

Abstract
N-methyl-D-aspartate (NMDA) receptors exist on noradrenergic axon terminals and mediate enhancement of noradrenaline (NA) release. We here investigated modulation by somatostatin (SRIF, somatotropin release inhibiting factor) of the NMDA-induced release of NA using superfused hippocampal synaptosomes.
The NMDA response was increased by SRIF-28 and SRIF-14, but not SRIF-28(1–14), whereas the release of [3H]-NA elicited by α-amino-3-hydroxy-5-methylisoxazide-4-propionic acid (AMPA) was unaffected. SRIF-14 did not mimic glycine at the NMDA receptor but activated SRIF receptors sited on noradrenergic terminals.
The SRIF-14 effect was blocked by pertussis toxin but mimicked by mastoparan, a G-protein activator. BIM-23056, but not Cyanamid 154806, antagonized the SRIF-14 effect. This effect was mimicked by L362855, a partial agonist at the sst5 subtype, but not by the new selective sst1–sst4 receptor agonists L797591, L779976, L796778 and L803087.
Protein kinase C (PKC) inhibitors (H7, staurosporine, GF 209103X, cheleritrine and sphingosine) prevented the SRIF-14 effect, while phorbol 12-myristate 13-acetate enhanced the NMDA response.
SRIF-14 permitted NMDA receptor activation in the presence of 1.2 mM Mg2+ ions, both in hippocampal synaptosomes and slices. Blockade of inositol-1,4,5-trisphosphate (InsP3) receptors with heparin abolished the effect of SRIF-14.
It is concluded that SRIF receptors, possibly of the sst5 subtype, can exert a permissive role on NMDA receptors colocalized on hippocampal noradrenergic terminals: activation of sst5 receptors is coupled to pertussis toxin-sensitive G proteins enhancing phosphoinositide metabolism with activation of InsP3 receptors and PKC; NMDA receptor subunits might be phosphorylated with consequent removal of the Mg2+ block in absence of depolarization.
Keywords: Somatostatin receptor subtypes; NMDA receptors; noradrenaline release; protein kinase C; inositol-1,4,5-trisphosphate; Mg2+ block; receptor–receptor interactions
Introduction
Several neurotransmitter/neuromodulator systems have been implicated in cognitive processes. The glutamatergic transmission in the hippocampus and, in particular, the glutamate receptors of the NMDA type are thought to play a major role in learning and memory. Long-term potentiation, that is regarded as a neuronal model to study memory function, depends on the activation of NMDA receptors (Bliss & Collingridge, 1993). Antagonists at the NMDA receptors can produce impairments in a wide variety of memory-related tasks in animals (Bannermann et al., 1995). The noradrenergic neurons of the locus coeruleus also have long been known to be involved in the regulation of attention and memory (Cahill et al., 1994). Besides classical transmitters, some neuropeptides seem to be positively involved in cognitive processes, possibly as neuromodulators. Among these, somatostatin (SRIF, somatotropin release inhibiting factor), a neuropeptide especially concentrated in hippocampus and cerebral cortex (for review see Epelbaum et al., 1994), has attracted considerable interest due to its implication in various behavioural functions, particularly learning and memory (for review see Rubinow et al., 1995).
Interactions between these different systems seem to occur in relevant brain regions. Glutamate-noradrenaline (NA) interactions have been described (see Huang & Kandel, 1996 and references therein). In particular, it is well known that NMDA receptors can mediate enhancement of the exocytotic release of NA in hippocampus and neocortex (Jones et al., 1987; Raiteri et al., 1992; Fink et al., 1992). In the hippocampus, NMDA receptors mediating enhancement of NA release are localized on noradrenergic axon terminals and non-NMDA receptors of the AMPA type, also mediating augmentation of NA release, coexist with NMDA receptors on the same noradrenergic terminals (Raiteri et al., 1992).
Modulations of NMDA responses by a transmitter other than glutamate or by glutamate itself acting at non-NMDA receptors have been identified and characterized in some detail. These studies, largely carried out using electrophysiological techniques, show acute potentiation of NMDA-mediated currents following activation of various receptors including μ opioid (Chen & Huang, 1991), glutamate metabotropic (Harvey & Collingridge, 1993) and cholinergic muscarinic (Calabresi et al., 1998) receptors.
In the current work we investigated the modulation of NMDA receptor function by SRIF using a biochemical approach, in which the NMDA-induced NA release from superfused rat hippocampal synaptosomes was taken as a functional response. We find that SRIF receptors, probably of the sst5 subtype, coexist with NMDA receptors on noradrenergic terminals. SRIF receptors are coupled to enhancement of phosphoinositide metabolism resulting into inositol-1,4,5-trisphosphate (InsP3) receptor and protein kinase C (PKC) activation. NMDA receptors, possibly phosphorylated by PKC, can finally be activated in presence of physiological concentrations of Mg2+ ions, without depolarization of the terminal membrane.
Methods
Animals and brain area used
Adult male rats (Sprague Dawley; 200–250 g) were housed at constant temperature (22±1°C) and relative humidity (50%) under a regular light–dark schedule (lights on 0700 to 1900 h). Food and water were freely available. The animals were killed by decapitation and the hippocampi rapidly dissected.
Release experiments from synaptosomes
Crude synaptosomes were prepared as previously described (Raiteri et al., 1984), with some modifications. Briefly, the hippocampi were homogenized in 40 volumes of 0.32 M sucrose buffered at pH 7.4 with phosphate (final concentration 0.01 M). The homogenate was centrifuged at 1000×g for 5 min, to remove nuclei and cellular debris, and crude synaptosomes were isolated from the supernatant by centrifugation at 12,000×g for 20 min. The synaptosomal pellet was then resuspended in a physiological medium having the following composition (mM): NaCl, 125; KCl, 3; MgSO4, 1.2; CaCl2, 1.2 NaH2PO4, 1; NaHCO3, 22; glucose, 10 (aeration with 95% O2 and 5% CO2); pH 7.2–7.4. In a set of experiments, when indicated, the hippocampi were homogenized in 0.32 M sucrose containing 5 nM pertussis toxin (PTx) or 40 μM heparin in order to entrap these agents into subsequently isolated synaptosomes (see Åkerman & Heinonen, 1983; Raiteri et al., 2000). In all experiments synaptosomes were incubated 15 min at 37°C with [3H]-NA (final concentration 0.03 μM) in presence of 6-nitroquipazine (final concentration 0.1 μM) to avoid false labelling of serotonergic terminals. Identical portions of the synaptosomal suspension were then layered on microporous filters at the bottom of parallel superfusion chambers (Raiteri et al., 1974). Superfusion (0.5 ml min−1) was started with standard physiological solution aerated with 95% O2 and 5% CO2, at 37°C. The system was first equilibrated during 36 min of superfusion; subsequently, four consecutive 3-min fractions (from t=36 min to t=48 min) were collected. Samples collected and superfused synaptosomes were then counted for radioactivity. When indicated, the physiological solution was replaced at t=20 min with a Mg2+-free medium or with a medium containing 0.6 mM Mg2+. Synaptosomes were exposed to NMDA, AMPA, glycine, mastoparan and SRIF receptor agonists at the end of the first fraction collected (t=39 min) until the end of the superfusion period (t=48 min), for a total of 9 min. When studying the time-dependency of the releasing stimulus, agonists were applied for 9 or 4 min, starting at t=39 min. Antagonists were added 8 min before agonists and remained present throughout the superfusion; PKC inhibitors were added concomitantly with agonists till the end of the superfusion.
