

Abstract
The influence of the receptor-G protein coupling state and the guanine nucleotide ligation state of the G protein on the binding mechanism of A1 adenosine receptor ligands has been investigated in [3H]-1,3-dipropyl-8-cyclopentylxanthine ([3H]-DPCPX) binding studies in rat brain membranes. Thermodynamic parameters of binding of A1 adenosine receptor ligands of different intrinsic activities were determined in the absence or presence of GDP and compared to the binding mechanism after receptor-G protein uncoupling.
In agreement with previous studies, it was found that xanthine and non-xanthine antagonists showed an enthalpy- or enthalpy- and entropy-driven binding mechanism under all conditions.
In contrast to antagonists, the binding mechanism of agonists was strongly affected by the G protein coupling state or the absence or presence of guanine nucleotides. Binding of full and partial agonists to the high-affinity state of the A1 receptor was entropy-driven in the absence of GDP, and a good correlation between intrinsic activities and the contribution of entropy was observed. In the absence of GDP, binding of full and partial agonists and antagonists to the high affinity state of the receptor was thermodynamically discriminated. In contrast, no such discrimination was found in the presence of GDP.
The binding mechanism of agonists to the low-affinity state of the receptor was identical to that of antagonists only after uncoupling of the receptor from G proteins by pretreatment with N-ethylmaleimide or guanosine-5′-(γ-thio)-triphosphate (GTPγS).
These results indicate the existence of two thermodynamically distinct high- and low-affinity states of the A1 adenosine receptor.
Keywords: A1 adenosine receptor, partial agonist, affinity, intrinsic activity, radioligand binding, binding thermodynamics
Introduction
The binding of ligands to receptors is a prerequisite for induction of signalling. The nature of the interactions between ligands and receptors defines if a ligand acts as an agonist, a partial agonist, an antagonist, or an inverse agonist. The underlying mechanisms which contribute to binding have been characterized in numerous thermodynamic studies. For a number of receptors, a correlation between thermodynamic parameters and the intrinsic activity of ligands has been described. At β-adrenergic receptors (Weiland et al., 1979; Contreras et al., 1986; Miklavc et al., 1990), M2 muscarinic receptors (Waelbroeck et al., 1993), γ-aminobutyric acidA receptors (Maksay, 1994) and 5-HT3 receptors (Borea et al., 1996), binding of agonists and antagonists is thermodynamically distinct. In contrast, agonist and antagonist binding to D2 dopamine (Kilpatrick et al., 1986) and 5-HT1A receptors (Dalpiaz et al., 1996) is not thermodynamically discriminated, and the thermodynamic characteristics of these ligands are better interpreted in accordance with their structural characteristics.
Initial thermodynamic analysis of ligand binding to A1 adenosine receptors indicated an entropy-driven mechanism of binding of agonists to the high-affinity state of the receptor, whereas binding to the low-affinity state was enthalpy-driven and thus similar to the binding of antagonists to this receptor (Murphy & Snyder, 1982; Lohse et al., 1984). These findings were extended to a larger number of agonists and xanthine antagonists (Borea et al., 1992). Based on this evidence, it was predicted that partial agonists of the A1 receptor should exhibit a binding mechanism intermediate between full agonists and antagonists. This prediction has been confirmed experimentally in one study of adenylate cyclase inhibition and receptor binding thermodynamics (Borea et al., 1994). Contradictory results have been determined when intrinsic activity was assessed as the ability of A1 receptor ligands to activate G proteins, and receptor binding characteristics were studied under identical conditions (Lorenzen et al., 1996). In this study, thermodynamic parameters of partial agonist binding did not correlate with intrinsic activities. Moreover, within the investigated group of partial agonists, a marked heterogeneity of the relative contributions of changes in entropy and enthalpy was observed. This suggests that binding of structurally distinct partial agonists of A1 receptors is driven by qualitatively distinct mechanisms of interaction with the receptor.
The reason for these differences concerning the binding mechanisms of partial agonists to A1 receptors might be the different incubation conditions employed in the binding studies. In the first study (Borea et al., 1994), [3H]-N6-cyclohexyladenosine binding was performed in Tris buffer. In contrast, the second study (Lorenzen et al., 1996) used identical incubation conditions for assessment of intrinsic activity and [3H]-DPCPX binding. Incubations were performed in the presence of NaCl, MgCl2 and GDP. In order to characterize the influence of the coupling state of the receptor to G proteins and the importance of the guanine nucleotide ligation state of G proteins in the present study, we investigated the relative contribution of enthalpic and entropic forces to the binding of adenosine receptor ligands comparing different binding conditions. Ligand binding to the high- and low-affinity state of the receptor was studied under control conditions and after uncoupling of the receptor from the G protein with sulphydryl alkylating agent N-ethylmaleimide (NEM). NEM alkylates the same cysteine residue in Gi and Go α subunits which is ADP-ribosylated by pertussis toxin (Böhm et al., 1993). Ligand binding mechanisms under control conditions and after receptor-G protein-uncoupling were compared to the binding mechanism in the presence of GDP, MgCl2 and NaCl as described previously (Lorenzen et al., 1996). The influence of the GDP ligation state of the G protein on ligand binding was investigated by omission of GDP from the incubation medium. We have further extended the thermodynamic characterization of adenosine receptor ligands to non-xanthine receptor antagonists, which have not been examined previously.
Methods
Materials
[3H]-DPCPX (80–120 Ci mmol−1) was obtained from New England Nuclear (Bad Homburg, Germany). 2-chloro-N6-cyclopentyladenosine (CCPA), 5-amino-9-chloro-2-(2-furyl)-[1,2,4]triazolo[1,5-c]quinazoline (CGS 15943), N6-cyclopentyl-9-methyladenine (CPMA), 2-phenylaminoadenosine (CV 1808), etazolate, and 1-methylisoguanosine (MIG) came from Research Biochemicals Inc. (Cologne, Germany). Adenosine deaminase (from calf intestine; 200 U mg−1), CHAPS, dithiothreitol and GDP were purchased from Boehringer (Mannheim, Germany). 2-Chloro-2′-deoxyadenosine (cladribine), 5′-deoxy-5′-methylthioadenosine (MeSA), NEM, bovine serum albumin and theophylline were from Sigma (Deisenhofen, Germany). All other chemicals were obtained from standard sources and were of the highest purity commercially available.
