressed as a percentage of the maximal peak hyperpolarization. The number of animals indicated in parentheses. *Indicates significant difference between Control (either L-NAME (a,c,e,g) alone or indomethacin (b,d,f,h) alone) and glibenclamide; ×significant difference between Control and both blockers+glibenclamide; ×significance between one blocker (either L-NAME or indomethacin) +glibenclamide and both blockers+glibenclamide.
Figure 6.
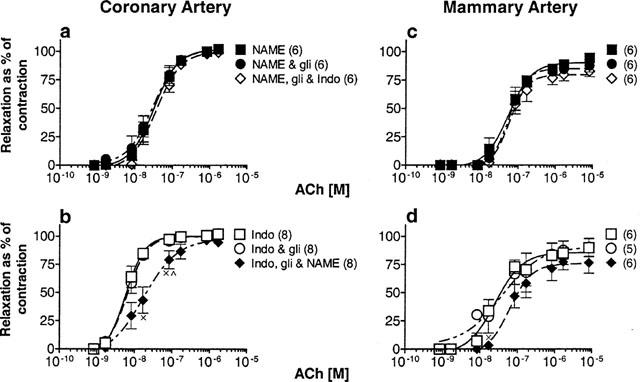
Effect of L-NAME (100 μM), glibenclamide (1 μM) and indomethacin (1 μM) on the peak amplitude of relaxation evoked by ACh in coronary and mammary arteries constricted with U 46619. These data correspond to those presented in Figure 5 and the same sequence of application of inhibitors applies. The relaxations were expressed as a percentage of the contraction evoked by U 46619. Shown are responses recorded in coronary (a,b) and mammary (c,d) arteries. The number of animals indicated in parentheses.×significant difference between indomethacin and indomethacin+glibenclamide+L-NAME; ×significant difference between indomethacin+glibenclamide and indomethacin+glibenclamide+L-NAME.
Table 3.
Effect of glibenclamide on the pD2's for peak amplitude of hyperpolarization and relaxation evoked by ACh
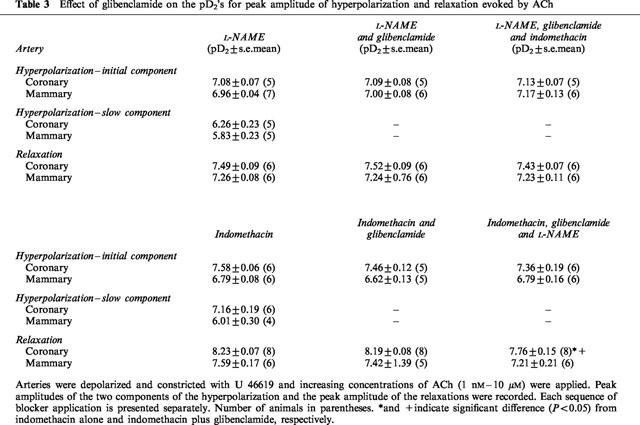
Although the amplitude of the slow component of the hyperpolarization was considerably reduced, glibenclamide was without effect on the absolute peak amplitude of the accompanying relaxation in either the coronary (Figure 6a,b) or mammary (Figure 6c,d) arteries. The pD2's for peak relaxation were not changed in glibenclamide (Table 3), neither were the maxima of the curves. The second blocker was then added in the presence of glibenclamide. When indomethacin was the second blocker there was no further effect on the amplitude of the relaxation in either artery (Figure 6a,c). However, when L-NAME was the second blocker to be applied, this caused a shift in the concentration-response curve for peak relaxation to the right in both arteries (Figure 6b,d), although this reached significance only in the coronary artery (Figure 6b) (P<0.05) (Table 3), but the maxima of the curves were not different.
Integrated responses
Since the action of glibenclamide was focused on the slow component of the hyperpolarization (Figure 5c,d,g,h) it had a greater impact on the duration than on the amplitude of relaxation, as can be seen in Figure 4b,e. Thus, the membrane potential and tension responses were integrated to take this aspect into consideration in the analysis.