Release experiments from slices
Slices (400 μm) were prepared from the ventro-medial portion of the hippocampus by a McIlwain tissue chopper and labelled with 0.08 μM [3H]-NA, 20 min at 37°C, in presence of 0.1 μM 6-nitroquipazine. After washing with tracer-free medium, slices were transferred to parallel superfusion chambers (1 slice/chamber) and superfusion was started at 1 ml min−1, at 37°C (t=0 min). The superfusion medium contained 0.1% of dialyzed bovine serum albumin to prevent sticking of peptides onto plastic tubings and the glass of the superfusion chambers. After 60 min of superfusion, seven 5-min fractions were collected. Fractions collected and superfused slices (solubilized with Soluene®) were then counted for radioactivity. Antagonists were added to the medium after 30 min of superfusion and were present till the end of the experiment (from t=30 min to t=95 min). Slices were exposed to NMDA and SRIF-14 for 10 min, starting at min 73 of superfusion; under this condition, the NMDA-evoked release of tritium was contained within the fourth and the fifth fractions.
Calculations and statistics
The amount of radioactivity released into each superfusate fraction was expressed as a percentage of the total tissue content at the start of the fraction collected (fractional efflux). In the experiments of release from synaptosomes, drug effects were evaluated from the ratio between the per cent release in the fraction corresponding to the maximal effect (third fraction) and that in the first fraction collected. This ratio was compared with the corresponding ratio obtained under control conditions (no drug added). In the experiments of release from slices, effects were expressed as agonist-induced tritium overflow taken as the difference between the total tritium efflux in the combined fourth and fifth fractions collected and the basal efflux present in the fractions collected immediately before (third fraction) and after (sixth fraction) those containing the agonist-evoked overflow. Direct comparison between two means was performed with the two-tailed Student's t-test. Significance of difference between means was analysed by two-way ANOVA followed by Newman-Keuls multiple comparison.
Chemicals
1-[7,83H]-noradrenaline (specific activity 39 ci mmol−1) was from Amersham Radiochemical Center (Buckinghamshire, U.K.). Somatostatin-28(1–14) (SRIF-28(1–14)), somatostatin-28 (SRIF-28) and somatostatin (SRIF-14) were from Peninsula Lab. Inc. (Merseyside, U.K.); N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methylisoxazide-4-propionic acid (AMPA) and 7-chloro-kynurenic acid from Tocris-Cookson (Bristol, U.K.); glycine, 1-(5-isoquinolinylsulphonyl)-2-methylpiperazine (H7), staurosporine, phorbol 12,13-didecanoate (PDD), phorbol 12-myristate 13-acetate (PMA) and heparin from Sigma Chemicals Co. (St. Louis, MO, U.S.A.). Pertussis toxin was from Calbiochem-Novabiochem (Nottingham, U.K.). Cheleritrine chloride, sphingosine, mastoparan and dihydrochloride3-[1-[3-(dimethylamino)propyl] -1H-indol-3yl] -4- (1H-indol-3-yl) -1H- pyrrole-2,5-dione (GF 209103X) were purchased from RBI (Natick, MA, U.S.A.). The following drugs are gifts from the companies indicated: 6-nitroquipazine maleate (Duphar, Amsterdam, The Netherlands), CGS 19755 (Novartis Pharmaceutical Corporation, Summit, NJ, U.S.A.) and L797591, L779976, L796778 and L803087 (Merck Research Laboratories, Rahway, NJ, U.S.A.). BIM-23056 and L362855 were a kind gift from Dr P.P.A. Humphrey (Cambridge, U.K.), while Cyanamid 154806 was kindly donated by Dr D. Hoyer (Basel, Switzerland).
Results
Effect of somatostatin peptides on the release of [3H]-NA induced by NMDA or AMPA
Exposure of rat hippocampal synaptosomes, prelabelled with [3H]-NA, to 1 nM SRIF-28, SRIF-14 or SRIF-28(1–14) caused no significant change of the basal tritium release (Figure 1). It is known that, in absence of Mg2+ ions, the glutamate receptor agonist NMDA plus its coagonist glycine or D-serine (Kleckner & Dingledine, 1987) can augment NA release from various brain tissue preparations. The release of NA that can be observed following addition of NMDA alone to a thin layer of synaptosomes up-down superfused, a condition in which any endogenous glycine/D-serine is removed, is clearly due to glycine present as contaminant in the superfusion medium (see, for details, Paudice et al., 1998). Based on frequent h.p.l.c. monitoring of the solutions here used, the glycine contamination amounted to ⩽50 nM, a concentration sufficient to permit significant NA release by 100 μM NMDA (Figure 1); this effect can, however, be further enhanced by glycine up to a maximum obtained with ∼1 μM glycine (Pattarini et al., 1998; Table 3).
Figure 1.
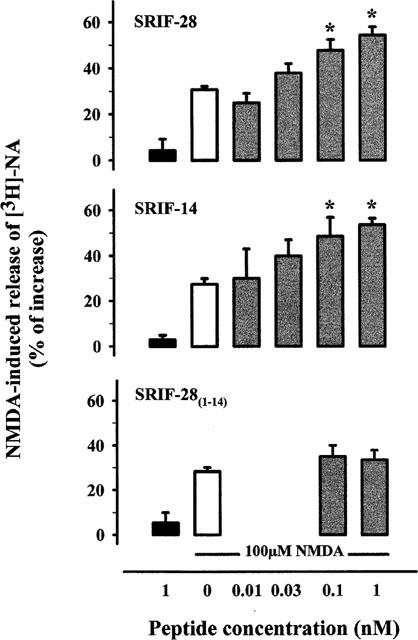
SRIF-28 and SRIF-14, but not SRIF-28(1–14) potentiates the NMDA-induced [3H]-NA release from superfused rat hippocampal synaptosomes. Superfusion was carried out with Mg2+-free medium. NMDA and the different peptides were added concomitantly. Results are expressed as per cent increase over basal release. Solid bars: peptides; empty bars: 100 μM NMDA; grey bars: 100 μM NMDA plus peptides. Data are means±s.e.mean from 3–23 experiments run in triplicate (three superfusion chambers for each condition). *P<0.01 versus NMDA.
Table 3.
Pertussis toxin prevents the potentiating effect of SRIF-14 on the NMDA-induced release of [3H]-NA from hippocampal synaptosomes

Figure 1 shows that the release of [3H]-NA elicited by 100 μM NMDA alone, in Mg2+-free medium, was enhanced when SRIF-28 or SRIF-14 was added to the superfusion medium; the maximal potentiation (about 100%) occurred between 0.1 and 1 nM of the peptides which, in our experimental conditions, appeared roughly equipotent. In contrast, SRIF-28(1–14) (0.1 or 1 nM) was without effect on the NMDA-evoked [3H]-NA release.
