

Abstract
Among the five human 5-HT4 (h5-HT4) receptor isoforms, the h5-HT4(a) receptor was studied with a particular emphasis on the molecular interactions involved in ligand binding. For this purpose, we used site-directed mutagenesis of the transmembrane domain. Twelve mutants were constructed with a special focus on the residue P4.53 of helix IV which substitutes in h5-HT4 receptors the highly conserved S residue among the rhodopsin family receptors. The mutated receptors were transiently expressed in COS-7 cells.
Ligand binding or competition studies with two h5-HT4 receptor agonists, serotonin and ML10302 and two h5-HT4 receptor antagonists, [3H]-GR113808 and ML10375 were performed on wild type and mutant receptors. Functional activity of the receptors was evaluated by measuring the ability of serotonin to stimulate adenylyl cyclase.
Ligand binding experiments revealed that [3H]-GR113808 did not bind to mutants P4.53A, S5.43A, F6.51A, Y7.43A and to double mutant F6.52V/N6.55L. On the other hand mutations D3.32N, S5.43A and Y7.43A appeared to promote a dramatic decrease of h5-HT4(a) receptor functional activity. From these studies, S5.43 and Y7.43 clearly emerged as common anchoring sites to antagonist [3H]-GR113808 and to serotonin.
According to these results, we propose ligand-receptor complex models with serotonin and [3H]-GR113808. For serotonin, three interaction points were selected including ionic interaction with D3.32, a stabilizing interaction of this ion pair by Y7.43 and a hydrogen bond with S5.43. [3H]-GR113808 was also docked, based on the same type of interactions with S5.43 and D3.32: the proposed model suggested a possible role of P4.53 in helix IV structure allowing the involvement of a close hydrophobic residue, W4.50, in a hydrophobic pocket for hydrophobic interactions with the indole ring of [3H]-GR113808.
Keywords: Serotonin, 5-HT4 receptor, human, binding site, directed mutagenesis, molecular modeling
Introduction
The neurotransmitter serotonin (5-HT) elicits a wide variety of physiological responses mediated by distinct receptor subtypes (Saxena, 1995). To the exception of 5-HT3, all other 5-HT receptors are members of the seven transmembrane spanning G protein-coupled family of receptors. Like 5-HT1, 5-HT6 and 5-HT7 receptors, h5-HT4 receptors are associated to an adenylyl cyclase second messenger system (Hedge & Eglen, 1996). They are expressed in a wide variety of mammalian tissues including brain (Eglen et al., 1995), gastrointestinal tract, heart (Hedge & Eglen, 1996) and are thought to be involved in various central and peripheral disorders including cardiac arrhythmias (Kaufmann, 1994) and neurodegenerative diseases (Reynolds et al., 1995; Wong et al., 1996). Several human h5-HT4 receptor isoforms with alternative splicing in the C-terminal tail were recently identified (Blondel et al., 1997; 1998; Claeysen et al., 1999) (Figure 1) and expressed transiently in COS-7 cells in order to analyse their pharmacological properties (Blondel et al., 1998; Claeysen et al., 1999). The different splice variants were shown to display an identical 5-HT4 pharmacological profile and a similar ability to stimulate adenylyl cyclase activity in the presence of 5-HT. In addition, tissue distribution studies revealed some degree of specificity in the pattern of expression of the different isoforms within the human body (Blondel et al., 1998).
Figure 1.
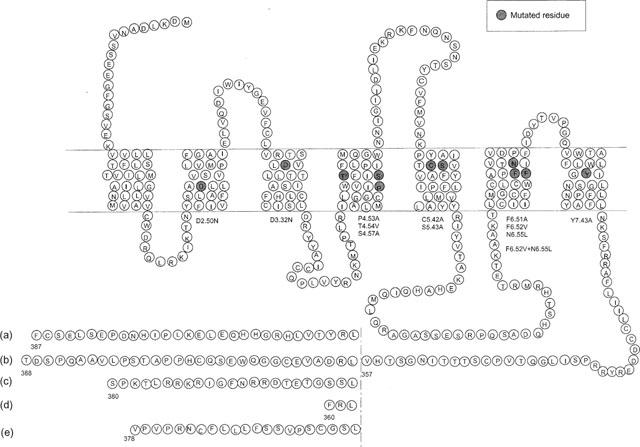
Snake representation of the five isoforms of h5-HT4 receptor. The amino acid sequences diverge after Leu 358. Site-directed mutagenesis studies were performed on the h5-HT4(a) receptor isoform.
To allow the development of selective drugs for the treatment of the various disorders in which h5-HT4 receptors might be involved, it is necessary to understand the molecular interactions involved in ligand binding to the receptor. Several studies have focused on the molecular interaction of endogenous serotonin with different 5-HT receptor subtypes (Hibert et al., 1991; Trumpp-Kallmeyer et al., 1992; Savarese & Fraser, 1992; Baldwin 1994). The molecular requirement for serotonin binding to its receptor should include the electrostatic interaction between the receptor and the amino group of the ligand, one hydrogen bond between donor-acceptor site of the receptor and the hydroxyl group of serotonin and finally van der Walls attractive interactions.
Mutagenesis studies of serotonin receptors (Ho et al., 1992; Wang et al., 1993; Boess et al., 1998) suggested that the serotonin amino group makes an electrostatic interaction with the carboxylate of the highly conserved D3.32 (for numbering of residues we have followed either the convenient method proposed by Van Rhee & Jacobson (1996) which includes both numbering in the sequence and position in a particular helix referred to its most conserved residue, or only the position in this helix). This interaction is usually found in biogenic amine receptors, generally involved in binding of agonists and in receptor activation. On the other hand, there is evidence that catecholamines, dopamine (Cavalli et al., 1996) and also serotonin (Ho et al., 1992; Almaula et al., 1996) hydroxyl groups interact with a donor-acceptor hydrogen bond residue present in transmembrane domain (TMD) V. However these results concerned only few serotonin receptor subtypes including 5-HT1A, 5-HT2 and 5-HT6 receptors. Furthermore, the role of aspartate D3.32 was not evidenced for antagonists in 5-HT1A receptor (Ho et al., 1992) and the role of the hydrogen bond donor residue of the TMDV was not unambiguously identified both in 5-HT1A and 5-HT2 receptors (Ho et al., 1992; Almaula et al., 1996). All these results have clearly shown that the role of individual residues in receptor-ligand molecular interactions may vary among different serotonin receptor subtypes. Consequently, the results obtained on serotonin interactions with one receptor cannot be directly extrapolated to another serotonin receptor subtype. In addition, despite the fact that in aminergic receptors, helix IV has been shown to be weakly or not involved in the receptor-ligand interaction, the exclusive presence in h5-HT4 receptor of a proline residue located at position 4.53, occupied by the highly conserved serine in rhodopsin family, could undergo conformational changes of the helical bundle, which might be important for the receptor-ligand interaction selectivity of this receptor (Figure 2). This type of structural modification has been shown to play an important role in the activity of several receptors (Ballesteros & Weinstein, 1995) including ion channel receptors (Ri et al., 1999).