Under conditions where NO synthesis had been blocked, subsequent addition of glibenclamide reduced both the integrated hyperpolarization (Figure 7a,d) and relaxation (Figure 7b,e) in coronary and mammary arteries. The reduction in the hyperpolarization and relaxation were equivalent in the coronary artery and for the lower concentrations of ACh in the mammary artery. However, at higher concentrations of ACh in the mammary artery, the reduction in relaxation (by 34±10%, n=6, at 8 μM, Figure 7e) was not as extensive as that of the hyperpolarization (by 53±15%, n=6, Figure 7d). Subsequent blockade of PG production with indomethacin was without further significant effect on the hyperpolarization or the relaxation in the coronary artery (Figure 7a,b). In the mammary artery indomethacin had no further effect on the hyperpolarization (Figure 7d) and only significantly reduced the relaxation evoked by the highest concentration of ACh (Figure 7e).
Figure 7.
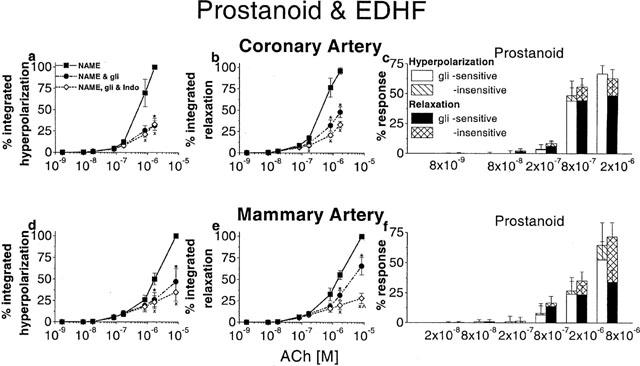
The involvement of a PG and EDHF in endothelium-dependent hyperpolarization and relaxation in coronary and mammary arteries of the guinea-pig. Arteries were depolarized and constricted with U 46619 in the presence of L-NAME (100 μM). Increasing concentrations of ACh were applied. The effects of glibenclamide (1 μM) and glibenclamide+indomethacin (1 μM) on the responses are shown. Integrated hyperpolarization and relaxation were expressed as a percentage of the maximum response in coronary (a,b) and mammary arteries (d,e). The raw data integrated were the same as those used for the peak measurements described in Figures 5 and 6. The numbers of animals are thus the same as indicated in Figures 5 and 6. The actions of each sequence of blockers on the integrated hyperpolarization and relaxation are shown in the left and middle panels, respectively. The bar graphs in the panels on the right indicate the components of the PG-dependent hyperpolarization and relaxation that were sensitive and insensitive to glibenclamide in coronary (c) and mammary (f) arteries. *Significance between L-NAME and L-NAME+glibenclamide;×significance between L-NAME and L-NAME+glibenclamide+indomethacin; ×significance between L-NAME+glibenclamide and L-NAME+glibenclamide+indomethacin.
In the second series of experiments where the inhibitors were added in the reverse order, that is, exposure to indomethacin first, so as to isolate the responses to NO and EDHF, glibenclamide reduced the integrated hyperpolarization in the coronary artery (Figure 8a), but it had little effect on the relaxation (Figure 8b). In the mammary artery glibenclamide was without effect on either the integrated hyperpolarization (Figure 8d) or relaxation (Figure 8e). Subsequent blockade of NO synthesis caused only a small further reduction in the integrated hyperpolarization in both arteries that was not significant except in the mammary at the highest concentration of ACh, 8 μM (Figure 8a,d). In both arteries application of L-NAME in the continued presence of indomethacin and glibenclamide caused a significant reduction in the integrated relaxations (Figure 8b,e).
Figure 8.