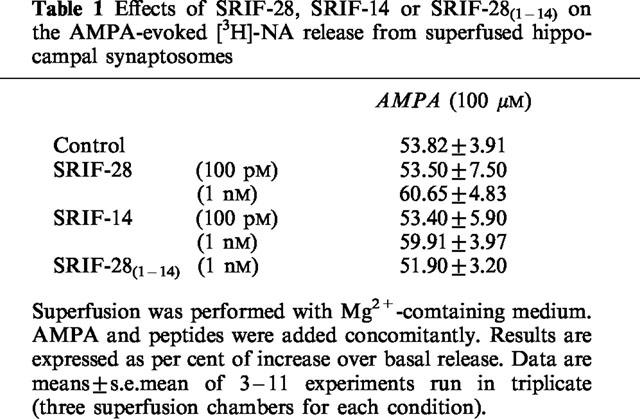
Since release of NA from brain tissue preparations can also be evoked by activation of glutamate AMPA receptors (Desai et al., 1994; Raiteri et al., 1992), we tested the effect of SRIF peptides on the AMPA-evoked release of [3H]-NA. Table 1 shows that the release of [3H]-NA elicited by 100 μM AMPA remained unchanged in the presence of 1 nM SRIF-28, SRIF-14 or SRIF-28(1–14). Since 100 μM AMPA released [3H]-NA to the same extent as 100 μM NMDA plus 1 nM SRIF-14 (cf. Figure 1 and Table 1), a lower concentration of AMPA (10 μM) was tested. Also in this case SRIF-14 (1 nM) was unable to potentiate the AMPA effect: AMPA=43.09±9.15%; AMPA+SRIF-14=45.67±15.39%.
Table 1.
Effects of SRIF-28, SRIF-14 or SRIF-28(1–14) on the AMPA-evoked [3H]-NA release from superfused hippocampal synaptosomes
Where does SRIF act to enhance NMDA responses?
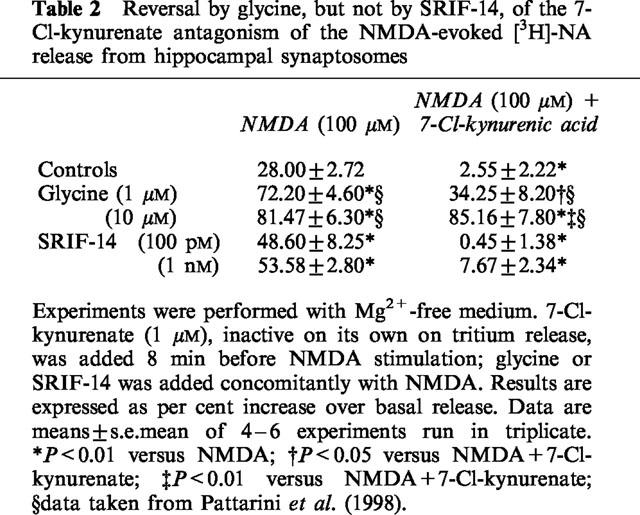
Glycine was found to potentiate the NMDA-induced release of [3H]-NA from superfused rat hippocampal synaptosomes, being inactive on its own (Pittaluga & Raiteri, 1990). Recently, some peptides have been reported to mimic glycine by potently activating the glycine site on the NMDA receptor that mediates the release of NA (Pattarini et al., 1998). Thus SRIF-14 might behave as a glycinomimetic agent at these receptors. To test this idea we compared the ability of glycine and SRIF-14 to reverse and surmount the receptor block brought about by 7-Cl-kynurenic acid, a selective antagonist at the glycine site of the NMDA receptor. The antagonist, added at 1 μM, abolished the release of [3H]-NA elicited by 100 μM NMDA alone (Table 2). This antagonism could be prevented in part by 1 μM glycine and surmounted by 10 μM glycine. In contrast, SRIF-14 (0.1 or 1 nM) failed to significantly attenuate the 7-Cl-kynurenate antagonism (Table 2).
Table 2.
Reversal by glycine, but not by SRIF-14, of the 7-Cl-kynurenate antagonism of the NMDA-evoked [3H]-NA release from hippocampal synaptosomes
Involvement of G protein-coupled somatostatin receptors
Somatostatin receptors in the CNS are frequently, but not always, linked to PTx-sensitive GTP binding G proteins (see Hoyer et al., 1994; Bell & Reisine, 1995; Siehler & Hoyer, 1999a). It has so far been difficult to study effects of PTx with synaptosomes because the prolonged incubations required reduce the viability of isolated nerve endings. For this reason we acutely entrapped PTx into synaptosomes by homogenizing the hippocampi in the presence of buffered sucrose to which the toxin was added at the final concentration of 5 nM. Table 3 shows that entrapping of PTx did not modify either the basal tritium release or the release of [3H]-NA elicited by NMDA alone, in Mg2+-free medium. In PTx-entrapped synaptosomes, SRIF-14 (1 nM) lost its ability to potentiate the NMDA response. On the other hand, glycine (1 μM) enhanced the effect of NMDA in PTx-entrapped synaptosomes to the same extent as in control synaptosomes.
The possible involvement of a G protein-linked mechanism was further investigated by superfusing synaptosomes with mastoparan, a wasp venom peptide known to activate G proteins (Perianin & Snyderman, 1989). The effect of 100 μM NMDA on [3H]-NA release (25.12±2.55; n=3) was increased by about 80% by 0.3 μM mastoparan (45.01±6.83; n=3; P<0.05). At the concentration used, mastoparan had no effect, on its own, on the basal release of tritium (not shown).
Pharmacological characterization of the SRIF receptor subtype involved
Five distinct SRIF receptor genes have been described, encoding five receptors called sst1 through sst5. Recently, selective non-peptide agonists have been introduced, showing high affinity for sst1–4 receptors (Rohrer et al., 1998). We tested the effect of L797591 (sst1-selective), L779976 (sst2-selective), L796778 (sst3-selective) and L803087 (sst4-selective) on the release of NA elicited by NMDA. All the compounds, inactive on their own on the spontaneous release of tritium (data not shown), failed to affect the 100 μM NMDA-induced release of [3H]-NA when tested at 1–1000 nM (Figure 2). On the contrary, the selective sst5 peptidergic partial agonist/antagonist L362855 (Wilkinson et al., 1997), added at 0.1–10 nM, concentration-dependently potentiated the NMDA-induced release of NA, without affecting the basal outflow of tritium (Figure 2).
Figure 2.
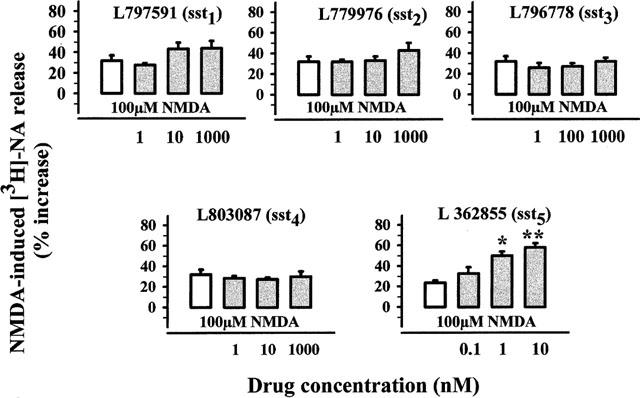
Effects of SRIF receptor agonists on the NMDA-evoked [3H]-NA release from hippocampal synaptosomes. Superfusion was carried out with Mg2+-free medium. Agonists were added concomitantly with NMDA. Results are expressed as per cent increase over basal release. Data are means±s.e.mean of 15 experiments run in triplicate. *P<0.05 versus respective control; **P<0.01 versus respective control.