Figure 2.
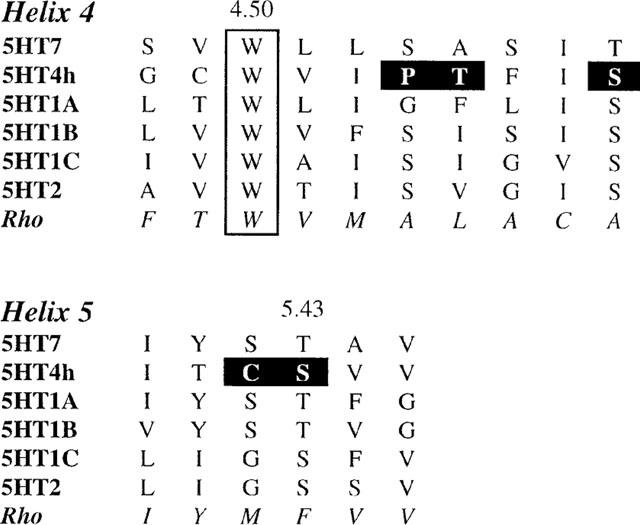
Sequence alignment of helices IV and V of 5-HT receptors. In bold are indicated the mutated residues of h5-HT4 receptor.
To determine the interactions which contribute to ligand binding in h5-HT4 receptor subtype we used site-directed mutagenesis. For a best exploration of the ligand pocket, we focused both on several highly conserved amino acids which are known to be involved in the ligand binding site of other 5-HT receptors, and on amino acids which are exclusively expressed in h5-HT4 receptor subtypes. For this purpose, we have successively examined 11 single point mutations (D2.50, D3.32, P4.53, T4.54, S4.57, C5.42, S5.43, F6.51, F6.52, N6.55 and Y7.43), and one double point mutation (F6.52, N6.55) of the h5-HT4(a) wild type receptor (Figure 1). These mutants were transiently expressed in COS-7 cells. The effects of these mutations were assessed by radioligand binding analysis using the 5-HT4 specific antagonist [3H]-GR113808 and by competition analysis with (i) the natural h5-HT4 receptor agonist 5-HT, (ii) a specific potent agonist, ML10302, and (iii) a potent h5-HT4 receptor antagonist, ML10375 (Figure 3) (Yang et al., 1997). The mutants were also tested for their ability to induce cyclic AMP production in response to 5-HT.
Figure 3.
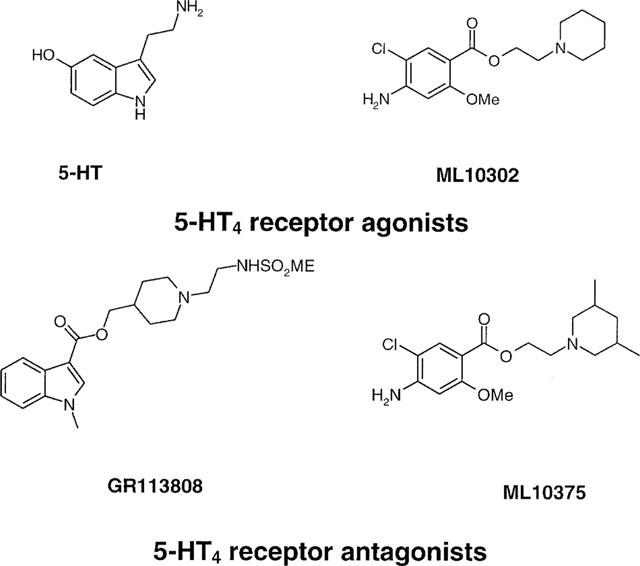
Chemical structures of h5-HT4 receptor agonists and antagonists selected for these studies.
Our results indicate a crucial role for residues D3.32, S5.43 and Y7.43 in h5-HT4 receptor functional activity. Furthermore, we show that residues S5.43, P4.53 and F6.51 are critical for the binding of the highly specific receptor antagonist GR113808. A model for h5-HT4 receptor was built, which is a combination of the α-carbon template for the transmembrane helices in the rhodopsin family of G-protein coupled receptors established by Baldwin et al. (1997) and of docking experiments of 5-HT and GR113808 taking into account our site-directed mutagenesis data.
Methods
Materials
Complementary mutagenesis oligonucleotides were purchased from Gibco Life Technologies. Polyethylenimine (PEI, MW 800,000) was from Fluka (L'Isle d'Abeau Chesnes, France). Dulbecco's modified Eagle's medium (DMEM) was obtained from Gibco Life Technologies. (1-(2-((methylsulphonyl)amino)ethyl)-4-piperidinyl)methyl-1-methyl-1H-indole-3-carboxylate (GR113808) was a gift from Glaxo Research Group (Ware, Hertfordshire, U.K.), and [3H]-GR113808 was purchased from Amersham (Arlington Heights, IL, U.S.A.). 2-piperidinoethyl 4-amino-5-chloro-2-methoxybenzoate (ML10302) and 2-(cis-3,5-dimethylpiperidino)ethyl 4-amino-5-chloro-2-methoxybenzoate (ML10375) were synthesized as previously described (Yang et al., 1997). Serotonin (5-HT) was from Aldrich (L'Isle d'Abeau Chesnes, France).
Site-directed mutagenesis
The full coding region of the h5-HT4(a) cDNA was subcloned in the mammalian expression vector pRC/CMV (Invitrogen, Carlsbad, CA, U.S.A.) as previously described (Blondel et al., 1998). Mutations in the human 5-HT4(a) receptor were introduced using QuickChange Site-directed Mutagenesis kit (Stratagene, Montigny-le-Bretonneux, France). Mutants were analysed by restriction enzymes and the authenticity of each mutation was confirmed by dideoxy chain termination DNA sequencing using T7 DNA polymerase kit (Pharmacia, Saclay, France).
Transient transfection of COS-7 cells
One day before transfection, COS-7 cells were seeded either into cell culture flasks at a density of 107 cells per flask for radioligand binding assays or into 12-well plates at a density of 4.5×105 cells per well for measurement of cyclic AMP formation. Transfections were performed using the cationic vector PEI as previously described (Boussif et al., 1995), with a ratio of 22 nmoles PEI per μg of DNA in 150 mM NaCl. Cells were incubated for 6 h with plasmid DNA (150 μg per flask or 4 μg per well in 12-well plates) and analysed 24 h after transfection for cyclic AMP measurement or 48 h after transfection for radioligand binding assays.