Preparation of rat brain membranes
Membrane preparation from rat forebrains was performed according to a previously described protocol (Lorenzen et al., 1993). Protein content was determined according to Peterson (1977), using bovine serum albumin as standard.
Treatment of membranes with NEM
Uncoupling of A1 adenosine receptors from G proteins with the sulphydryl alkylating agent NEM was performed as described (Lorenzen et al., 1993).
Binding of [3H]-DPCPX to rat brain membranes
Equilibrium binding to A1 adenosine receptors was performed as 25, 20, 10 and 0°C. Incubation times were chosen according to previous time course experiments. All buffers were adjusted to pH 7.4 at the incubation temperature used in the experiment. Forty μg of membrane protein were incubated with [3H]-DPCPX in the presence of 0.2 U ml−1 adenosine deaminase. Separation of bound from free radioligand was performed by rapid filtration through Whatman GF/B filters, which were washed twice with 4 ml of ice-cold 50 mM Tris-HCl, pH 7.4, containing 0.02% 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate (CHAPS). Nonspecific binding was determined in the presence of 10 μM R-N6-phenylisopropyladenosine (R-PIA).
Four different conditions were used: (A) Membranes were incubated in 50 mM Tris-HCl, pH 7.4, containing 0.02% CHAPS in a total volume of 1 ml for 2 h at 25°C (0.50 nM [3H]-DPCPX in competition experiments), 2.5 h at 20°C (0.25 nM [3H]-DPCPX), 3 h at 10°C (0.18 nM [3H]-DPCPX), or overnight at 0°C (0.14 nM [3H]-DPCPX). (B) 40 μg of membranes were incubated in 2 ml at 25°C for 2 h (1 nM [3H]-DPCPX in competition experiments), 20°C for 3 h (0.8 nM [3H]-DPCPX), 10°C for 6 h (0.4 nM [3H]-DPCPX), or at 0°C for 10 h (0.3 nM [3H]-DPCPX) as previously described (Lorenzen et al., 1996). The incubation medium contained (mM): Tris-HCl 50, pH 7.4, triethanolamine 2, EDTA 1, MgCl2 5, dithiothreitol 1, NaCl 100, and 0.5% bovine serum albumin. (C) Incubations were done as described in B, but in the presence of 10 μM GDP. Samples were incubated with 0.8 nM [3H]-DPCPX for 2 h at 25°C, 0.6 nM [3H]-DPCPX for 2.5 h at 20°C, 0.4 nM [3H]-DPCPX for 3 h at 10°C and 0.3 nM [3H]-DPCPX for 8 h at 0°C. (D) Membranes had been pretreated with 1 mM NEM. Experiments were carried out in 50 mM Tris-HCl, pH 7.4, 0.02% CHAPS in a total volume of 1 ml. Incubation times were 2 h at 25°C (0.31 nM [3H]-DPCPX in competition experiments), 2.5 h at 20°C (0.26 nM [3H]-DPCPX), 3 h at 10°C (0.22 nM [3H]-DPCPX) and 15 h at 0°C (0.19 nM [3H]-DPCPX).
Data analysis
Binding data were analysed by nonlinear curve fitting using the programs SCTFIT and LIGAND. The mathematical basis and equations used in SCTFIT (de Lean et al., 1982) and LIGAND (Munson & Rodbard, 1980) have been described previously in detail. Results were fitted to a one site model if curve fitting to two sites did not improve the fit significantly (P<0.05, f-test). KD, KH and KL values (Ki values for the high and low affinity states of the receptor for agonists) are given as geometric means with 95% confidence limits derived from 3–6 independent experiments. Bmax values are given as arithmetic means±s.e.mean. Standard free energy was calculated as ΔG°=−RTlnKA (T=298.15 K, R=8.314 J K−1 mol−1, KA=KD−1 or Ki−1, respectively). Standard enthalpy ΔH° was calculated from van't Hoff plots (lnKA versus T−1; slope=−ΔH° R−1), and standard entropy ΔS° as (ΔH°-ΔG°)×T−1. −TΔS was obtained by multiplying −T (T=298.15 K) and ΔS°. A binding reaction is driven by enthalpy alone when ΔH° is negative and ΔS° is also negative or close to zero. An interaction is enthalpy- and entropy-driven when ΔH° is negative and ΔS° is positive.
Results
In this study, equilibrium binding experiments with [3H]-DPCPX were performed at four different incubation temperatures under four different incubation conditions. Data for two full agonists (CCPA and MIG), three partial agonists (MeSA, CV 1808 and cladribine), two xanthine ([3H]-DPCPX, theophylline) and three non-xanthine antagonists (CPMA, etazolate and CGS 15943) are reported. Binding parameters representative for one antagonist ([3H]-DPCPX saturation experiments; Table 1), one full agonist (CCPA; competition for [3H]-DPCPX binding; Table 2A) and one partial agonist (MeSA; competition for [3H]-DPCPX binding, Table 2B) are shown. Binding parameters from competition experiments with other purine derivatives are depicted as lnKA values (KA=Ki−1) in van't Hoff plots (Figure 1).
Table 1.