The linear peptide BIM-23056 has been reported to behave as a competitive antagonist of the sst5 receptor-mediated activation of phosphoinositide metabolism (Wilkinson et al., 1997; Siehler & Hoyer, 1999b), but controversial results exist in literature as to the action of BIM-23056 on adenylate cyclase activity. The compound was a full agonist in the sst5 receptor-mediated inhibition of cyclic AMP formation in CHO-K1 cells (Carruthers et al., 1999), while it behaved as a partial agonist or close to full antagonist in inhibiting the forskolin-stimulated adenylate cyclase activity mediated by the five human SRIF receptors expressed in CCL39 cells (Siehler & Hoyer, 1999c). In our experimental paradigm, the SRIF-14 effect was concentration-dependently prevented by BIM-23056 (0.5–10 nM; Figure 3). On the other hand, the linear peptide Cyanamid 154806, a compound known to act as a preferential antagonist at the sst2 receptor (Bass et al., 1996), did not affect significantly the enhancement by SRIF-14 (1 nM) of the NMDA (100 μM)-evoked release of [3H]-NA (Figure 3). At the concentrations used, the two compounds did not modify, on their own, the basal or the NMDA-induced release of [3H]-NA (100 μM NMDA=28.35±2.37; 10 nM BIM 23056=4.25±0.35; 100 μM NMDA+10 nM BIM 23056=26.87±4.37; 100 nM Cyanamid 154806=2.71±0.61; 100 μM NMDA+100 nM Cyanamid 154806=25.68±5.23).
Figure 3.
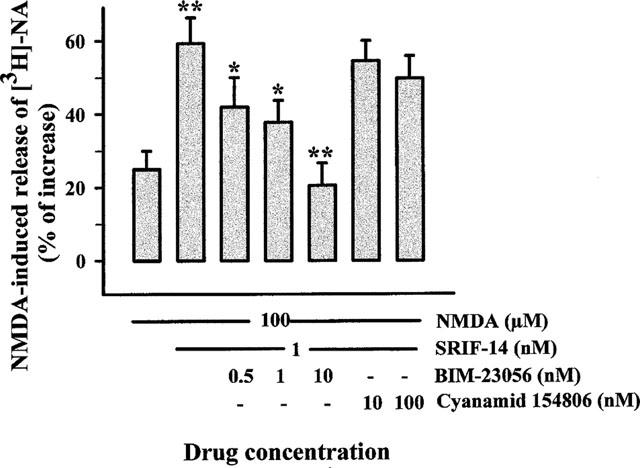
Effects of SRIF receptor antagonists on the SRIF-14 potentiation of the NMDA-evoked [3H]-NA release from hippocampal synaptosomes. Superfusion was carried out with Mg2+-free medium. Antagonists were added 8 min before agonists and maintained till the end of superfusion period. Results are expressed as per cent increase over basal release. Data are means±s.e.mean of 12 experiments run in triplicate. *P<0.05 versus respective control; **P<0.01 versus respective control.
Involvement of protein kinase C
Somatostatin receptors have been reported to couple to a diversity of second messenger transducing systems. One of the classical actions of SRIF is the inhibition of adenylyl cyclase activity (see Bell & Reisine, 1995). On the other hand, SRIF receptors of the sst2 and sst5 subtypes (Akbar et al., 1994; Wilkinson et al., 1997) and, more recently, of the sst3 subtype (Siehler & Hoyer, 1999b) when expressed in heterologous cells have also been found to mediate activation of phosphoinositide metabolism in a PTx-sensitive manner. As the SRIF receptors involved in the potentiation of NMDA function seems to be sst5 subtype, the effect of SRIF-14 was studied in the presence of several PKC inhibitors acting at different domains of the enzyme (see Gordge & Ryves, 1994).
Three inhibitors at the ATP binding site of PKC, H7 (10 μM), staurosporine (0.1 μM) and GF 209103X (0.1 μM), largely prevented the SRIF-14 potentiation of the NMDA-induced [3H]-NA release from hippocampal synaptosomes (Table 4). The SRIF-14 effect was completely prevented by 1 μM cheleritrine, a PKC inhibitor selective for the substrate binding site and, to a large extent, by 10 μM sphingosine which blocks PKC at the diacylglycerol-binding site. On their own, at the concentrations used, the inhibitors had no significant effects on the NMDA-evoked release of [3H]-NA (Table 4) or the basal tritium release (not shown).
Table 4.
Blockade by PKC inhibitors of the potentiation of the NMDA-induced release of [3H]-NA produced by SRIF-14 in hippocampal synaptosomes

To strengthen the idea that PKC activation in noradrenergic terminals is involved in the potentiation of the NMDA-induced NA release, synaptosomal PKC was activated by a phorbol ester. As shown in Figure 4, phorbol 12-myristate 13-acetate (PMA; 1 μM) strongly enhanced the NMDA-induced [3H]-NA release. Figure 4 also shows that phorbol 12,13-didecanoate (PDD) was ineffective both on the basal release of tritium and on the NMDA-evoked release of [3H]-NA. Similar results were obtained in neocortical slices by Wang & White (1998).
Figure 4.
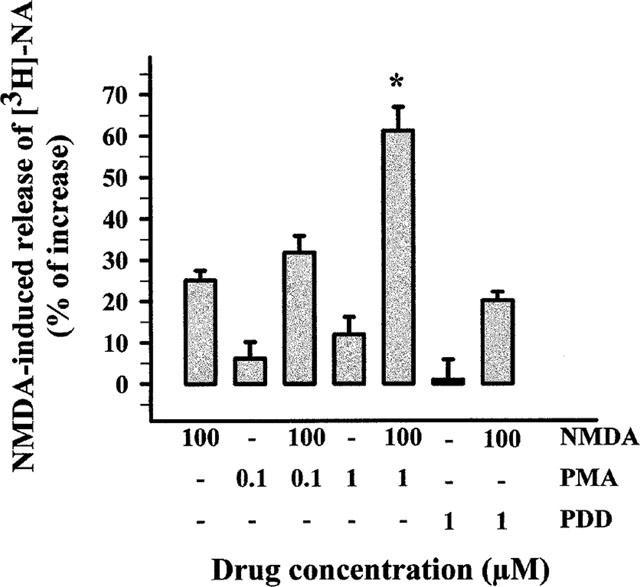
Effects of phorbol esters on the NMDA-evoked [3H]-NA release from hippocampal synaptosomes. Superfusion was carried out with Mg2+-free medium. Phorbol 12-myristate 13-acetate (PMA) or phorbol 12,13-didecanoate (PDD) was added concomitantly with NMDA. Results are expressed as per cent increase over basal release. Data are means±s.e.mean of 3 experiments run in triplicate. *P<0.01 versus respective control.