Binding assays
Transfected COS-7 cells were washed twice with phosphate-buffered saline, scraped, collected and centrifuged at 3000 × g for 5 min. The pellet was resuspended in 10 volumes of ice-cold HEPES buffer (50 mM, pH 7.4) and homogenized by an Ultraturax tissue grinder. The lysate was subsequently centrifuged at 40,000 × g for 15 min at 4°C. The resulting pellet was resuspended in 10 volumes of HEPES buffer (50 mM, pH 7.4). Protein concentrations were determined by the method of Bradford et al. (1976) using bovine serum albumin as a standard.
Radioligand binding assays were performed in 500 μl buffer (50 mM HEPES, pH 7.4) containing 20 μl of [3H]-GR113808, 50 μl (∼50 μg) of membrane preparation and 20 μl of displacing drug or assay buffer. Non-specific binding was measured in the presence of 20 μM GR113808. Saturation experiments were performed using [3H]-GR113808 at nine concentrations ranging from 0.01 to 4 nM. Competition assays were performed in the presence of nine concentrations of the displacing ligands (10−12–10−4 M) and a concentration of [3H]-GR113808 corresponding to the KD value of each mutant receptor. Incubations were performed at 25°C for 30 min and terminated by rapid filtration through Whatman GF/B filter paper using the Brandel model 48R cell harvester. Filters were pre-soaked in a solution of PEI (0.1%) to reduce non-specific binding. After filtration, they were subsequently washed with ice-cold buffer (50 mM Tris-HCl, pH 7.4) and placed overnight in 4 ml of Ready-Safe scintillation cocktail (Beckman, Fullerton, CA, U.S.A). Radioactivity was measured using a Beckman model LS 6500C liquid scintillation counter. Binding data were analysed by computer-assisted non-linear regression analysis (Prism, GraphPad Software, San Diego, CA, U.S.A.).
Antibody production
A peptide (sequence: G I I D L I E K R K F N Q N S N S T Y C V) corresponding to the second extracellular loop of the 5HT4 receptor (G21V) was synthesized in an automated synthesizer using the Fmoc technique (Neimark & Briand, 1993). The peptide was then purified by HPLC and checked by mass spectrophotometry.
The first injections were performed in Complete Freund's adjuvant and three booster injections were given in Incomplete Freund's adjuvant. Serum was collected 1 week after the last booster injection. Rabbit sera were precipitated at 40% (NH4)2SO4 saturation and dissolved in PBS (10 mM phosphate, 140 mM NaCl, pH 7.4) at 1 : 1 dilution.
The G21V peptide was coupled to activated EAH-Sepharose (Pharmacia diagnostics AB, Uppsala, Sweden) according to the standard procedure. The rabbit immunoglubin fractions were diluted 10 times with PBS and centrifuged at 200 × g for 5 min. They were then passed through the column with a flow of 6 ml h−1 for 3 h at 4°C. Finally the adsorbed antibodies were eluted with 3 ml 3 M KSCN, dialysed directly against 6 1 PBS overnight at 4°C. Specificity of antibodies were checked by enzyme immunoassay as described before (Lebesgue et al., 1998).
Plasma membrane preparation and immunoblotting
COS-7 cells transfected with wild-type or mutant h5-HT4 receptors were washed twice with phosphate-buffered saline, scraped, collected and centrifuged at 300 × g for 5 min. Pure plasma membranes were isolated according to the protocol of Persson & Jergil (1992). Briefly, total cellular membrane was first separated from other cellular compounds in an aqueous polyethyleneglycol-dextran two-phase system. Then, an affinity ligand (Wheat Germ Agglutinin, WGA) conjugated with dextran was introduced to bind specifically extracellular residues of the plasma membrane. Under these conditions, WGA selectively pulled plasma membranes into the dextran-rich phase. In the final step, WGA-dextran bottom phase was diluted 10 fold in N-acetyl-glucosamine 0.1 M and centrifuged at 45,000 r.p.m. for 1 h at 4°C. Pellet was resuspended in a buffer containing 50 mM Tris, 0.5 mM EDTA and 1 mM PMSF. The protein content of each sample was determined with the Bio-rad protein reagent (Bio-Rad; Ivry sur Seine; France) with Bovine Serum Albumin as a standard.
Thirty μg of membrane protein expressing wild-type or mutant receptors were diluted in a 2× loading buffer, boiled 5 min and subjected to 10% SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a nitrocellulose membrane (Hybond-P, Amersham Pharmacia Biotech, Orsay, France) for 1.5 h at 150 V. Nitrocellulose membrane was then blocked in a buffer containing 3% powdered milk and incubated overnight at 4°C with a 1 : 1000 dilution of rabbit anti-h5-HT4 antiserum raised against the second extracellular loop. The membranes were then incubated 1.5 h with a rabbit anti-horseradish peroxidase antibody (Amersham Pharmacia Biotech, Orsay, France) at room temperature. Immunoreactive bands were visualized by light emitting non-radioactive method (ECL, Amersham Pharmacia Biotech, Orsay, France).
Measurement of cyclic AMP formation
Transiently transfected COS-7 cells were incubated 24 h after transfection in DMEM containing 5 mM theophylline, and 10 μM pargyline for 15 min at 37°C in 5% CO2. 5-HT was then added and incubated for an additional 15 min at 37°C in 5% CO2. The reaction was stopped by aspiration of the medium and addition of 500 μl of ice-cold ethanol. After 30 min at room temperature, the ethanol fraction was collected and lyophilized. The pellet was reconstituted, and cyclic AMP was quantified using a radioimmunoassay kit (cyclic AMP competitive radioimmunoassay, Immunotech, Marseille, France).
Molecular modeling
Molecular modeling experiment were performed using a Silicon Graphics O2 (R10 000), and the Sybyl 6.5 program from Tripos (St-Louis, Missouri, U.S.A.). Energy calculations were done using Amber for protein, Tripos force fields and Gasteiger-Marsilii charges for complexes with a dielectric constant of 1, and distance as dielectric function. Minimizations were terminated with a gradient convergence of 0.1 Kcal mol−1 Å−1. Molecular dynamics were performed for 50 ps at 300°K with steps of 1 fs and non-bonded update every 25 fs using backbone as aggregate. Average structures were taken for the last 25 ps trajectory. For the ligand-receptor complexes resulting from docking experiments, molecular dynamics were performed with two supplementary distance constraints (i) from the carboxy carbon of D3.32 to the amino nitrogen of ligand (10% range) and (ii) from the hydroxyl oxygen of S5.43 to the oxygen atom of serotonin or to the carbonyl oxygen of GR113808 (20% range).