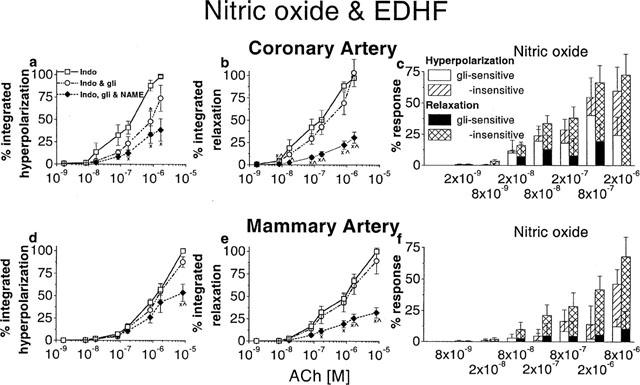
The involvement of NO and EDHF in endothelium-dependent hyperpolarization and relaxation was investigated in coronary and mammary arteries of the guinea-pig. Arteries were depolarized and constricted with U 46619 in the presence of indomethacin (1 μM). Increasing concentrations of ACh were applied. The effects of glibenclamide (1 μM) and glibenclamide+L-NAME (100 μM) on the responses are shown. Integrated hyperpolarization and relaxation were expressed as a percentage of maximum response in coronary (a,b) and mammary arteries (d,e). The raw data integrated were the same as those used for the peak measurements described in Figures 5 and 6. The numbers of animals are thus the same as indicated in Figures 5 and 6. The actions of each sequence of blockers on the integrated hyperpolarization and on the relaxation is shown in the left and middle panels, respectively. The bar graphs (c,f) indicate the components of the NO-dependent hyperpolarization and relaxation that were sensitive and insensitive to glibenclamide. *Significance between indomethacin and indomethacin+glibenclamide;×significance between indomethacin and indomethacin+glibenclamide+L-NAME; ×significance between indomethacin+glibenclamide and indomethacin+glibenclamide+L-NAME.
Relationship between hyperpolarization and relaxation
The relationship between hyperpolarization and relaxation for each of NO, PG and EDHF was explored by subtracting the integrated responses shown in Figures 7 and 8. For both arteries there was a tight relationship between the hyperpolarization and relaxation attributable to EDHF, that is, there was no difference in the extent of these parameters for each artery (2-way ANOVA, coronary P=0.44; mammary, P=0.45) (Figure 9). Thus hyperpolarization appears to play an important role in relaxation attributed to EDHF.
Figure 9.
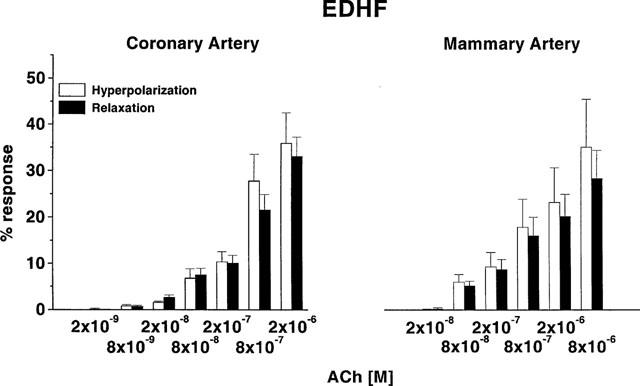
Relationship between integrated hyperpolarization and relaxation evoked in coronary (n=11) and mammary (n=6) arteries by EDHF, that is, in the presence of L-NAME (100 μM) and indomethacin (1 μM), and glibenclamide (1 μM), in coronary and mammary arteries. Responses were expressed as a percentage of the maximum integrated response (hyperpolarization or relaxation) recorded in the presence of only one blocker (either L-NAME or indomethacin, i.e. the data represented by the squares in Figures 7 and 8).
Most of the response attributed to endogenous PG was sensitive to glibenclamide in both arteries (Figure 7c,f). There was no difference between the magnitude of the hyperpolarization and the associated relaxation, indicating the important role of hyperpolarization in the relaxation evoked by PG (2-way ANOVA, coronary, P=0.59; mammary P=0.64) (Figure 7c,f). The residual, glibenclamide-insensitive components of the PG response were comparatively small. Nevertheless, there was no difference between the magnitude of the hyperpolarization and relaxation for this residual response either (Figure 7c,f).