SRIF causes reduction of the Mg2+ block of NMDA receptors
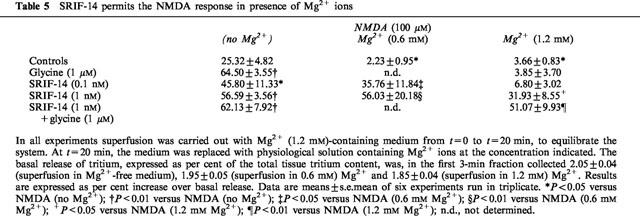
The effects of NMDA, either alone or with added glycine, observed in Mg2+-free medium are known to be prevented when Mg2+ ions are added to the medium (Mayer & Westbrook, 1985; see also Table 5). Interestingly, the releasing effects of NMDA plus 0.1 nM SRIF-14, in Mg2+-free medium, was largely resistant to the addition of 0.6 mM Mg2+, while that of NMDA plus 1 nM SRIF-14 was totally unaffected by 0.6 mM Mg2+ and largely resistant to 1.2 mM Mg2+ (Table 5). Furthermore, in Mg2+-free medium, SRIF-14 could potentiate the NMDA response occurring in presence of ⩽50 nM contaminating glycine, but not when glycine was present at its maximally effective concentration (1 μM). Of particular interest are the observations made in the presence of physiological concentrations of Mg2+ (1.2 mM). As expected, under these conditions, NMDA plus glycine (up to 1 μM) failed to evoke any response. However, addition of 1 nM SRIF-14 was able to counteract the Mg2+ block thus permitting activation of the NMDA receptors in the presence of glycine contamination and further potentiation of the NMDA response in the presence of glycine. The effect of NMDA plus 1 nM SRIF-14, in Mg2+ (1.2 mM)-containing medium, was strongly (about 90%) prevented by 10 μM CGS 19755, an antagonist at the NMDA receptor glutamate site, confirming that the release of [3H]-NA is essentially due to activation of NMDA receptors (not shown).
Table 5.
SRIF-14 permits the NMDA response in presence of Mg2+ ions
It was found previously that ionotropic AMPA receptors coexist with NMDA receptors on noradrenergic terminals of the hippocampus; moreover, activation of these AMPA presynaptic receptors permit activation of NMDA receptors in the presence of physiological concentrations (1.2 mM) of Mg2+ ions (Raiteri et al., 1992). Table 6 shows that, in the presence of 1.2 mM Mg2+, AMPA but not NMDA, could elicit release of [3H]-NA; moreover, when added in combination, AMPA and NMDA could release more [3H]-NA than AMPA alone. Glycine, unable to potentiate the effect of AMPA, did potentiate that of AMPA plus NMDA. On the contrary SRIF-14 was ineffective. Table 6 also shows that the effect of AMPA or of AMPA plus NMDA was insensitive to the PKC inhibitor H7.
Table 6.
Effects of glycine, of SRIF-14 and of the PKC inhibitor H7 on the release of [3H]-NA induced by AMPA, alone or in combination with NMDA, from hippocampal synaptosomes

The SRIF-NMDA receptor–receptor interaction also occurs in slices
It was important to ascertain if SRIF could enhance the NMDA function in a more intact brain tissue preparation, in the presence of a physiological concentration of Mg2+. Figure 5 illustrates the results obtained when hippocampal slices, prelabelled with [3H]-NA, were superfused with NMDA in the absence or in the presence of SRIF-14. NMDA, added alone at 100 μM, was unable to release tritium. On its own, 1 nM SRIF-14 also failed to elevate basal tritium outflow. When added together to the superfusion medium, 100 μM NMDA and 1 nM SRIF-14 significantly augmented the outflow of tritium from the slices. This effect was clearly due to activation of NMDA receptors, since it was abolished by CGS 19755. Furthermore, the effect of SRIF-14 plus NMDA could be prevented by the PKC inhibitor GF 209103X.
Figure 5.
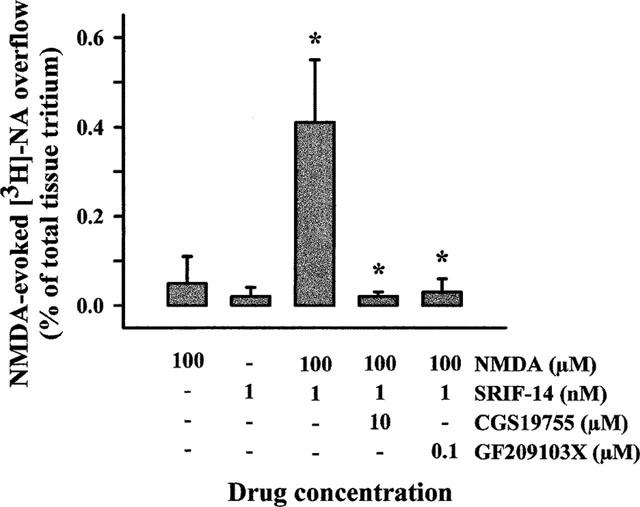
SRIF-14 permits activation of NMDA receptors regulating [3H]-NA release from hippocampal slices in the presence of a physiological concentration of Mg2+ ions. Superfusion of slices was carried out with physiological medium containing 1.2 mM Mg2+. Results are expressed as per cent increase over basal release and represent means±s.e.mean of 7 experiments run in triplicate. *P<0.01 versus respective control.
Involvement of an InsP3-induced Ca2+ release
To shed light on the involvement of intraterminal Ca2+ stores, the effect of NMDA plus SRIF-14 was investigated in synaptosomes prepared in the presence of 40 μM heparin, a competitive antagonist at the InsP3 receptors able to block the release of Ca2+ from InsP3-sensitive stores (Taylor & Broad, 1998). Synaptosomes entrapped with heparin released much less [3H]-NA than control synaptosomes when exposed to 100 μM NMDA plus 1 nM SRIF-14. Heparin had no effect on the release of [3H]-NA elicited by NMDA plus AMPA (Table 7).
Table 7.
Heparin prevents the effect of SRIF-14, but not of AMPA, on the NMDA-induced release of [3H]-NA from hippocampal synaptosomes

Discussion
SRIF-14, but not SRIF-28(1–14), was able to potentiate the NMDA-evoked [3H]-NA release from hippocampal synaptosomes. SRIF-14 is the peptide predominantly released in a Ca2+-dependent, probably exocytotic, manner from brain tissue preparations subjected to depolarizing stimuli (Bonanno et al., 1988).
The possibility that SRIF-14 is a glycinomimetic agent that, similarly to some other peptides (Pattarini et al., 1998), can activate the glycine site of the NMDA receptor on noradrenergic terminals should be excluded because: (a) unlike glycine, SRIF-14 was unable to reverse the antagonism of the NMDA effect caused by 7-Cl-kynurenate, a selective antagonist at the glycine site; (b) the effect of SRIF-14 depends on a PTx-sensitive mechanism; and (c) SRIF-14 permits activation of the NMDA receptor in presence of Mg2+.
The maximally effective concentrations of SRIF-14 ranged between 0.1 and 1 nM, well consistent with those reported to activate SRIF receptors in various systems (Epelbaum et al., 1994; Matsuoka et al., 1995; Rubinow et al., 1995). The block of the SRIF-14 effect by the SRIF receptor antagonist BIM-23056 indicates that the peptide acts through specific receptors. Our experimental set up (a very thin layer of synaptosomes up-down perfused in which endogenous compounds released are rapidly removed by the superfusing medium before they can act on the synaptosomes) has often been shown to prevent indirect effects; therefore SRIF receptors and NMDA receptors are co-localized on noradrenergic terminals of the hippocampus and the effect of SRIF-14 on the NMDA response (the peptide is inactive on its own) implicates a receptor–receptor cross-talk occurring in these noradrenergic terminals.