Equilibrium binding parameters of [3H]-DPCPX at A1 adenosine receptors at four different temperatures and under different incubation conditions
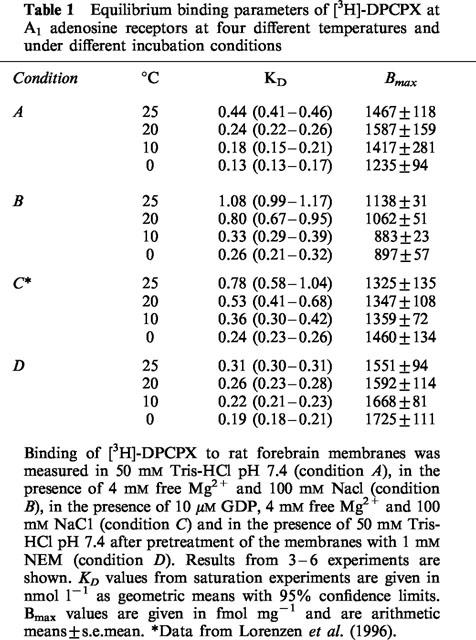
Table 2.
Equilibrium binding parameters of the full agonist CCPA and the partial agonist MeSA at A1 adenosine recptors at four different temperatures and under different incubation conditions
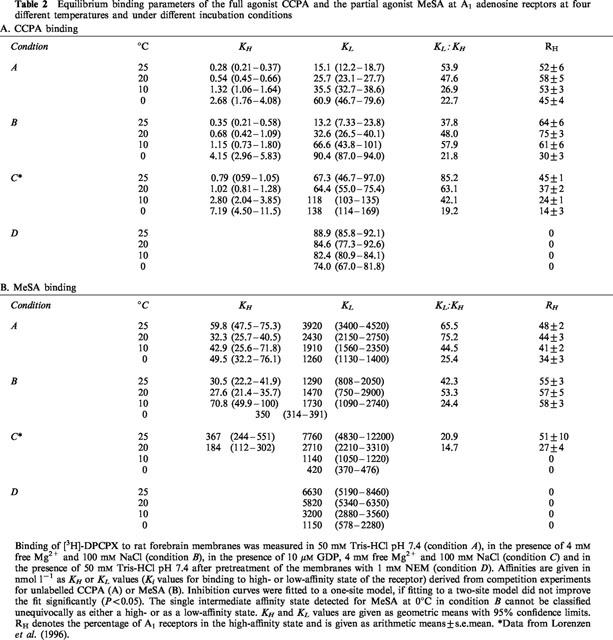
Figure 1.
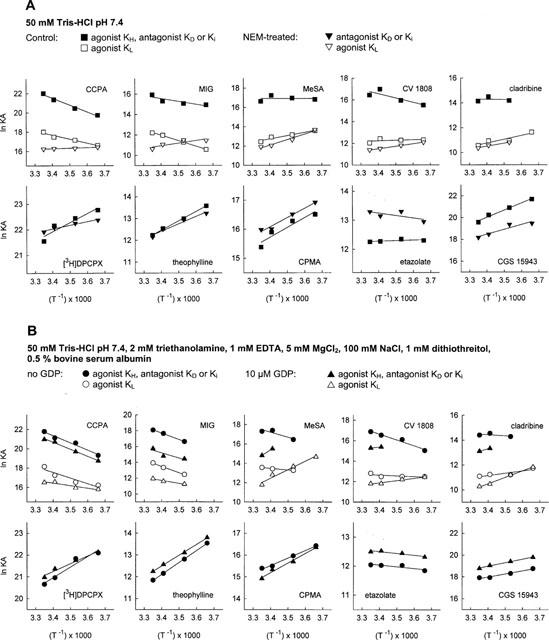
van't Hoff plots for ligand binding to A1 adenosine receptors in rat brain membranes. (A) Binding experiments were conducted in 50 mM Tris-HCl pH 7.4 (A) with membranes not pretreated or pretreated with NEM. (B) Experiments were performed in 50 mM Tris-HCl pH 7.4, 2 mM triethanolamine, 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl, 1 mM dithiothreitol and 0.5% bovine serum albumin or, alternatively, in the same incubation medium containing in addition 10 μM GDP. KD values for [3H]-DPCPX are from saturation experiments. KH values for agonists, KD and Ki values for antagonists are indicated by filled symbols, KL values for agonists are shown as open symbols. The lines in the figure are the linear regression lines of lnKA from 3–6 experiments for each compound versus (T−1).
[3H]-DPCPX binding was saturable in a single component under all incubation conditions. The affinity of [3H]-DPCPX was consistently higher at lower incubation temperatures (Table 1) with KD values ranging from 0.13–1.08 nM. [3H]-DPCPX was bound with highest affinities when the incubation was performed in Tris buffer (condition A) or when membranes had been pretreated with NEM (condition D). In the presence of 4 mM free Mg ions and 100 mM NaCl (condition B) or 4 mM free Mg2+, 100 mM NaCl and 10 μM GDP (condition C), the affinity of this compound was slightly lower than in conditions A or D. The temperature dependence of the affinity of [3H]-DPCPX (KA=Ki−1) is depicted in the van't Hoff plots in Figure 1. All plots for [3H]-DPCPX appear linear with a positive slope regardless of the incubation conditions. Thermodynamic parameters are reported in Table 3 and do not greatly vary when results from saturation experiments under different conditions are compared except for a greater relative contribution of entropic forces after receptor-G protein uncoupling with NEM. The plots of ΔH° versus −TΔS (Figure 2) shows that binding of [3H]-DPCPX, under all circumstances, is enthalpy- and entropy-driven with only minor variations in these parameters.
Table 3.
Thermodynamic parameters of ligand binding to A1 adenosine receptors
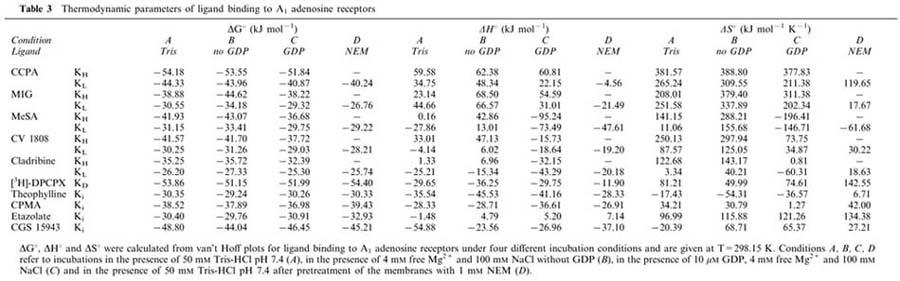
Figure 2.