In contrast, the glibenclamide-sensitive hyperpolarization and relaxation attributed to NO constituted a much smaller proportion of the overall, total NO-dependent response (Figure 8c,f). For the glibenclamide-sensitive component, the magnitude (in %) of the integrated hyperpolarization and relaxation were the same (2-way ANOVA, coronary, P=0.08; mammary, P=0.96) (Figure 8c,f) indicating that hyperpolarization contributed to relaxation. In the mammary artery the hyperpolarization attributable to NO was small in amplitude as indicated by the closeness of the curves represented by the squares and the diamonds in Figure 8d. The hyperpolarization in this artery appeared to be largely insensitive to glibenclamide (Figure 8d,f). In both arteries, there was a significant difference between the extent of the hyperpolarization and the relaxation for the glibenclamide-insensitive components of the NO response (Figure 8c,f; 2-way ANOVA, coronary P=0.0007; mammary P=0.0198). This indicates that for the glibenclamide-insensitive component of the NO response, which accounts for the majority of the NO response, the hyperpolarization probably does not play an important role in relaxation.
Discussion
In the guinea-pig coronary and mammary arteries EDHF, NO and PG each contribute to the endothelium-dependent hyperpolarization and relaxation evoked by ACh, but between the two arteries, the relative contribution of these factors to endothelial responses is different. However, the relationship between hyperpolarization and relaxation for each factor is preserved in the two arteries: for EDHF, the hyperpolarization and relaxation is closely linked in both vessels, as expected; NO causes considerable relaxation that is independent of hyperpolarization in the coronary and mammary arteries; the relaxation attributed to a PG is considerably dependent on hyperpolarization, a new revelation that has emerged from this study.
A novelty of the present investigation was the detailed dissection of hyperpolarization and relaxation and the ability to assign responses to individual factors. This was only possible because of the discrete application of each concentration of ACh, and the integration and subsequent subtraction of the data. This study revealed the extent of the disparity between results when the same responses were analysed using the traditional method of peak measurement as compared with integration. Using the peak method of analysis heterogeneities between vessels and agonists for the different types of endothelial factors released have been reported in the past (e.g. Nagao et al., 1992; Zygmunt et al., 1994; 1995; 1998). The ability to detect these differences using peak analysis relies on the contributing endothelial factors producing responses of different amplitude. Alternatively, cumulative application of ACh may lead to a ‘functional rundown' of the response to endothelial vasodilators, such as EDHF, so that its contribution to responses later in the protocol is reduced. Using discrete application of ACh in arteries such as used in our study, indomethacin or L-NAME alone had no effect on the peak amplitude relaxation or hyperpolarization. This indicated that each of the factors can produce maximal amplitude responses in their own right and therefore the actions of L-NAME and indomethacin cannot be properly assessed from peak analysis alone. However, it was clear from the raw data that the durations of the endothelial responses were substantially affected by the blockers, L-NAME and indomethacin, in both arteries and it was possible to quantify this using the integration method.
Contribution of NO, PG and EDHF to endothelium-dependent hyperpolarization and relaxation
By substracting the integrated data (Figure 3), the complete contribution of each factor to the endothelium-dependent responses could be evaluated. Comparison of the two arteries revealed that the overall integrated responses were strikingly smaller in the mammary artery. Although the amplitude of the EDHF hyperpolarization is significantly smaller in the mammary artery (Hammarstöm et al., 1999; Table 1), the difference in the integrated responses is likely to reflect the markedly shorter duration of the responses attributed to NO and a PG in the mammary compared with the coronary artery (Figure 1). This could be explained by a reduced release of NO and PG from the mammary artery endothelium or to enhanced phosphodiesterase or phosphatase activity.
The profiles for the contributions of NO, PG and EDHF to the integrated responses were different in the two arteries. NO and PG dominated both the hyperpolarization and relaxation in the coronary artery at all but the lowest concentrations of ACh. The contributions of these two vasodilators to the integrated hyperpolarization and relaxation were around 4 fold greater than that of EDHF. This is explained by the comparatively short duration of the EDHF response and the persistence of the responses to NO and PG. This contrasts with the mammary artery, in which all three factors contributed approximately equally to the integrated hyperpolarization and relaxation. This was largely due to the much shorter duration of the NO and PG responses in the mammary artery. Only at the highest concentration of ACh used did the contribution of PG begin to exceed those of the other two factors.