The pharmacological characterization of the SRIF receptors involved suggests that SRIF-14 activates a receptor of the sst5 subtype. In fact, agonists selective for the sst1, sst2, sst3 or sst4 subtype could not enhance the NMDA receptor function, whereas the effect of SRIF-14 was mimicked by L362855, a peptidergic partial agonist/antagonist displaying selectivity for recombinant sst5 receptors (Wilkinson et al., 1997). Furthermore, the native receptor here characterized appears to be coupled to the phosphoinositide pathway, a characteristic that has been previously attributed to the sst2 and the sst5 subtypes (Akbar et al., 1994; Wilkinson et al., 1997) and more recently also to the sst3 receptor (Siehler & Hoyer, 1999b). Finally, the effect of SRIF-14 was prevented by BIM-23056, a compound shown to behave as a competitive antagonist of the sst5 receptor-mediated activation of phosphoinositide metabolism (Wilkinson et al., 1997; Siehler & Hoyer, 1999a,1999b), but not by Cyanamid 154806, a sst2-preferring antagonist (Bass et al., 1996; Feniuk et al., 1998), It has to be added that the expression of sst5 receptors in the brain is currently a subject of controversy (Bruno et al., 1993; Thoss et al., 1996). However, as shown in other receptor systems, the method used here has a very high sensitivity in detecting receptors; therefore the present results (obviously based on the reported selectivity of the drugs used) favour the existence of functional sst5 receptors in the hippocampus.
To establish the involvement of PTx-sensitive G-proteins, we tested the sensitivity of the SRIF-14 effect to PTx. In order to preserve the viability of synaptosomes, prolonged incubation with the toxin was avoided; PTx was entrapped into synaptosomes by homogenizing the hippocampal tissue in the presence of 5 nM PTx, according to a technique originally set up for small impermeant molecules (Åkerman & Heinonen, 1983) and recently shown to permit entrapping of compounds of high molecular weight (Raiteri et al., 2000). Based on estimates made by entrapping [3H]-sucrose, the intrasynaptosomal concentration of PTx should be ⩽0.25 nM, i.e. the 5% of the original concentration in the homogenization medium; this concentration was in any case well sufficient to block the effect of SRIF-14 in PTx-staffed synaptosomes leaving that of glycine unaffected (Table 3). Furthermore, the G-protein activator mastoparan augmented on its own the NMDA-evoked release of [3H]-NA. These results suggest that SRIF-14 acts through a PTx-sensitive G-protein, in keeping with the reported properties of SRIF receptors (see, for reviews, Hoyer et al., 1994; Bell & Reisine, 1995).
As to the transduction mechanism involved, it is well known that one of the classical effects of SRIF is the inhibition of adenylyl cyclase activity; actually, all five recombinant SRIF receptors known so far have been shown to negatively couple to this enzyme (Hoyer et al., 1994; Bell & Reisine, 1995). On the other hand, it has been reported that recombinant receptors of the sst2, sst3 and sst5 subtype can also couple to PTx-sensitive transduction mechanisms linked to InsP3 formation and to increase in intracellular Ca2+ concentration (Akbar et al., 1994; Wilkinson et al., 1997; Siehler & Hoyer, 1999b). There are only a few studies which describe the coupling of SRIF receptors to stimulation of the InsP3-Ca2+ signalling pathway in native tissues (Munoz-Acedo et al., 1995). The present finding that the effect of SRIF-14 on the NMDA-evoked NA release was prevented by PKC inhibitors indicates an important role for this enzyme. Furthermore, the effect of SRIF-14 also was largely reduced when heparin, an antagonist at the InsP3 receptor (Taylor & Broad, 1998), was entrapped into hippocampal synaptosomes, suggesting that PKC could be, at least in part, activated by Ca2+ ions originating from InsP3-sensitive intrasynaptosomal stores.
Several kinases, including PKC, can positively modulate the function of NMDA receptors (Zukin & Bennett, 1995; Bi et al., 1998 for reviews). Biochemical analysis has demonstrated that the NR1 subunit can be directly phosphorylated by PKC in the intracellular carboxyl terminus (Tingley et al., 1993; Logan et al., 1999). Such a phosphorylation increased the ionic response of the NMDA receptor channel. Our results show that the potentiation of the NMDA-evoked NA release by SRIF-14 is a PKC-dependent process, sensitive to PKC inhibitors acting at the ATP-binding site, like H7, staurosporine and GF 209103X, at the substrate binding site, like cheleritrine and at the diacylglycerol binding site, like sphingosine. Altogether it seems that activation of SRIF receptors on hippocampal noradrenergic terminals stimulates diacylglycerol production and InsP3-dependent increase of [Ca2+]i, which, in turn, enhance presynaptic PKC activity. Moreover, as recently shown by Wang & White (1998) in neocortex slices and here confirmed in hippocampal synaptosomes (Figure 5), PKC activators phorbol esters enhanced the NMDA-evoked NA release. Unfortunately, the extremely small percentage (<1%) of noradrenergic terminals in the synaptosomal preparations used permits to monitor NA release but not to ascertain if the possible increases of InsP3 and internal Ca2+ occurring in the whole synaptosomal preparation reflect release potentiation.
When added in Mg2+ (1.2 mM)-containing medium SRIF-14, but not glycine, could permit the NMDA-mediated response. On the other hand, glycine could potentiate the effect of NMDA plus SRIF-14. It seems that activation of SRIF receptors on noradrenergic terminals can reduce the voltage-dependent Mg2+ block of NMDA receptor channels, in spite of the fact that the terminal membrane is not depolarized. It was reported that PKC intracellularly applied in isolated trigeminal neurons potentiated NMDA-activated currents by increasing the probability of channel openings and by reducing the Mg2+ block of the channels (Chen & Huang, 1992). Differently from SRIF, membrane depolarization and consequent reduction of the Mg2+ block appear to be prerequisites for the coagonist action of glycine.
Ionotropic glutamate AMPA receptors are known to coexist with NMDA receptors on hippocampal noradrenergic terminals. When the former are activated, the latter respond to NMDA even in the presence of 1.2 mM Mg2+ (Raiteri et al., 1992). The release of NA elicited by AMPA plus NMDA, differently from that of SRIF-14 plus NMDA, was insensitive to PKC inhibition. This is compatible with the idea that SRIF-14 acting at SRIF receptors permits NMDA receptor activation in the presence of Mg2+ through the phosphoinositide system and the PKC-mediated reduction of the Mg2+ block, whereas glutamate acting at AMPA receptors exerts a permissive role on NMDA receptors, in the presence of Mg2+, by depolarizing the terminal membrane with consequent reduction of the Mg2+ block. Interestingly, glycine, but not SRIF-14, can potentiate the NA release evoked by AMPA plus NMDA, supporting the view that the glutamate coagonist requires previous removal of the Mg2+ block by depolarization, whereas SRIF causes by itself reduction of the Mg2+ block and permits the action of both NMDA and glycine (see Table 5).
In conclusion, SRIF receptors are present on noradrenergic terminals in the hippocampus; these receptors seem to coexist with NMDA receptors mediating enhancement of NA release; SRIF receptors can interact with NMDA receptors by a mechanism involving pertussis toxin-sensitive G-proteins, activation of InsP3 receptors and of PKC; phosphorylation of NMDA receptor subunits might finally remove the Mg2+ block without membrane depolarization.