Results
To determine the role of several transmembrane residues of the h5-HT4(a) receptor, a series of mutated h5-HT4(a) receptors were generated (Figure 1). These mutant receptors were examined by comparing the radioligand binding properties and agonist-induced cyclic AMP production, using transient transfected COS-7 cells.
[3H]-GR113808 binding
Saturation analysis
[3H]-GR113808 was used to determine the dissociation constant (KD) and maximum binding capacity (Bmax) for the wild-type and mutant receptors (Figure 4). Affinity of [3H]-GR113808 for the D66(2.50)N (KD=0.42 nM), D100(3.32)N (KD=0.32 nM), T150(4.54)V (KD=0.50 nM), S153(4.57)A (KD=0.34 nM) and F276(6.52)V (KD=0.45 nM) mutant h5-HT4(a) receptors was similar to that observed for the wild-type receptor (KD=0.23 nM) (Table 1). However, the density of mutant receptors transiently expressed in COS-7 cells varied greatly, from 345 fmol mg−1 protein for the D3.32N mutant to 3453 fmol mg−1 protein for the D2.50N mutant receptor (Table 1). These variations were probably due to different efficiency of the transfection method. As shown in Table 1, [3H]-GR113808 had a 3 fold higher affinity for the C196(5.42)A mutant receptor (KD=0.07 nM) compared with the wild-type receptor. On the contrary, mutation N279(6.55)L reduced 5 fold the affinity for [3H]-GR113808 (KD=1.2 nM). When this mutation was combined with mutation F276(6.52)V, no specific binding was detected. Likewise no specific binding of [3H]-GR113808 was detected for the P149(4.53)A, S197(5.43)A, F275(6.51)A and Y302(7.43)A mutations.
Figure 4.
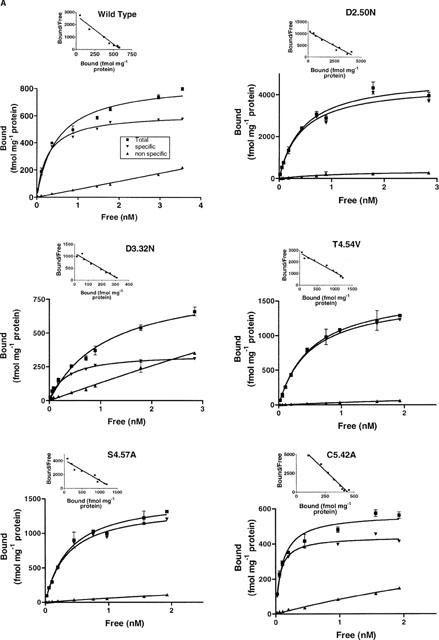
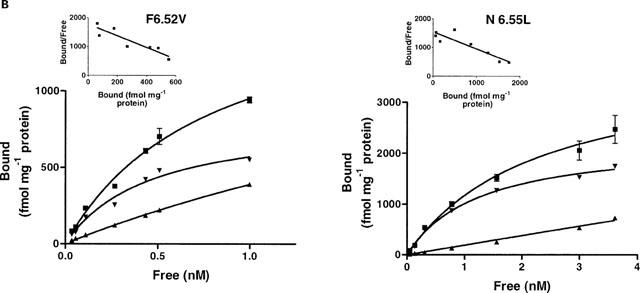
Saturation analysis of [3H]-GR113808 binding to COS-7 cells transfected with wild-type or mutant human h5-HT4(a) receptors. Non-specific binding was determined with 10 μM ML10375. Experiments were performed in triplicate using a range of nine concentrations of radioligand.
Table 1.
Results of binding experiments on wild-type and mutant receptors

Competition analysis
The affinities of 5-HT, ML10302, and ML10375 for the mutant receptors were compared with the wild-type h5-HT4(a) receptor. The results are presented in Table 1. As a consequence of the absence of any radioligand binding for the mutations P149(4.53)A, S197(5.43)A, F275(6.51)A, Y302(7.43)A and F276(6.52)V-N279(6.55)L, the corresponding mutants were not submitted to displacement experiments. We observed that mutations T150(4.54)V, S153(4.57)A and F276(6.52)V did not affect the binding affinities for 5-HT, ML10302 and ML10375 (Table 1). On the other hand, competition curve analysis with the endogenous agonist 5-HT showed a remarkable reduced affinity for the D100(3.32)N and the N279(6.55)L mutant receptors (Ki>10000 nM) compared with the wild-type receptor (Ki=772 nM) (Figure 5). In contrast, residues D66(2.50) and C196(5.42) did not affect 5-HT binding when mutated to asparagine (Ki=6.72 nM) and alanine (Ki=378 nM) respectively. In addition, the selective partial agonist ML10302 had a 6 fold and 3 fold reduced affinity respectively for the D66(2.50)N (Ki=57.5 nM) and D100(3.32)N (Ki=29.6 nM) mutant receptors and a 5 fold higher affinity for the C196(5.42)A (Ki=1.8 nM) mutant receptor, as compared to the wild-type form (Ki=8.5 nM). In contrast, no change in the ML10302 affinity was observed when residue N279(6.55) was mutated to leucine. D66(2.50)N and D100(3.32)N mutant receptors also displayed a reduction of the affinity for the antagonist ML10375, which was more pronounced than that observed for ML10302 (26 fold and 63 fold respectively) (Table 1).
Figure 5.
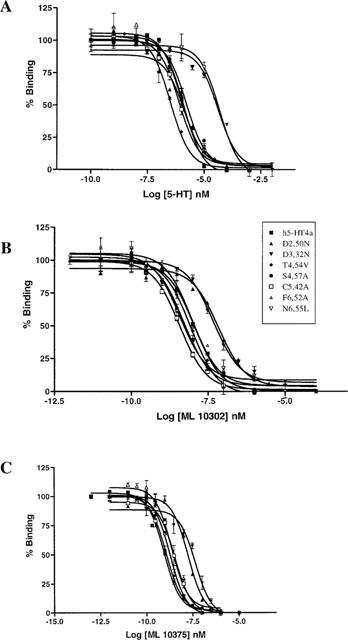
Competition binding of agonists (5-HT (A), ML10302 (B)) and antagonist (ML10375 (C)) in COS-7 cells transfected with wild-type and mutant h5-HT4(a) receptors. Concentration of [3H]-GR113808 was 0.3 nM. Experiments were performed in triplicate using a range of nine concentrations of displacing ligand.