Relationship between hyperpolarization and relaxation
Of the three endothelial factors, the hyperpolarization attributed to EDHF was the largest in amplitude, although shortest in duration in the coronary artery. In the mammary artery its amplitude was on par with that of the other factors. This factor was capable of producing complete relaxation in both coronary and mammary arteries. There was a close relationship between the hyperpolarization and relaxation produced by EDHF for all concentrations of ACh. This is consistent with previous observations that a reduction or abolition of EDHF-mediated hyperpolarization, effected by K+ channel blockers or elevated extracellular K+, is accompanied by a corresponding reduction in tension (Zygmunt et al., 1994; Parkington et al., 1995; Chen & Cheung, 1997), indicating a strong dependence of relaxation on the hyperpolarization. The combination of charybdotoxin and apamin abolished the relaxation attributed to EDHF in circumflex coronary arteries of guinea-pigs (Yamanaka et al., 1998) and we also found that these blockers abolished the EDHF hyperpolarization and relaxation in main coronary and mammary arteries (unpublished observations).
The role of the hyperpolarization in the relaxation evoked by endogenous NO and PG is less completely understood and was evaluated here using glibenclamide, as this selectively blocks the hyperpolarization produced by these factors in the smooth muscle of many arteries (Garland & McPherson 1992; Murphy & Brayden, 1995; Parkington et al., 1995; Plane et al., 1995; Corriu et al., 1996), including coronary and mammary arteries of guinea-pigs. As reported previously (Parkington et al., 1995), abolition of the glibenclamide sensitive hyperpolarization was without effect on the peak amplitude of the relaxation produced by NO or PG in coronary artery was confirmed in the present study. However, using the integration methods of quantification in this study, we have revealed a significant effect of glibenclamide on the duration of the relaxation and importantly, differences between NO and PG in terms of the dependence of relaxation on hyperpolarization.
Endogenous NO produced hyperpolarization and relaxation in both the coronary and mammary arteries. A small proportion of the hyperpolarization and relaxation was sensitive to glibenclamide in both arteries and notwithstanding, there was a close relationship between hyperpolarization and relaxation for this glibenclamide-sensitive component of the NO response. However, the majority of the relaxation was glibenclamide-insensitive and there was no relationship between the small residual hyperpolarization and the substantial relaxation in either artery for this component of the NO response. We have previously shown that, although exogenous NO evokes hyperpolarization in the guinea-pig coronary artery, the correlation between hyperpolarization and relaxation was not strong (Parkington et al., 1995). In the rabbit isolated carotid artery, although there appeared to be a good correlation between hyperpolarization and relaxation to exogenous NO in control conditions, when extracellular K+ was raised, the hyperpolarization and relaxation were dissociated (Plane et al., 1998). In the rat mesenteric artery (Cheung et al., 1999) and rabbit femoral artery (Plane et al., 1995) the hyperpolarization evoked by exogenous NO was not correlated with relaxation. Thus, for exogenous NO there seems to be considerable inter vessel variation in the presence of hyperpolarization and in some arteries it is entirely absent (see Garland et al., 1995), and we have confirmed this in the present study for endogenous NO. Thus it appears that in both coronary and mammary arteries the predominant pathways underpinning NO-dependent relaxation involve mechanisms that are independent of membrane potential, most likely involving the second messenger-mediated, cyclic GMP-dependent actions on extrusion pumps and the sensitivity of the contractile apparatus to Ca2+. When NO-dependent hyperpolarization occurs it may facilitate relaxation but plays only a minor role compared with the membrane potential independent pathways.
In both coronary and mammary arteries the PG-dependent hyperpolarization is largely sensitive to glibenclamide and there was a close relationship between hyperpolarization and relaxation for this component of the response. Interestingly, there was also a good relationship between hyperpolarization and relaxation for the glibenclamide-insensitive components of the PG response in both arteries. Thus, a new revelation of the present study is that the hyperpolarization produced by endogenous PGs appears to play an important role in relaxation. Since the PGs involved, prostacyclin and PGE2, stimulate cyclic AMP synthesis and the associated relaxatory pathways, it is intriguing that the hyperpolarization appears to play such a significant role in the relaxation induced by this endogenous factor. A recent report in veins suggests that cyclic AMP formed in endothelial cells spreads to the smooth muscle cells via myoendothelial gap junctions to contribute to relaxation of the smooth muscle and may also enhance hyperpolarization (Griffith & Taylor, 1999). This could be one explanation for the intimate link between the PG hyperpolarization and relaxation. An earlier study (Parkington et al., 1995) reported on the lack of effect of glibenclamide on the peak amplitude of relaxation evoked by exogenously applied iloprost, a stable analogue of prostacyclin, despite obvious actions of glibenclamide on the duration of the relaxation. This reinforces the notion that a thorough quantitative evaluation of the effects of the blocker may be made possible by the integrative methods of analysis.