Considering the involvement of SRIF, NMDA receptors and NA in memory and learning, the enhancement by SRIF of the NMDA-mediated NA release in the hippocampus deserves further investigation. As previously mentioned, SRIF receptors are highly heterogenenous (Hoyer et al., 1994; Bell & Reisine, 1995) and non-peptidergic agonists selective for different subtypes of the somatostatin receptor have recently been introduced. The use of these selective ligands in biochemical and behavioural studies would certainly help in understanding the role of the transmitter interaction, here characterized in cognitive processes.
Acknowledgments
This work was supported by MURST-CNR Biotechnology Program L. 95/95 and by ISS–‘Programma nazionale di ricerca sull'AIDS–Progetto Patologia, clinica e terapia dell‘AIDS'. We are grateful to Maura Agate for editorial assistance.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methylisoxazide-4-propionic acid
- BIM-23056
D-Phe-Phe-Tyr-D-TRP-Lys-Val-Phe-D-Nal-NH2
- Cyanamid 154806
AcNH-4-NO2-Phe-c[D-Cys-Tyr-D-Trp-Lys-Thr-Cys]-Tyr-NH2
- GF 209103X
dihydrochloride3-[1-[3-(dimethylamino)propyl]-1H-indol-3yl]-4-(1H-indol-3-yl)-1H-pyrrole-2,5-dione
- H7
1-(5-isoquinolinylsulphonyl)-2-methylpiperazine
- InsP3
inositol-1,4,5-trisphosphate
- NA
noradrenaline
- NMDA
N-methyl-D-aspartate
- PDD
phorbol 12,13-didecanoate
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PTx
pertussis toxin
- SRIF
somatotropin release inhibiting factor (somatostatin)
References
- AKBAR M.F., OKAJIMA H., TOMURA M.A., MAJID Y., YAMADA S., SEINA S., KONDO Y. Phospholipase C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1–5, in transfected COS-7 cells. FEBS Lett. 1994;348:192–196. doi: 10.1016/0014-5793(94)00603-2. [DOI] [PubMed] [Google Scholar]
- ÅKERMAN K.E.O., HEINONEN E. Qualitative measurements of cytosolic calcium ion concentration within isolated guinea pig nerve endings using entrapped arsenazo III. Biochim. Biophys. Acta. 1983;732:117–121. doi: 10.1016/0005-2736(83)90193-1. [DOI] [PubMed] [Google Scholar]
- BANNERMANN D.M., GOOD M.A., BUTCHER S.P., RAMSAY M., MORRIS R.G.M. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature. 1995;378:182–186. doi: 10.1038/378182a0. [DOI] [PubMed] [Google Scholar]
- BASS R.T., BUCKWALTER B.L., PATEL B.P., PAUSCH M.K., PRICE L.A., STRNAD J., HADCOCK J.R. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996;50:709–715. [PubMed] [Google Scholar]
- BELL G.I., REISINE T. Molecular biology of somatostatin receptors. Trends Neurosci. 1995;16:34–38. doi: 10.1016/0166-2236(93)90050-v. [DOI] [PubMed] [Google Scholar]
- BI X., STANDLEY S., BAUDRY M. Posttranslational regulation of ionotropic glutamate receptors and synaptic plasticity. Int. Rev. Neurobiol. 1998;42:227–284. doi: 10.1016/s0074-7742(08)60612-1. [DOI] [PubMed] [Google Scholar]
- BLISS T.V.P., COLLINGRIDGE G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- BONANNO G., RAITERI M., EMSON P.C. In vitro release of somatostatin from cerebral cortical slices: characterization of electrically evoked release. Brain Res. 1988;447:92–97. doi: 10.1016/0006-8993(88)90968-7. [DOI] [PubMed] [Google Scholar]
- BRUNO J.F., XU Y., SONG J., BERELOWITZ M. Tissue distribution of somatostatin receptor subtype messenger ribonucleic acid in the rat. Endocrinology. 1993;133:2561–2567. doi: 10.1210/endo.133.6.8243278. [DOI] [PubMed] [Google Scholar]
- CAHILL L., PRINS B., WEBER M., MCGAUGH J.L. Beta-adrenergic activation and memory for emotional events. Nature. 1994;371:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., CENTONZE D., GUBELLINI P., PISANI A., BERNARDI G. Endogenous ACh enhances striatal NMDA-responses via M1-like muscarinic receptors and PKC activation. Eur. J. Neurosci. 1998;10:2887–2895. doi: 10.1111/j.1460-9568.1998.00294.x. [DOI] [PubMed] [Google Scholar]
- CARRUTHERS A.M., WARNER A.J., MICHEL A.D., FENIUK W., HUMPHREY P.P.A. Activation of adenylate cyclase by human recombinant sst5 receptors expressed in CHO-K1 cells and involvement of Gαs proteins. Br. J. Pharmacol. 1999;126:1221–1229. doi: 10.1038/sj.bjp.0702401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN L., HUANG L.Y.M. Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by μ opioid. Neuron. 1991;7:319–326. doi: 10.1016/0896-6273(91)90270-a. [DOI] [PubMed] [Google Scholar]
- CHEN L., HUANG L.Y.M. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- DESAI M.A., BURNETT J.P., SCHOEPP D.D. Cyclothiazide selectively potentiates AMPA and kainate-induced [3H]norepinephrine release from rat hippocampal slices. J. Neurochem. 1994;63:231–237. doi: 10.1046/j.1471-4159.1994.63010231.x. [DOI] [PubMed] [Google Scholar]
- EPELBAUM J., DOURNAUD P., FODOR M., GRENET C. The neurobiology of somatostatin. Crit. Rev. Neurobiol. 1994;8:25–44. [PubMed] [Google Scholar]
- FINK K., SCHULTHEISS R., GÖTHERT M. Stimulation of noradrenaline release in human cerebral cortex mediated by N-methyl-D-aspartate (NMDA) and non-NMDA receptors. Br. J. Pharmacol. 1992;106:67–72. doi: 10.1111/j.1476-5381.1992.tb14294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENIUK W., JARVIE E.M., LUO J., HUMPHREY J.A., HUMPHREY P.P.A. Functional studies with the novel somatostatin (SRIF) sst2 receptor blocking drug AcNH-4-NO2-Phe-c[D-Cys-Tyr-D-Trp-Lys-Thr-Cys]-Tyr-NH2 (CYANAMID 154806) Br. J. Pharmacol. 1998;123:111P. [Google Scholar]
- GORDGE P.C., RYVES W.J. Inhibitor of protein kinase C. Cell Signal. 1994;6:871–882. doi: 10.1016/0898-6568(94)90020-5. [DOI] [PubMed] [Google Scholar]
- HARVEY J., COLLINGRIDGE G.L. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br. J. Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOYER D., LÜBBERT H., BRUNS C. Molecular pharmacology of somatostatin receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1994;350:441–453. doi: 10.1007/BF00173012. [DOI] [PubMed] [Google Scholar]
- HUANG Y.Y., KANDEL E.R. Modulation of both the early and the late phase of mossy fiber LTP by the activation of β-adrenergic receptors. Neuron. 1996;16:611–617. doi: 10.1016/s0896-6273(00)80080-x. [DOI] [PubMed] [Google Scholar]
- JONES S.M., SNELL L.D., JOHNSON K.M. Phencyclidine selectively inhibits N-methyl-D-aspartate-induced hippocampal [3H]norepinephrine release. J. Pharmacol. Exp. Ther. 1987;240:492–497. [PubMed] [Google Scholar]