Western blotting
Plasma membrane expression of mutant receptors deficient in specific [3H]-GR113808 binding was monitored by Western blotting using a polyclonal antibody directed against the second extracellular loop of the h5-HT4(a) receptor (Figure 6). Purification of plasma membrane from COS-7 cells was achieved by affinity partitioning in a polyethyleneglycol-dextran two-phase system (see methods). Plasma membrane from COS-7 cells expressing wild-type receptor displayed a specific band around 60 kDa which was displaced by incubation with a peptide (G21V) corresponding to the extracellular loop of the h5-HT4(a) receptor (Figure 6a).
Figure 6.
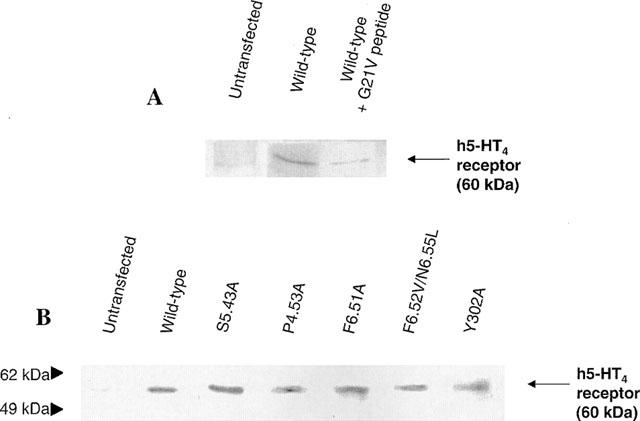
Protein immunoblotting of the mutant and wild type h5-HT4(a) receptors expressed in COS-7 cells. Plasma membrane protein extracts were separated on a polyacrylamide gel and analysed by immunoblotting with antiserum against the second extracellular loop of h5-HT4(a) receptor (see methods). (A) COS-7 cells were transfected with the wild-type h5-HT4(a) receptor (lane 2 and lane 3) or the corresponding control (lane 1). The immunoreactivity of wild-type receptor was blocked by incubation with a peptide (G21V) corresponding to the second extracellular loop of the receptor (lane 3). (B) In COS-7 cells transfected with the wild-type h5-HT4 receptor (lane 2) and different mutants (lane 3–7), a specific band migrating to the level of 60 kDa was detected whereas in non transfected control cells (lane 1) no labelling was detected at this position.
The same immunoreactive band of 60 kDa was detected in plasma membranes of COS-7 cells transfected with mutant receptors S197(5.43)A, P149(4.53)A, F275(6.51)A, F276(6.52)V/N279(6.55)L and Y302(7.43)A. Therefore, we can conclude that mutant receptors for which no [3H]-GR113808 binding was observed, are well expressed at the plasma membrane of COS-7 cells.
Functional properties
We next examined the effect of different mutations on the transductional response by measuring cyclic AMP production after 15 min serotonin stimulation (1 μM). Basal cyclic AMP levels varied between 6 and 31 pmoles per well. Since these values were not correlated to a particular mutation, the variation in basal cyclic AMP concentration was rather due to variable efficiency of the transfection method. As a control, 5-HT (1 μM) did not induce cyclic AMP formation in COS-7 cells transfected with a crude pRC/CMV vector (Figure 7).
Figure 7.
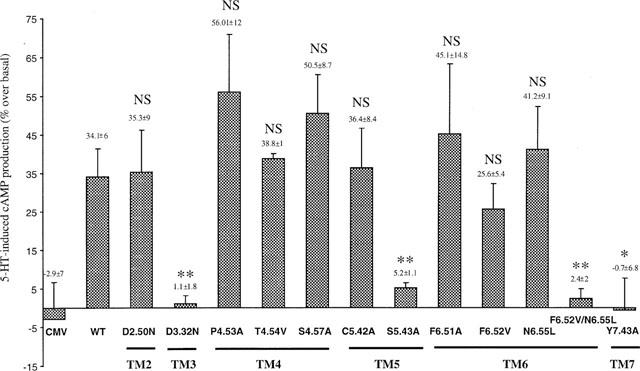
Functional response of wild-type and mutant human 5-HT4(a) receptors. Cyclic AMP accumulation in COS-7 cells expressing wild-type and mutant 5-HT4(a) receptors after stimulation with 1 μM of 5-HT. Values are expressed as percentage of basal cyclic AMP. Data are the mean±s.e.mean of 3–4 experiments performed in triplicate. CMV represents COS-7 cells transfected with crude pRC/CMV vector. WT, Wild Type; NS, Not Significant; *P<0.05, **P<0.01 with t-test between wild-type and mutant receptors.
Mutant receptors D66(2.50)N, T149(4.54)V, S153(4.57)A, C196(5.42)A and F276(6.52)V stimulated cyclic AMP production with the same efficiency as the wild-type receptor (34.1±6.0%) (Figure 7). Moreover, mutant receptors P149(4.53)A (56.0±12.0%) and F275(6.51)A (45.1±14.8%) displayed a significant ability to stimulate cyclic AMP formation like the wild-type receptor despite the absence of any radioligand [3H]-GR113808 binding (Table 1), indicating that the mutated receptors were functional and properly folded into the plasmic membrane. COS-7 cells transfected with D100(3.32)N mutant 5-HT4(a) receptor showed no response to 5-HT (Figure 7) and this was correlated with a drastic decrease in the affinity of 5-HT (Ki>10000, Table 1). On the contrary, COS-7 cells transfected with N279(6.55)L mutant showed appreciable response to 5-HT despite the decrease in the affinity of 5-HT (Ki>10000 nM, Table 1) and of [3H]-GR113808 (KD=1.2 nM, Table 1). On the other hand, COS-7 cells expressing S197(5.43)A, Y302(7.43)A and F276(6.52)V-N279(6.55)L mutant receptors were unable to induce cyclic AMP production in response to 5-HT (1 μM) (Figure 7). Since no binding with [3H]-GR113808 was observed in COS-7 cells expressing these three mutant receptors, we did not proceed to any further competition studies with 5-HT.
Discussion
In the present study 11 single point mutations and one double point mutation of the human 5-HT4(a) receptor were constructed, and the twelve mutant receptors were examined for their ability to bind h5-HT4 ligands and to induce cyclic AMP production.