At very high concentrations of ACh, a component of the integrated hyperpolarization was resistant to the effects of glibenclamide which was abolished when production of NO or PG was inhibited in both arteries. It may well be that this residual hyperpolarization reflects the action of NO and PG or their respective second messengers, on a different class of K+ channel or electrogenic pump. NO, PG and their associated cyclic nucleotides have also been reported to activate Ca2+-activated K+ channels in some vessels (Archer et al., 1994; Bolotina et al., 1994; Schubert et al., 1997). Nishiyama et al. (1998) recently reported that the slow component of the ACh induced hyperpolarization attributed to a PG in the circumflex coronary artery of the guinea-pig was considerably reduced by glibenclamide. They found that 4-aminopyridine also reduced this component and they suggested that a combination of KATP and possibly delayed rectifier K+ channels were activated by endothelial prostanoids.
In summary, the individual contributions of EDHF, NO and PG to endothelium-dependent responses are markedly different in the coronary and mammary arteries. The overall integrated hyperpolarization and relaxation in coronary artery is dominated equally by NO and PG, with EDHF making a proportionally smaller contribution. Thus the total response in the coronary artery is dominated by factors that produce prolonged vasodilatation which favours maintained perfusion of this important vascular bed. In diseases where the EDHF response is compromised, such as diabetes (Fukao et al., 1997) and hypertension (Fujii et al., 1992), NO and/or PG may thus maintain tissue perfusion. In the mammary artery all three factors contribute equally to the endothelial responses. There is some question as to the precise role of EDHF in some arteries, with studies on larger arteries suggesting that the NO pathway is central to relaxation and the EDHF pathway only assumes significance when NO is compromised (Kilpatrick & Cocks, 1994; Kemp et al., 1995; McCulloch et al., 1997) and that NO may interfere with the EDHF response (Bauersachs et al., 1996). In other arteries EDHF is not merely a ‘backup' system for NO, and NO does not interfere with the EDHF response (Zygmunt et al., 1998). A recent report suggests that prostacyclin may inhibit hyperpolarization attributed to EDHF (Yajima et al., 1999). It is likely that the roles of each of the endothelial factors in the final response and the possible interaction between factors will vary with vessel type, size and location. However, unlike in larger arteries that are mainly NO dependent, EDHF is likely to be an important player in smaller arteries. When other factors such as NO and PG are also involved in the response, loss of EDHF still leaves the artery with these other factors to maintain some level of perfusion.
Despite the considerable variation in the extent of the responses between the coronary and mammary arteries, the role of the hyperpolarization in relaxation produced by each of the three factors has elements of commonality in the two arteries. In both arteries the relaxation produced by EDHF and PG are strongly dependent on hyperpolarization whilst a significant proportion of the NO relaxation is independent of membrane potential. Thus, endothelium-dependent vasodilatation in response to ACh in guinea-pig coronary and mammary arteries relies on three different factors that can invoke relaxation via a combination of pathways that are both dependent upon and independent of membrane potential. This may confer a type of ‘protective' mechanism ensuring some measure of vasorelaxation should one of these pathways be compromised, or the balance of membrane potential-dependent and -independent cellular mechanisms mediating vascular contraction be altered in disease.
Acknowledgments
The authors gratefully acknowledge the support of the National Heart Foundation of Australia and the National Health and Medical Research Council of Australia. The authors thank Dr Neela Kotecha for discussion of the manuscript. Glibenclamide was a kind gift from Hoechst.