- KLECKNER N.W., DINGLEDINE R. Requirement for glycine in activation of N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Science. 1987;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- LOGAN S.M., RIVERA F.E., LEONARD J.P. Protein kinase C modulation of recombinant NMDA receptor currents: roles for the C-terminal C1 exon and calcium ions. J. Neurosci. 1999;19:974–986. doi: 10.1523/JNEUROSCI.19-03-00974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUOKA N., YAMAZAKI M., YAMAGUCHI I. Changes in brain somatostatin in memory-deficient rats: comparison with cholinergic markers. Neuroscience. 1995;66:617–626. doi: 10.1016/0306-4522(94)00628-i. [DOI] [PubMed] [Google Scholar]
- MAYER M.L., WESTBROOK G.L. The action of N-methyl-D-aspartic acid on mouse spinal neurones in culture. J. Physiol. (Lond.) 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNOZ-ACEDO G., IZQUIERDO-CLAROS R.M., SANCHEZ-ALONSO J.A., DEL-HOYO N., PEREZ-ALBARSANZ A., ARILLA E. Effect of somatostatin on the mass accumulation of inositol-1,4,5-trisphosphate in rat hypothalamus, striatum, frontoparietal cortex and hippocampus. Neurosci. Lett. 1995;197:41–44. doi: 10.1016/0304-3940(95)11896-5. [DOI] [PubMed] [Google Scholar]
- PATTARINI R., PITTALUGA A., RAITERI M. The human immunodeficiency virus-1 envelope protein gp120 binds through its V3 sequence to the glycine site of N-methyl-D-aspartate receptors mediating noradrenaline release in the hippocampus. Neuroscience. 1998;87:147–157. doi: 10.1016/s0306-4522(98)00125-0. [DOI] [PubMed] [Google Scholar]
- PAUDICE P., GEMIGNANI A., RAITERI M. Evidence for functional native NMDA receptors activated by glycine or D-serine alone in the absence of glutamatergic coagonist. Eur. J. Neurosci. 1998;10:2934–2944. doi: 10.1046/j.1460-9568.1998.00302.x. [DOI] [PubMed] [Google Scholar]
- PERIANIN A., SNYDERMAN R. Mastoparan, a wasp venome peptide, identifies two discrete mechanisms for elevating cytosolic calcium and inositol triphosphates in human polymorphonuclear leukocytes. J. Immunol. 1989;143:1669–1673. [PubMed] [Google Scholar]
- PITTALUGA A., RAITERI M. Release-enhancing glycine-dependent presynaptic NMDA receptors exist on noradrenergic terminals of hippocampus. Eur. J. Pharmacol. 1990;191:231–234. doi: 10.1016/0014-2999(90)94153-o. [DOI] [PubMed] [Google Scholar]
- RAITERI M., ANGELINI F., LEVI G. A simple apparatus for studying the release of neurotransmitters from synaptosomes. Eur. J. Pharmacol. 1974;25:411–414. doi: 10.1016/0014-2999(74)90272-6. [DOI] [PubMed] [Google Scholar]
- RAITER M., BONANNO G., MARCHI M., MAURA G. Is there a functional linkage between neurotransmitter uptake mechanisms and presynaptic receptors. J. Pharmacol. Exp. Ther. 1984;231:671–677. [PubMed] [Google Scholar]
- RAITERI M., GARRONE B., PITTALUGA A. N-Methyl-D-aspartic acid (NMDA) and non-NMDA receptors regulating hippocampal norepinephrine release. II. Evidence for functional cooperation and for coexistence on the same axon terminal. J. Pharmacol. Exp. Ther. 1992;260:238–242. [PubMed] [Google Scholar]
- RAITERI M., SALA R., FASSIO A., ROSSETTO O., BONANNO G. Entrapping of impermeant probes of different size into non-permeabilized synaptosomes as a method to study presynaptic mechanisms. J. Neurochem. 2000;74:423–431. doi: 10.1046/j.1471-4159.2000.0740423.x. [DOI] [PubMed] [Google Scholar]
- ROHRER S.P., BIRZIN E.T., MOSLEY R.T., BERK S.C., HUTCHINS S.M., SHEN D.-M., XIONG Y., HAYES E.C., PARMAR R.M., FOOR F., MITRA S.W., DEGRADO S.J., SHU M., KLOPP J.M., CAI S.-J., BLAKE A., CHAN W.W.S., PASTERNAK A., YANG L., PATCHETT A.A., SMITH R.G., CHAPMAN K.T., SCHAEFFER J.M. Rapid identification of subtype-selective agonists of the somatostatin receptor through combinatorial chemistry. Science. 1998;282:737–740. doi: 10.1126/science.282.5389.737. [DOI] [PubMed] [Google Scholar]
- RUBINOW D.R., DAVIS C.L., POST R.M.Somatostatin in the central nervous system Psychopharmacology: The Fourth Generation of Progress 1995New York: Raven Press; 553–596.ed. Bloom, F.E. & Kupfer, D.J. pp [Google Scholar]
- SIEHLER S., HOYER D. Characterisation of human recombinant somatostatin receptors. 2. Modulation of GTPγS binding. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999a;360:500–509. doi: 10.1007/s002109900142. [DOI] [PubMed] [Google Scholar]
- SIEHLER S., HOYER D. Characterisation of human recombinant somatostatin receptors. 4. Modulation of phospholipase C activity. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999b;360:522–532. doi: 10.1007/s002109900144. [DOI] [PubMed] [Google Scholar]
- SIEHLER S., HOYER D. Characterisation of human recombinant somatostatin receptors. 3. Modulation of adenylate cyclase activity. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999c;360:510–521. doi: 10.1007/s002109900143. [DOI] [PubMed] [Google Scholar]
- TAYLOR C.W., BROAD L.M. Pharmacological analysis of intracellular Ca2+ signalling: problems and pitfalls. Trends Pharmacol. Sci. 1998;19:370–375. doi: 10.1016/s0165-6147(98)01243-7. [DOI] [PubMed] [Google Scholar]
- THOSS V.S., PEREZ J., PROBST A., HOYER D. Expression of five somatostatin receptor mRNAs in the human brain and pituitary. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:411–419. doi: 10.1007/BF00168430. [DOI] [PubMed] [Google Scholar]
- TINGLEY W.G., ROCHE K.W., THOMPSON A.K., HUGANIR R.L. Regulation of NMDA receptor phosphorylation by alternative splicing of the C-terminal domain. Nature. 1993;364:70–73. doi: 10.1038/364070a0. [DOI] [PubMed] [Google Scholar]
- WANG Y., WHITE T.D. Effect of protein kinase C activation on N-methyl-D-aspartate-evoked release of adenosine and [3H]norepinephrine from rat cortical slices. J. Pharmacol. Exp. Ther. 1998;285:105–109. [PubMed] [Google Scholar]
- WILKINSON G.F., FENIUK W., HUMPHREY P.P.A. Characterization of human recombinant somatostatin sst5 receptors mediating activation of phosphoinositide metabolism. Br. J. Pharmacol. 1997;121:91–96. doi: 10.1038/sj.bjp.0701116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZUKIN R.S., BENNETT M.V.L. Alternatively spliced isoforms of the NNDARI receptor subunit. Trends Neurosci. 1995;18:306–313. doi: 10.1016/0166-2236(95)93920-s. [DOI] [PubMed] [Google Scholar]