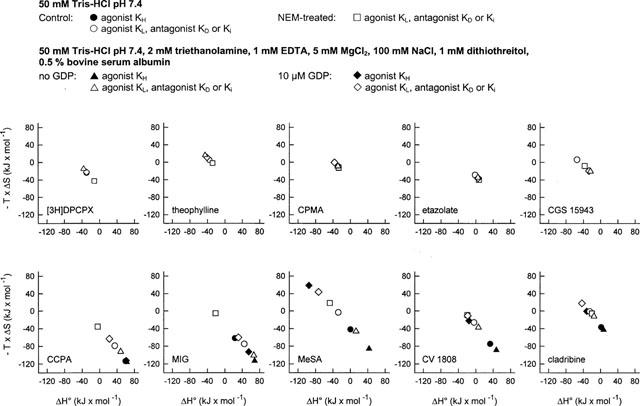
Contribution of enthalpy and entropy changes to ligand binding to A1 adenosine receptors under different incubation conditions. Equilibrium binding experiments with A1 adenosine receptor ligands had been conducted in 50 mM Tris-HCl pH 7.4 with membranes not pretreated or pretreated with NEM or in 50 mM Tris-HCl pH 7.4, 2 mM triethanolamine, 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl, 1 mM dithiothreitol and 0.5% bovine serum albumin or, alternatively, in the same incubation medium containing in addition 10 μM GDP. Thermodynamic parameters were calculated from van't Hoff plots. Data for KH values are represented by filled symbols, data for KD and KL values are shown as open symbols.
The maximum number of binding sites detected by [3H]-DPCPX (Table 1) was identical when conditions A (Tris), B (no GDP; 100 mM NaCl, 4 mM free Mg2+) and C (addition of 10 μM GDP, NaCl and MgCl2) were compared. However when GDP was omitted from the incubation mixture (condition B), the Bmax values were significantly lower at 10°C (P<0.01) and 0°C (P<0.05) from the results obtained in the presence of 10 μM GDP. The radioligand labelled significantly (P<0.05) less receptors at these temperatures compared to 25 and 20°C. The reason for these differences is presently not known.
The antagonists theophylline, CPMA, etazolate and CGS 15943 induced monophasic displacement of [3H]-DPCPX from A1 adenosine receptors. The xanthine derivative theophylline and the non-xanthine antagonists CPMA and CGS 15943, in a manner similar to [3H]-DPCPX, displayed an increase of affinity for the A1 receptor at lower incubation temperatures, and slopes of van't Hoff plots were positive under all conditions studied (Figure 1). In contrast, the Ki values of the non-xanthine etazolate did not differ greatly between the different incubation temperatures from 0–25°C, pointing to a slightly different binding mechanism (Figure 1). Thermodynamic parameters of all antagonists, which were determined from van't Hoff plots, were hardly affected by differences in incubation conditions (Table 3). The binding of theophylline, CPMA and CGS 15943 was either enthalpy- and entropy-driven or merely enthalpy-driven (Figure 2). The binding of etazolate was mainly entropy-driven (Figure 2).
CCPA and MIG have been characterized as full agonists of the A1 adenosine receptor in G protein activation studies in rat brain membranes (Lorenzen et al., 1996). Displacement of [3H]-DPCPX by increasing concentrations of CCPA (Table 2A) and MIG (not shown) revealed two binding sites for these agonists when the experiments were conducted in Tris buffer (A), in the presence of Na+ and Mg2+ ions (B), or in the presence of Na+, Mg2+ and 10 μM GDP (C). After pretreatment of the membranes with NEM (D), only one low-affinity binding site was detected by CCPA (Table 2A) and MIG (not shown), indicating successful uncoupling of the A1 receptor-G protein-complex. Both agonists displayed higher affinities at higher incubation temperatures for the high- and low-affinity states of the A1 receptor under all incubation conditions except after pretreatment of the membranes with NEM (Figure 1). In contrast to antagonist binding, the slopes of van't Hoff plots for CCPA and MIG were negative when the membranes had not been subjected to NEM pretreatment (Figure 1). Thermodynamic parameters for agonist binding to the high- and low-affinity state of the A1 receptor differed markedly between incubation conditions A, B, C and D (Table 3). The relative contribution of entropic and enthalpic forces to the binding is depicted in Figure 2. It is obvious that binding of MIG, but not of CCPA to the high-affinity state is highly dependent on incubation conditions. In addition, the contribution of enthalpic or entropic forces to agonist binding to the low affinity state was markedly influenced by Na+, Mg2+, GDP and NEM pretreatment. In untreated membranes, binding of CCPA and MIG to the high- and low-affinity state was entropy-driven. After uncoupling the A1 adenosine receptor from G proteins by NEM treatment, binding of CCPA and MIG to a single low-affinity state was enthalpy- and entropy-driven and thus revealed an identical thermodynamic mechanism as antagonist binding (Figure 3B). Further experiments addressed the possibility that the distinct thermodynamic parameters of full agonist binding to the low affinity state between NEM-treated and untreated membranes might be due to damage of the A1 receptor protein by NEM rather than to receptor-G protein-uncoupling. As an alternative uncoupling agent, we used 25 μM GTPγS. The thermodynamic parameters of CCPA and MIG binding to the low affinity state were compared to those parameters in NEM-treated membranes. As already determined in NEM-treated membranes (Table 3), an enthalpy- and entropy-driven binding mechanism was found for these agonists in the presence of 25 μM GTPγS (CCPA: ΔG°=−40.53 kJ mol−2, ΔH°=−12.12 kJ mol−1, −TΔS=−28.40 kJ mol−1; MIG: ΔG°=−28.02 kJ mol−1, ΔH°=−19.70 kJ mol−1, −TΔS=−8.32 kJ mol−1; curves not shown). Because NEM treatment as well as GTPγS induced identical changes in thermodynamic parameters, we conclude that the change in the thermodynamic binding mechanism of agonists to the low affinity state induced by NEM is due to receptor-G protein-uncoupling.