Abbreviations
- ACh
acetylcholine
- cyclic AMP
adenosine 3′:5′ cyclic monophosphate
- cyclic GMP
guanosine 3′ : 5′ cyclic monophosphate
- EDHF
endothelium-derived hyperpolarizing factor
- L-NAME
Nω-nitro-L-arginine methyl ester
- NO
nitric oxide
- PG
prostanoid
References
- ARCHER S.L., HUANG J.M.C., HAMPL V., NELSON D.P., SHULTZ P.J., WEIR E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUERSACHS J., POPP R., HECKER M., SAUER E., FLEMING I., BUSSE R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. doi: 10.1161/01.cir.94.12.3341. [DOI] [PubMed] [Google Scholar]
- BÉNY J.-L. Electrical coupling between smooth muscle cells and endothelial cells in pig coronary arteries. Pflügers Arch. 1997;433:364–367. doi: 10.1007/s004240050289. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- CHEN G., CHEUNG D.W. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. Am. J. Physiol. 1997;272:H2306–H2312. doi: 10.1152/ajpheart.1997.272.5.H2306. [DOI] [PubMed] [Google Scholar]
- CHEUNG D.W., CHEN G., MACKAY M.J., BURNETTE E. Regulation of vascular tone by endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 1999;26:172–175. doi: 10.1046/j.1440-1681.1999.03008.x. [DOI] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., JUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit coronary artery. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRIU C., FÉLÉTOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., COLE W.C., TRIGGLE C.R. Roles of calcium-activated and voltage-gated delayed rectifier potassium channels in endothelium-dependent vasorelaxation of the rabbit middle cerebral artery. Br. J. Pharmacol. 1998;123:821–832. doi: 10.1038/sj.bjp.0701680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Endothelium-derived hyperpolarizing factor – a critical appraisal. Prog. Drug Res. 1998;50:108–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FUJII K., TOMINAGA M., OHMORI S., KOBAYASHI K., KOGA T., TAKATA Y., FUJISHIMA M. Decreased endothelium-dependent hyperpolarization to acetylcholine in smooth muscle of the mesenteric artery of spontaneously hypertensive rats. Circ. Res. 1992;70:660–669. doi: 10.1161/01.res.70.4.660. [DOI] [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA I., KITABATAKE A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1997;121:1383–1391. doi: 10.1038/sj.bjp.0701258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARLAND C.J., MCPHERSON G.A. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br. J. Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B., COCKS T. Endothelium-derived hyperpolarizing factor. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GRIFFITH T.M., TAYLOR H.J. Cyclic AMP mediates EDHF-type relaxations of rabbit jugular vein. Biochem. Biophys. Res. Commun. 1999;263:52–57. doi: 10.1006/bbrc.1999.1313. [DOI] [PubMed] [Google Scholar]
- HAMMARSTRÖM A.K.M., PARKINGTON H.C., TARE M., COLEMAN H.A. Endothelium-dependent hyperpolarization in resting and depolarized mammary and coronary arteries of guinea-pigs. Br. J. Pharmacol. 1999;126:421–428. doi: 10.1038/sj.bjp.0702313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLZMANN S., KUKOVETZ W.R., SCHMIDT K. Mode of action of coronary arterial relaxation by prostacyclin. J. Cyc. Nucl. Res. 1980;6:451–460. [PubMed] [Google Scholar]
- IGNARRO L.J., BURKE T.M., WOOD K.S., WOLIN M.S., KADOWITZ P.J. Association between cyclic GMP accumulation and acetylcholine-elicited relaxation of bovine intrapulmonary artery. J. Pharmacol. Exp. Ther. 1984;228:682–690. [PubMed] [Google Scholar]