The two negatively charged amino acids D66(2.50) and D100(3.32) were both replaced with the homologous neutral N residue, which cannot establish any ionic interaction but is a hydrogen bond donor-acceptor residue. The hydrogen bond donor-acceptor residues S153(4.57), C196(5.42) and S197(5.43) were replaced with homologous neutral A, T150(4.54) with homologous neutral V, and N279(6.55) with homologous neutral L. The aromatic residues F275(6.51) and Y302(7.43) were substituted with A and F276(6.52) with hydrophobic V. Finally P149(4.53) was replaced with the helix structure favorable residue A.
Mutation of the aspartate D100 in TMDIII : D(3.32)N
From the literature, there is strong evidence for the direct involvement of D100(3.32) residue in ligand-aminergic receptor interaction through an ionic interaction between the negatively charged carboxylate of aspartate and the positively charged nitrogen of the ligand (Van Rhee & Jacobson, 1996). The mutation D100(3.32)N totally impaired the ability of the h5-HT4(a) receptor to bind 5-HT and to stimulate cyclic AMP production. However, only a weak decrease in affinity of the agonist ML10302 was observed. Likely, antagonist binding varied according to the ligands with a clear decrease in affinity of the antagonist ML10375 and no effect on the binding of the antagonist [3H]-GR113808. If we consider that N mutation of residue D100(3.32) abolishes the possibility of a strong ionic interaction with the ligand, but maintains the occurrence of a weaker hydrogen bond interaction, we could make the assumption that the effect of the replacement of an ionic interaction by a hydrogen bond is sufficient to modify the affinity of ligands, depending on the strength of the other receptor-ligand interactions.
Mutation of donor-acceptor residues C196 and S197 in TMDV: C196(5.42)A and S197(5.43)A
In serotonin receptors, these positions are generally occupied by respectives hydrogen bond donor-acceptor residues S and T. It was shown that S5.42 and T5.43 of other serotonin receptors play an important role in the binding of serotonin and in activity (Ho et al., 1992). Apparently the 5.42 residue is not involved in binding pindolol-like 5-HT1A antagonist (Ho et al., 1992). In h5-HT4 receptor, these two positions are occupied by hydrogen bond donor-acceptor residues C196(5.42) and S197(5.43). Mutation of these residues by A clearly shows that only S197(5.43) is involved in antagonist [3H]-GR113808 binding and serotonin-induced activity. Furthermore, the mutation C196(5.42)A shows a higher affinity for the binding of [3H]-GR113808 and ML10302, clearly suggesting that this residue is not involved in the ligand binding site. These results strongly suggest that, similarly to other serotonin receptors, a hydrogen bond interaction is displayed between the hydroxyl group of serotonin and that of a residue present in TMDV and this residue is clearly identified as the S197(5.43) aminoacid. If we assume that S197(5.43) interacts with the antagonist [3H]-GR113808 also through a hydrogen bond, it is reasonable to propose the carbonyl oxygen of the ester group as the hydrogen bond acceptor.
Mutation of the proline P149 and of the two other residues T150 and S153 in TMDIV : P149(4.53)A, T150(4.54)V and S153(4.57)A
Among G-protein-coupled receptors of rhodopsin family, residue S4.53 is highly conserved, and with the most conserved residue W4.50 is part of a surface probably in contact with other helices or with the ligand binding pocket between the helices (Baldwin et al., 1997). This position is occupied by P only in the h5-HT4 receptor. As P is known to induce a kink of the alpha-helical structure, it may induce a particular modification of the helical bundle necessary for ligand binding and activity of this receptor. For this reason, this residue was mutated to A. Furthermore, in order to explore a possible structural involvement of the environment of this residue, vicinal residue T150(4.54) and one helical turn separated S153(4.57) were also mutated to their homologous neutral residues, respectively V and A. The loss of [3H]-GR113808 binding induced by P149(4.53)A mutation clearly indicates that the structural particularity of h5-HT4 receptor helix IV is necessary for fixation of this molecule. Nevertheless, this mutation appears to have no effect on serotonin-induced cyclic AMP production. These results could indicate that inter-helical contacts due to P of helix IV are weakly or not involved in serotonin binding and signal transduction but are necessary for the ligand selectivity of h5-HT4 receptor. Mutations T150(4.54)V and S153(4.57)A have no remarkable effect on the affinity of the studied ligands and on the coupling of the receptor to adenylyl cyclase indicating that these residues are not involved in the binding site.
Mutation of other aminoacids in TMD
To investigate the role of other aminoacids in agonist and antagonist binding sites, we mutated several highly conserved residues and others exclusively expressed in h5-HT4 receptor aminoacids which are proposed to reside near the binding site.
First, in TMDII, mutation D66(2.50)N does not affect the affinity of the antagonist [3H]-GR113808 and of 5-HT, and retains the same level of serotonin stimulated production of cyclic AMP as compared with the wild type receptor. However we observed a 6 fold decrease binding of the partial agonist ML10302 and a 26 fold decrease binding of the antagonist ML10375. These results indicate that this residue is weakly involved in 5-HT binding and in receptor activity in contrast to 5-HT1 and 5-HT2 receptors (Ho et al., 1992; Wang et al., 1993; Sealfon et al., 1995). They also suggest that D66(2.50)N mutation contributes to some changes in the three-dimensional structure of the receptor, affecting only the binding of some synthetic ligands.
Second, mutations in TMDVI of the highly conserved aromatic residues F275(6.51)A, F276(6.52)V and the largely present N279(6.55)L have no effect on the ability of serotonin to activate adenylyl cyclase. However, mutation F275(6.51)A abolishes the specific binding of the antagonist [3H]-GR113808, and mutation N279(6.55)L induces a clear decrease of serotonin affinity. Furthermore the double mutation F276(6.52)V/N279(6.55)L decreases both specific binding of [3H]-GR113808 and serotonin-induced adenylyl cyclase activity. Taken together these results are consistent with the fact that like in 5-HT2 receptors (Roth et al., 1997), these highly conserved aromatic and hydrophobic residues of helix VI form a hydrophobic pocket necessary for ligand binding, with some specificity for [3H]-GR113808 induced by residue F6.51.
Finally, mutation Y302(7.43)A in TMDVII confirms the finding that residue Y302 is a prominent anchoring site (Roth et al., 1997). Indeed, both specific binding of [3H]-GR113808 and serotonin-induced adenylyl cyclase activity are lost upon mutation Y302(7.43)A. The membrane expression of the mutated receptors (P149(4.53)A, S197(5.43)A, F275(6.51)A, Y302(7.43)A and F276(6.52)A/N279(6.55)L) where no specific binding of [3H]-GR113808 was observed, was similar to that of the wild-type h5-HT4 receptor as shown by Western blotting of transiently transfected COS-7 cells membranes, using a polyclonal antibody directed against the second extracellular loop of the h5-HT4 receptor (Figure 6). These results demonstrate that the observed loss of binding of [3H]-GR113808 is mainly due to a lack of affinity of this molecule for the mutated h5-HT4 receptors.