Figure 3.
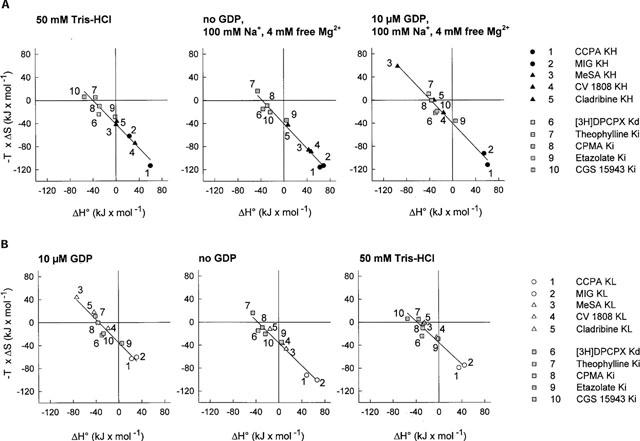
Scatter plots of ΔH° versus-TΔS. Thermodynamic parameters for binding of A1 adenosine receptor full agonists, partial agonists and antagonists were determined. Data for agonist binding to high affinity states of the receptor (filled symbols; A) and to low affinity states (open symbols; B) and antagonist binding are shown for experiments performed under different incubation conditions.
MeSA, CV 1808 and cladribine have been characterized previously as partial agonists of the A1 adenosine receptor. The intrinsic activities of these compounds were assessed as their ability to maximally stimulate [35S]-GTPγS binding, and were compared to the full agonist CCPA (Lorenzen et al., 1996). The intrinsic activities of MeSA, CV 1808 and cladribine are 45, 56 and 19% of the intrinsic activity of CCPA. Binding data representative of one partial agonist, MeSA, are reported in Table 2B. Affinities of all partial agonists depicted as lnKA are shown in Figure 1. Like the full agonists CCPA and MIG, all partial agonists detected A1 receptors in high- and low-affinity states (Table 2B, Figure 1). Only MeSA detected a single state of the A1 receptor when incubations were done in the presence of Mg2+ and Na+ ions at 0°C (condition B; Table 2B). This affinity state could neither be classified as a high- or nor as a low-affinity state, since the Ki value was intermediate between the values determined at 10–25°C (Table 2B). After NEM treatment, partial agonists bound to a single low-affinity state of the A1 receptor (Table 2B, Figure 1). The affinities (Table 2B, Figure 1) and thermodynamic parameters (Table 3, Figure 2) of partial agonists were strongly influenced by the incubation conditions used. In the presence of 10 μM GDP (condition C; Lorenzen et al., 1996), partial agonists detected a lower percentage of A1 receptors than full agonists in the high affinity state and exhibited a smaller difference between affinities to the low- and high-affinity states (KL : KH ratio). However, when incubations were performed in Tris buffer or in the presence of 100 mM NaCl and 4 mM free Mg2+ ions in the absence of GDP, partial agonists were not systematically discriminated from full agonists by the fractional occupancy of receptors in the high-affinity state or their KL : KH ratio (Table 2B). The affinities of MeSA, CV 1808 and cladribine to the high-affinity state of the A1 receptor were generally 3–12 fold lower in the presence of 10 μM GDP (C) than in its absence (A, B; Table 2B, Figure 1). The temperature dependence of affinities of partial agonists was markedly changed by different incubation conditions, especially when incubations with or without GDP are compared (Figure 1). Partial agonists displayed higher affinities at lower temperatures in the presence of 10 μM GDP, 100 mM NaCl and 4 mM free Mg2+. In the presence of 100 mM NaCl and 4 mM free Mg2+ or in Tris buffer, this was not observed for binding to the high affinity state (Table 2B). The slopes of van't Hoff plots were negative or neutral under these conditions in contrast to positive slopes in the presence of GDP (Figure 1). Affinities of partial agonists to the low-affinity state of the A1 receptor were mostly higher at lower temperatures (Table 2B, Figure 1).
The thermodynamic parameters of receptor binding of ligands of different intrinsic activities are compared in Figure 3. In the presence of 10 μM GDP, 100 mM NaCl and 4 mM free Mg2+ ions (condition C), binding to the low- as well as the high-affinity states of full agonists is entropy-driven, whereas binding of xanthine- as well as non-xanthine antagonists and also of partial agonists is enthalpy- and entropy-driven or merely enthalpy-driven. Partial agonists and antagonists are not thermodynamically discriminated (Figure 3A, right). However, when GDP was omitted from the incubation medium, partial agonists showed a binding mechanism at the high-affinity state which was intermediate between full agonists and antagonists (Figure 3A; centre panel). Incubations in Tris buffer yielded a similar thermodynamic discrimination of ligands of different intrinsic activities (Figure 3A, left). The intrinsic activity of A1 receptor ligands in the absence of GDP and in Tris buffer, but not in the presence of GDP correlates with the driving force in binding, ΔS°. The nonxanthine derivative etazolate displayed a binding mechanism which was intermediate between full agonists and antagonists under all conditions investigated (Figure 3A,B). The reason for this mechanism, which is somewhat distinct from that of the other antagonists, is not clear. Further studies might address the possibility that etazolate may exhibit weak partial agonistic properties.