- KEMP B.K., SMOLICH J.J., RITCHIE B.C., COCKS T.M. Endothelium-dependent relaxations in sheep pulmonary arteries and veins: resistance to block by NG-nitro-L-arginine in pulmonary hypertension. Br. J. Pharmacol. 1995;116:2457–2467. doi: 10.1111/j.1476-5381.1995.tb15096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUBO M., NAKAYA Y., MATSUOKA S., SAITO K., KURODA Y. Atrial natriuretic factor and isosorbide dinitrate modulate the gating of ATP-sensitive K+ channels in cultured vascular smooth muscle cells. Circ. Res. 1994;74:471–476. doi: 10.1161/01.res.74.3.471. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH A.I., BOTTRILL F.E., RANDALL M.D., HILEY C.R. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br. J. Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- MURPHY M.E., BRAYDEN J.E. Apamin-sensitive K+ channels mediate an endothelium-dependent hyperpolarization in rabbit mesenteric arteries. J. Physiol. 1995;489:723–734. doi: 10.1113/jphysiol.1995.sp021086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGAO T., ILLIANO S., VANHOUTTE P.M. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am. J. Physiol. 1992;263:H1090–H1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- NISHIYAMA M., HASHITANI H., FUKUTA H., YAMAMOTO Y., SUZUKI H. Potassium channels activated in the endothelium-dependent hyperpolarization in guinea-pig coronary artery. J. Physiol. 1998;510:455–465. doi: 10.1111/j.1469-7793.1998.455bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINGTON H.C., TARE M., TONTA M.A., COLEMAN H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINGTON H.C., TONTA M.A., COLEMAN H.A., TARE M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., PEARSON T., GARLAND C.J. Multiple pathways underlying endothelium-dependent relaxation in the rabbit isolated femoral artery. Br. J. Pharmacol. 1995;115:31–38. doi: 10.1111/j.1476-5381.1995.tb16316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., WILEY K.E., JEREMY J.Y., COHEN R.A., GARLAND C.J. Evidence that different mechanisms underlie smooth muscle relaxation to nitric oxide and nitric oxide donors in the rabbit isolated carotid artery. Br. J. Pharmacol. 1998;123:1351–1358. doi: 10.1038/sj.bjp.0701746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAYLE J.M., BONEV A.D., BRAYDEN J.E., NELSON M.T. Calcitonin gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth muscle via protein kinase A. J. Physiol. 1994;475:9–13. doi: 10.1113/jphysiol.1994.sp020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAPOPORT R.M., MURAD F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ. Res. 1983;52:352–357. doi: 10.1161/01.res.52.3.352. [DOI] [PubMed] [Google Scholar]
- SCHUBERT R., SEREBRYAKOV V.N., MEWES H., HOPP H.-H. Iloprost dilates rat small arteries: role of KATP- and Kca-channel activation by cAMP-dependent protein kinase. Am. J. Physiol. 1997;272:H1147–H1156. doi: 10.1152/ajpheart.1997.272.3.H1147. [DOI] [PubMed] [Google Scholar]
- SHIMOKAWA H., YASUTAKE H., FUJII K., OWADA M.K., NAKAIKE R., FUKUMOTO Y., TAKAYANAGI T., NAGAO T., EGASHIRA K., FUJISHIMA M., TAKESHITA A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- YAJIMA K., NISHIYAMA M., YAMAMOTO Y., SUZUKI H. Inhibition of endothelium-dependent hyperpolarization by endothelial prostanoids in guinea-pig coronary artery. Br. J. Pharmacol. 1999;126:1–10. doi: 10.1038/sj.bjp.0702254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMANAKA A., ISHIKAWA T., GOTO K. Characterization of endothelium-dependent relaxation independent of NO and prostaglandins in guinea-pig coronary artery. J. Pharmacol. Exp. Ther. 1998;285:480–489. [PubMed] [Google Scholar]
- ZYGMUNT P.M., PLANE F., PAULSSON M., GARLAND C.J., HÖGESTÄTT E.D. Interactions between endothelium-derived relaxing factors in the rat hepatic artery: focus on regulation of EDHF. Br. J. Pharmacol. 1998;124:992–1000. doi: 10.1038/sj.bjp.0701893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., RYMAN T., HÖGESTÄTT E.D.Regional differences in endothelium-dependent relaxation in the rat-contribution of nitric oxide and nitric oxide-independent mechanisms Endothelium-dependent hyperpolarization and relaxation of vascular smooth muscle 1995Lund University: Sweden; In: Zygmunt, P.M.: Ph.D. thesis [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., WALDECK K., HÖGESTÄTT E.D. The endothelium mediates a nitric oxide-independent hyperpolarization and relaxation in the rat hepatic artery. Acta Physiol. Scand. 1994;152:375–384. doi: 10.1111/j.1748-1716.1994.tb09819.x. [DOI] [PubMed] [Google Scholar]