Model of ligand-receptor complex
The mutagenesis data of this study suggest the presence of some ligand-receptor interaction sites which are common to the antagonist [3H]-GR113808 and to serotonin, allowing us to propose both receptor and ligand-binding complex models. First, residues D66(2.50), T150(4.54), S153(4.57) and C196(5.42) are involved neither in binding sites nor in h5-HT4 receptor activation process. Second, [3H]-GR113808 and serotonin have in common two anchoring sites: S197(5.43) and Y302(7.43). Residue D100(3.32) is more concerned in binding serotonin than [3H]-GR113808, contrary to residue P149(4.54) which probably plays a structural role at the helical bundle level favouring the binding of the antagonist [3H]-GR113808. Residue F275(6.51) of helix VI appears also to be involved only in the binding site of [3H]-GR113808. The other residues of helix VI participate in the antagonist and agonist site but not predominantly.
The starting point of our model receptor structure was based on the α-carbon template of the transmembrane helices in the rhodopsin family of GPCRs, proposed by Baldwin et al. (1997). The seven helices were constructed de novo as α-helices, and kinks typical of proline containing α-helix were introduced for helices II, IV to VII (Sankararamakrishnan et al., 1990; Nordvall & Hacksell 1993). Then, these helices were fitted on α-carbon template. Slight translations and rotations of the helices III, V, VI and VII were then performed to refine the topography of binding pocket defined by residues D100(3.32), S197(5.43), F275(6.51), F276(6.52) and Y302(7.43), following biological results. In this construct, helix IV appeared too deeply embedded in the intracellular side and as a consequence the highly conserved W146(4.50) was situated far from other helices and largely outside the binding pocket. For this reason, a translation of the helix IV of about 8 Å from the intracellular side was necessary to improve helix–helix packing in the W146(4.50) region. This movement is still compatible with the 16.5 Å resolution in the perpendicular to the membrane plane of the frog rhodopsin map (Baldwin et al., 1997). Molecular dynamics (MD) was performed using Amber force fields and average structure was used for further docking experiments.
Docking experiments were performed manually. For serotonin docking, three interaction points were selected following mutagenesis results: (i) ionic interaction between the ammonium group of serotonin and carboxylate group of residue D100(3.32) and (ii) stabilizing interaction of this ion pair by the residue Y302(7,43); the third interaction was reasonably assumed to be a hydrogen bond between the OH of serotonin and the OH of S197(5.43). After MD simulation, average structure of serotonin-h5-HT4 receptor complex (Figure 8) was characterized by distances d=2.11 Å (between the hydroxylic oxygen of S197(5.43) and the serotonin hydroxylic hydrogen) consistent with a hydrogen bond and d=3.4 Å (between the carboxylic carbon of D100(3.32) and the serotonin basic nitrogen) consistent with an ionic interaction. Furthermore, a short distance (1.7 Å) between one carboxylate oxygen of D100(3.32) and the phenolic hydrogen of Y302(7.43) is in agreement with our experimental results, showing that this residue appears to be essential in stabilizing the ionic interaction between the serotonin basic ammonium and the D100(3.32) carboxylate. This h5-HT4 receptor model was further used for GR113808 docking. Following mutagenesis results, P149(4,53), S197(5.43), F275(6.51) and Y302(7.43) residues are the major ones involved in the binding site of this antagonist. F276(6.52) and N279(6.55) play also a role in the binding site but with a reduced effect. Indeed, only the double mutation affects the affinity of the antagonist GR113808. Assuming that S197(5.43) is concerned through a hydrogen bond, the possible ligand counterpart of this interaction could be the oxygen of GR113808 carbonyl group. For that reason a distance between the OH hydrogen of S197(5.43) and the carbonyl oxygen of GR113808 of about 2 Å was set. Furthermore, according to biogenic amine-receptor interaction studies, an ionic interaction between the basic nitrogen of antagonist and the carboxylate function of D100(3.32) was taken into account, although binding of GR113808 was not modified upon mutation of this residue. Indeed we made the assumption that the contribution of the ionic interaction to the total binding interactions of GR113808 is less important than to those of serotonin, since the affinity of antagonist was three orders of magnitude higher than that of serotonin. Consequently, the replacement of this ionic interaction by a hydrogen bond should not significantly modify the total binding interactions of GR113808. The average MD simulation structure gave distances d=2.58 Å (between the hydroxylic hydrogen of S197(5.43) and the GR 113808 carbonyl oxygen) and d=4.26 Å (between the carboxylic carbon of D100(3.32) and the GR113808 basic nitrogen) consistent with respective hydrogen bond and ionic interaction (Figure 9). This model indicates a possible interaction through a hydrogen bond between the sulfamido group of GR113808 and Y302(7.43). Furthermore, this model suggests that the indole ring of GR113808 is embedded in a hydrophobic pocket defined by residues W146(4.50), F275(6.51), F276(6.52) and N279(6.55). This last interaction could explain the loss of binding promoted by mutation P149(4.53)A which could modify the helix IV structure preventing the formation of this hydrophobic pocket (Figure 10).
Figure 8.
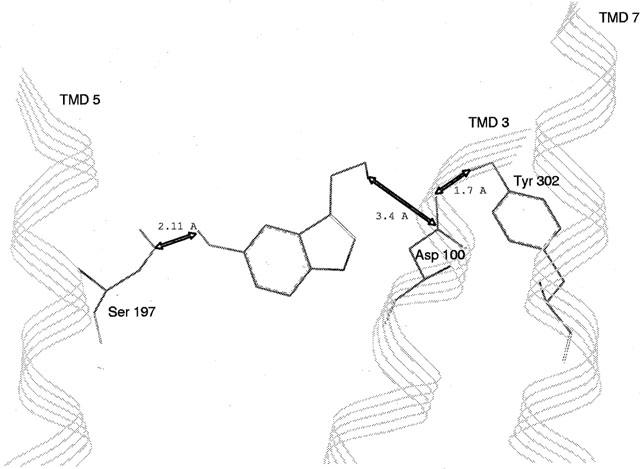
Serotonin-h5-HT4 receptor complex model. Obtained by docking experiment, this model is characterized by distances: (i) d=2.11 Å between the hydroxylic oxygen of S197(5.43) and the serotonin hydroxylic hydrogen, (ii) d=3.4 Å between the carboxylic carbon of D100(3.32) and the serotonin basic nitrogen, and (iii) d=1.7 Å between the carboxylate oxygen in D100(3.32) and the phenolic hydrogen of Y302(4.43).