Discussion
The mechanisms which contribute to the binding of ligands to G protein-coupled receptors have been investigated in a variety of thermodynamic studies in order to examine the possibility of a correlation or even a causal relationship between the binding mechanism–as characterized by thermodynamic parameters–and the intrinsic activity of receptor ligands. The experimental evidence concerning a possible thermodynamic discrimination of A1 adenosine receptor ligands is conflicting (Murphy & Snyder, 1982; Lohse et al., 1984; Borea et al., 1992; 1994; Lorenzen et al., 1996). The majority of studies, which describe a thermodynamic differentiation of A1 receptor ligands of distinct intrinsic activities, has been performed in 50 mM Tris buffer using [3H]-cyclohexyladenosine (Murphy & Snyder, 1982; Lohse et al., 1984; Borea et al., 1992; 1994). The contribution of the G protein coupling state of the A1 adenosine receptor has not been addressed in detail. In a more recent study, which assessed intrinsic activity as stimulation of [35S]-GTPγS binding and ligand affinities by inhibition of antagonist ([3H]-DPCPX) binding under identical conditions, we did not observe a correlation between thermodynamic parameters and intrinsic activity (Lorenzen et al., 1996). In the present study, the reasons for the discrepancies in these results were investigated. Importantly, high-affinity states observed in binding studies may not be indicative of a functional receptor state which induces G protein activation. The studies of Fraser (1989) and Hausdorff et al. (1990) have conclusively shown by site-directed mutagenesis of the β2-adrenergic receptor that mutated receptors, which are able to bind agonists with high affinity, may nevertheless be unable to induce activation of adenylate cyclase. Therefore, the states described in conflicting studies of the thermodynamics of A1 receptor binding may also represent functionally distinct states.
In the present study, experimental evidence indicates that the guanine nucleotide ligation state of the G protein exerts a major influence on the thermodynamic binding mechanism of A1 adenosine receptor ligands. When the binding experiments were performed under identical conditions as used for assessment of G protein activation (condition C) in the presence of 10 μM GDP, 100 mM NaCl and 4 mM free Mg2+, only full agonists were thermodynamically differentiated from partial agonists and antagonists. Non-xanthine antagonists (CPMA, CGS 15943 and etazolate) showed an identical binding mechanism as xanthine-derived antagonists (Figure 3A, left), indicating that the chemical structure was of minor importance compared to intrinsic activity. When GDP was omitted from the incubation medium (condition B), binding of ligands of increasing intrinsic activities showed an increasing contribution of entropic forces to binding to the high-affinity state of the receptor (Figure 3A, centre). A correlation between ΔS° and intrinsic activity was observed only in the absence and not in the presence of GDP. Similar results, albeit with a less clear-cut distinction of different intrinsic activities, were obtained when the binding had been performed in Tris buffer (condition A; Figure 3A, left). Therefore, the GDP ligation state of the α subunit of the G protein determines the binding mechanism of partial agonists.
The GDP ligation state of the α subunit also determines the activational state of the G protein. Agonists release prebound [3H]-GDP from G proteins specifically by receptor activation (Murayama & Ui, 1984), and this dissociation step is assumed to be rate-limiting in the consecutive association of GTP. Agonists may, in addition, also stabilize the α subunit in a GDP-free form by preventing GDP association, which in turn allows GTP binding (Florio & Sternweis, 1989). The selective decrease in the affinity for GDP, but not GTP for the α subunit allows agonist activation of receptors independent of the GDP : GTP ratio. A1 and A2a receptor agonists release [3H]-GDP from striatal membranes; this effect shows an absolute requirement for the addition of at least 10 nM guanylylimidodiphosphate (Marala & Mustafa, 1993). We have previously shown that A1 receptor ligands decrease the amount of membrane-bound [35S]-GDPβS without addition of a second, unlabelled guanine nucleotide (Lorenzen et al., 1996). Because the binding of GDP is an equilibrium reaction, our results are compatible both with the stimulation of GDP release and a decreased association of GDP to the G protein α subunit by A1 agonists.
Thermodynamic data indicate that binding to the receptor-G protein complex in the GDP-occupied form is not thermodynamically differentiated whereas there is a clear thermodynamic differentiation in the absence of GDP (Figure 3A). Ligands of different intrinsic activities can be distinguished on the basis of their thermodynamic parameters only when the receptor-G protein complex is in a GDP-free form. This finding is somewhat unexpected, since it is generally assumed that agonists act on GDP-ligated G proteins to induce the GDP release. According to the thermodynamic analysis, partial agonists and antagonists display identical binding mechanisms in the presence of GDP. This result would indicate that different intrinsic activities, if mediated through the GDP-ligated state, are not caused by different binding mechanisms. Thermodynamic analysis, in this case, would not be a meaningful instrument in the characterization of receptor-ligand interactions. However, if the mode of ligand-receptor-interaction, as quantitatively described by thermodynamic binding parameters, is relevant and indicative of the intrinsic activity of ligands, a significant contribution of agonist binding to the GDP-free state of the receptor-G protein complex in the activation process must be proposed, because a thermodynamic differentiation of ligands is found exclusively in the absence of GDP.
These experimental findings can be reconciled with the hypothetical model depicted in Figure 4, which is in agreement with the finding that agonists decrease the affinity of GDP both by an increased rate of dissociation and a decreased rate of association (Florio & Sternweis, 1989). Two high-affinity states of the receptor(R)-G protein-complex in the GDP-liganded or GDP-free form bind ligands. Binding to R-αGDPβγ is not thermodynamically differentiated. Agonists bound to this form may induce GDP release. Binding of ligands to R-αβγ is thermodynamically differentiated, and agonists may stabilize this form by preventing association of GDP, thereby facilitating association of GTP. Partial agonists bind to R-αGDPβγ by a thermodynamic mechanism not distinct from antagonists. In the presence of GDP, the intrinsic activity does not correlate with standard entropy ΔS°, whereas binding to R-αβγ is an entropy-dependent process. For partial agonists, stimulatory intrinsic activity is therefore probably more significantly mediated by a decreased association of GDP to R-αβγ than by a release of prebound GDP. In the interaction with R-αGDPβγ, partial A1 receptor agonists may not alter the guanine nucleotide ligation state of the α subunit, which corresponds to the partial antagonist quality of partial agonists. This is in agreement with the partially agonistic and partially antagonistic characteristics of partial A1 receptor agonists in [35S]-GTPγS binding studies (Lorenzen et al., 1996). The amount of membrane-bound GDP is decreased by partial agonists due to a decrease in GDP association rather than by GDP release. This proposed mechanism of action is in agreement with our previous finding that the efficacy of partial agonists relative to full agonists is favoured at low GDP concentrations (Lorenzen et al., 1996).
Figure 4.
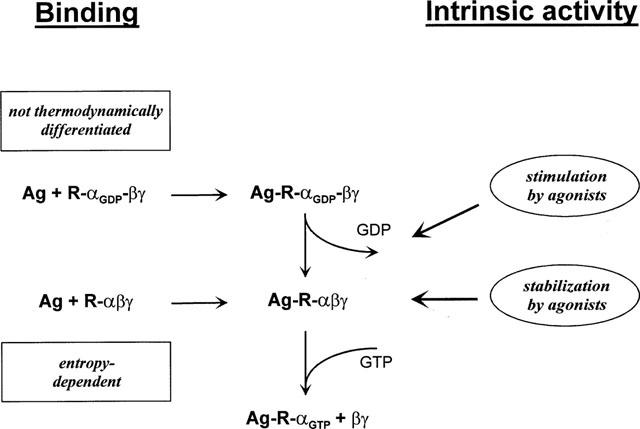
Proposed mechanism of agonist binding to and activation of A1 adenosine receptors. Agonists bind to receptors in the absence (R-αβγ) or presence of GDP (R-αGDPβγ) and may either stimulate the release of GDP or stabilize receptor-G protein-complexes in the GDP-free form. A1 adenosine receptor agonists of different intrinsic activities are not thermodynamically discriminated in binding in the presence of GDP. In the absence of GDP, ligands were thermodynamically discriminated, and a linear correlation between standard entropy and intrinsic activity was observed. This relationship between intrinsic activity and standard entropy only in the absence of GDP possibly indicates that agonist binding to the GDP-free state of the receptor-G protein complex determines the intrinsic activity of the ligand, rather than the stimulation of GDP release.
The significance of a GDP-free state of the G protein for A1 receptor agonists has also been pointed out by van der Ploeg et al. (1992), who showed that the A1-selective agonist N6-cyclopentyladenosine decreased the [32P]-ADP-ribosylation by pertussis toxin in brain cortex membranes. It should be noted that only the GDP-liganded holotrimeric G protein (R-αGDPβγ) is a substrate for pertussis toxin (Birnbaumer et al., 1990). Therefore, if the agonist, in the absence of GTP, but in the presence of GDP, decreases the amount of pertussis toxin substrates, it must be concluded that the agonist increases the proportion of the GDP-free form of R-αβγ over R-αGDPβγ (van der Ploeg et al., 1992).
The model in Figure 4 implies that the GDP-free complex R-αβγ is spontaneously active, since it can bind GTP, and the role of GDP would be to decrease agonist-independent ‘noise'. More direct experimental evidence is required to address the relative importance of the stimulation of GDP release and the inhibition of GDP association by agonist-activated G protein-coupled receptors.
Binding of full agonists to the low-affinity state was entropy-driven when the binding experiments were performed in 50 mM Tris buffer (condition A), in the presence of GDP, NaCl and MgCl2 (condition C) or in the absence of GDP (condition B). After uncoupling the receptor-G protein complex by membrane pretreatment with NEM or by incubation in the presence of 25 μM GTPγS, the full agonists CCPA and MIG displayed an entropy- and enthalpy-driven binding mechanism not distinct from the binding mechanism of antagonists (Figure 3B). These results are in agreement with the findings of Lohse et al. (1984), who described an antagonist-like binding mode of the agonist-R-N6-phenylisopropyladenosine in the presence of 100 μM GTP. Therefore, it has to be concluded that full agonists differentiate between two thermodynamically distinct low-affinity states of the A1 adenosine receptor. One of these states is the uncoupled state (induced by GTP or NEM), the second is the low affinity state observed in the absence of GTP, in the presence of GDP or in experiments performed in Tris buffer. A discrimination of high-, intermediate- and low-affinity states revealed by the addition of GDPβS, GTP or GTPγS has also been observed for agonists of the μ and δ opioid receptors (Werling et al., 1988). However, the functional significance of the presence of two thermodynamically distinct low-affinity states remains to be resolved.
Abbreviations
- CCPA
2-chloro-N6-cyclopentyladenosine
- CGS 15943
5-amino-9-chloro-2-(2-furyl)-[1,2,4]triazolo[1,5-c]quinazoline
- CHAPS
3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate
- cladribine
2-chloro-2′-deoxyadenosine
- CPMA
N6-cyclopentyl-9-methyladenine
- CV 1808
2-phenylaminoadenosine
- DPCPX
1,3-dopropyl-8-cyclopentylxanthine
- G protein
guanine nucleotide binding protein
- MeSA
5′-deoxy-5′-methylthioadenosine
- MIG
1-methylisoguanosine
- NEM
N-ethylmaleimide
- R-PIA
R-N6-phenylisopropyladenosine
References
- BIRNBAUMER L., ABRAMOWITZ J., BROWN A.M. Receptor-effector coupling by G proteins. Biochim. Biophys. Acta. 1990;1031:163–224. doi: 10.1016/0304-4157(90)90007-y. [DOI] [PubMed] [Google Scholar]
- BÖHM M., GRÄBEL C., KIRCHMAYR R., LENSCHE H., ERDMANN E., GIERSCHIK P. C-Terminal modifications of pertussis toxin-sensitive G-protein α-subunits differentially affect immunoreactivity. Biochem. Pharmacol. 1993;46:2145–2154. doi: 10.1016/0006-2952(93)90603-t. [DOI] [PubMed] [Google Scholar]
- BOREA P.A., DALPIAZ A., GESSI S., GILLI G. Thermodynamics of 5-HT3 receptor binding discriminates agonistic from antagonistic behaviour. Eur. J. Pharmacol. 1996;298:329–334. doi: 10.1016/0014-2999(95)00813-6. [DOI] [PubMed] [Google Scholar]
- BOREA P.A., VARANI K., DALPIAZ A., CAPUZZO A., FABBRI E., IJZERMAN A.P. Full and partial agonistic behaviour and thermodynamic binding parameters of adenosine A1 receptor ligands. Eur. J. Pharmacol. 1994;267:55–61. doi: 10.1016/0922-4106(94)90224-0. [DOI] [PubMed] [Google Scholar]
- BOREA P.A., VARANI K., GUERRA L., GILLI P., GILLI G. Binding thermodynamics of A1 adenosine receptor ligands. Mol. Neuropharmacol. 1992;2:273–281. [Google Scholar]
- CONTRERAS M.L., WOLFE B.B., MOLINOFF P.B. Thermodynamic properties of agonist interactions with the beta adrenergic receptor-coupled adenylate cyclase system. I. High- and low-affinity states of agonist binding to membrane-bound beta adrenergic receptors. J. Pharmacol. Exp. Ther. 1986;237:154–164. [PubMed] [Google Scholar]
- DALPIAZ A., BOREA P.A., GESSI S., GILLI G. Binding thermodynamics of 5-HT1A receptor ligands. Eur. J. Pharmacol. 1996;312:107–115. doi: 10.1016/0014-2999(96)00429-3. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., HANCOCK A.A., LEFKOWITZ R.J. Validation and statistical analysis of radioligand binding data for mixtures of pharmacological receptor subtypes. Mol. Pharmacol. 1982;21:5–16. [PubMed] [Google Scholar]
- FLORIO V.A., STERNWEIS P.C. Mechanisms of muscarinic receptor activation on Go in reconstituted phospholipid vesicles. J. Biol. Chem. 1989;264:3909–3915. [PubMed] [Google Scholar]
- FRASER C.M. Site-directed mutagenesis of β-adrenergic receptors. J. Biol. Chem. 1989;264:9266–9270. [PubMed] [Google Scholar]
- HAUSDORFF W.P., HNATOWICH M., O'DOWD B.F., CARON M.G., LEFKOWITZ R.J. A mutation of the β2-adrenergic receptor impairs agonist activation of adenylyl cyclase without affecting high affinity agonist binding. J. Biol. Chem. 1990;265:1388–1393. [PubMed] [Google Scholar]
- KILPATRICK G.J., EL TAYAR N., VAN DE WATERBEEMD H., JENNER P., TESTA B., MARSDEN C.D. The thermodynamics of agonist and antagonist binding to dopamine D-2 receptors. Mol. Pharmacol. 1986;30:226–234. [PubMed] [Google Scholar]
- LOHSE M.J., LENSCHOW V., SCHWABE U. Two affinity states of Ri adenosine receptors in brain membranes. Analysis of guanine nucleotide and temperature effects on radioligand binding. Mol. Pharmacol. 1984;26:1–9. [PubMed] [Google Scholar]
- LORENZEN A., FUSS M., VOGT H., SCHWABE U. Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding. Mol. Pharmacol. 1993;44:115–123. [PubMed] [Google Scholar]
- LORENZEN A., GUERRA L., VOGT H., SCHWABE U. Interaction of full and partial agonists of the A1 adenosine receptor with receptor/G protein complexes in rat brain membranes. Mol. Pharmacol. 1996;49:915–926. [PubMed] [Google Scholar]
- MAKSAY G. Thermodynamics of γ-aminobutyric acid type A receptor binding differentiate agonists from antagonists. Mol. Pharmacol. 1994;46:386–390. [PubMed] [Google Scholar]
- MARALA R.B., MUSTAFA S.J. Direct evidence for the coupling of A2-adenosine receptor to stimulatory guanine nucleotide-binding protein in bovine brain striatum. J. Pharmacol. Exp. Ther. 1993;266:294–300. [PubMed] [Google Scholar]
- MIKLAVC A., KOCJAN D., MAVRI J., KOLLER J., HADZI D. On the fundamental difference in the thermodynamics of agonist and antagonist interactions with β-adrenergic receptors and the mechanism of entropy-driven binding. Biochem. Pharmacol. 1990;40:663–669. doi: 10.1016/0006-2952(90)90299-z. [DOI] [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. Ligand: A versatile computerized approach for the characterization of ligand binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- MURAYAMA T., UI M. [3H]GDP release from rat and hamster adipocyte membranes independently linked to receptors involved in activation or inhibition of adenylate cyclase. J. Biol. Chem. 1984;259:761–769. [PubMed] [Google Scholar]
- MURPHY K.M.M., SNYDER S.H. Heterogeneity of adenosine A1 receptor binding in brain tissue. Mol. Pharmacol. 1982;22:250–257. [PubMed] [Google Scholar]
- PETERSON G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- VAN DER PLOEG I. , PARKINSON F.E., FREDHOLM B.B. Effect of pertussis toxin on radioligand binding to rat brain adenosine A1 receptors. J. Neurochem. 1992;58:1221–1229. doi: 10.1111/j.1471-4159.1992.tb11332.x. [DOI] [PubMed] [Google Scholar]
- WAELBROECK M., CAMUS J., TASTENOY M., LAMBRECHT G., MUTSCHLER E., KROPFGANS M., SPERLICH J., WIESENBERGER F., TACKE R., CHRISTOPHE J. Thermodynamics of antagonist binding to rat muscarinic M2 receptors: antimuscarinics of the pridinol, sila-pridinol, diphenidol and sila-diphenidol type. Br. J. Pharmacol. 1993;109:360–370. doi: 10.1111/j.1476-5381.1993.tb13578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEILAND G.A., MINNEMAN K.A., MOLINOFF P.B. Fundamental difference between the molecular interactions of agonists and antagonists with the β-adrenergic receptor. Nature. 1979;281:114–117. doi: 10.1038/281114a0. [DOI] [PubMed] [Google Scholar]
- WERLING L.L., PUTTFARCKEN P.S., COX B.M. Multiple agonist-affinity states of opioid receptors: regulation of binding by guanyl nucleotides in guinea pig cortical, NG108-15, and 7315c cell membranes. Mol. Pharmacol. 1988;33:423–431. [PubMed] [Google Scholar]