Figure 9.
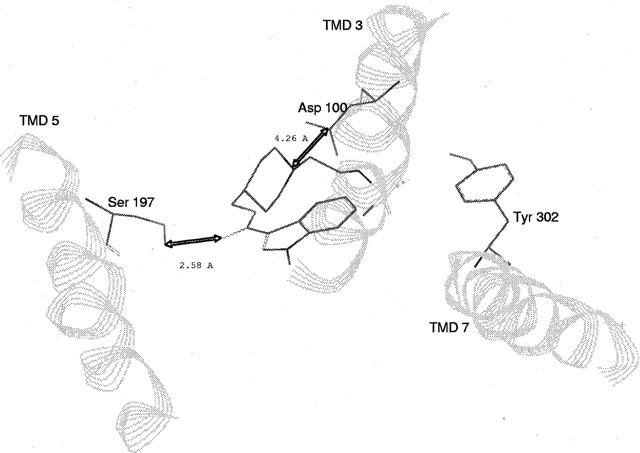
GR113808-h5-HT4 receptor complex model. It is characterized by distances: (i) d=2.58 Å between the hydroxylic hydrogen of S197(5.43) and the GR113808 carbonyl oxygen, (ii) d=4.26 Å between the carboxylic carbon of D100(3.32) and the GR113808 basic nitrogen.
Figure 10.
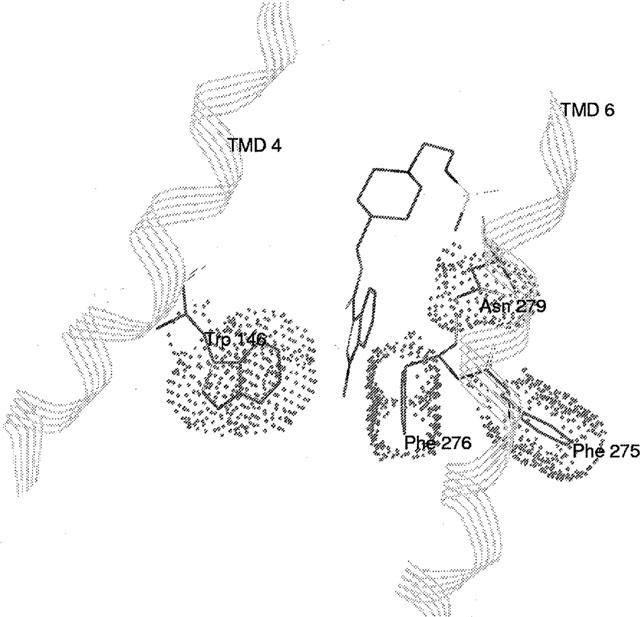
Representation of hydrophobic interactions between the indol ring of GR113808 and a pocket constituted by W146(4,46), F275(6.51), F276(6.52) and N279(6.55).
The docking experiments of the agonist ML10302 and the antagonist ML10375 were not further performed because of the lack of biological results. We could assume that like for the agonist 5-HT-receptor complex and the antagonist GR113808-receptor complex, three interaction sites are involved in the ligand binding site including D100(3.32) for an ionic interaction, S197(5.43) for a hydrogen bond and residues W146(4.50), F275(6.51), F276(6.52) and N279(6.55) for a hydrophobic pocket. However, in addition to these hypotheses, the ligand groups involved in the hydrogen bond with S197(5.43) cannot be clearly identified without further studies. Consequently it is too early to propose any ML10302- or ML10375-receptor complex but this work is in progress.
In conclusion, the ligand binding site of the h5-HT4 receptor had not been previously structurally explored. In our study, the combination of site-directed mutagenesis and structural analysis of the h5-HT4 receptor provides information about ligand-receptor interactions. Furthermore, our analysis gives strong evidence that h5-HT4 receptor presents particular features as compared with 5-HT1, 5-HT2 and 5-HT6 receptors specially the involvement of helix IV through a specific P residue in ligand selectivity. All these results may be used to generate clues for future molecular biology (i.e. further mutation) studies and for a more rational design of selective h5-HT4 receptor ligands.
Acknowledgments
We thank Olivier Blondel for participating in some of the initial experiments.
Abbreviations
- 5-HT
serotonin
- DMEM
Dulbecco's modified Eagle's medium
- ML10375
2-(cis-3,5-dimethylpiperidino)ethyl 4-amino-5-chloro-2-methoxybenzoate
- h5-HT4
human 5-HT4
- GR113808
(1-(2-((methylsulphonyl)amino)ethyl)-4-piperidinyl)methyl-1-methyl-1H-indole-3-carboxylate
- PMSF
phenylmethylsulphonyl fluoride
- ML10302
2-piperidinoethyl 4-amino-5-chloro-2-methoxybenzoate
- PEI
polyethyleneimine
- TMD
trans membrane domain
- WGA
wheat germ agglutinin
References
- ALMAULA N., EBERSOLE B.J., BALLESTEROS J.A., WEINSTEIN H., SEALFON S.C. Contribution of a helix 5 locus selectivity of hallucinogenic and non-hallucinogenic ligands for the human 5-Hydroxytryptamine2A and 5-Hydroxytryptamine2C receptors: direct and indirect effects on ligand affinity mediated by the same locus. Mol. Pharmacol. 1996;50:34–42. [PubMed] [Google Scholar]
- BALDWIN J.M. Structure and function of receptors coupled to G proteins. Curr. Opin. Cell. Biol. 1994;6:180–190. doi: 10.1016/0955-0674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- BALDWIN J.M., SCHERTLER G.F.X., UNGER V.M. An alpha-carbon template for the transmembrane helices in rhodopsin family of G-protein-coupled receptors. J. Mol. Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- BALLESTEROS J.A., WEINSTEIN H. Integrated methods for modelling G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- BLONDEL O., GASTINEAU M., DAHMOUNE Y., LANGLOIS M., FISCHMEISTER R. Cloning, expression, and pharmacology of four human 5-hydroxytryptamine4 receptor isoforms produced by alternative splicing in the carboxyl terminus. J. Neurochem. 1998;70:2252–2261. doi: 10.1046/j.1471-4159.1998.70062252.x. [DOI] [PubMed] [Google Scholar]
- BLONDEL O., VANDECASTEELE G., GASTINEAU M., LECLERC S., DAHMOUNE Y., LANGLOIS M., FISCHMEISTER R. Molecular and functional characterization of a 5-HT4 receptor cloned from human atrium. FEBS Lett. 1997;412:465–474. doi: 10.1016/s0014-5793(97)00820-x. [DOI] [PubMed] [Google Scholar]
- BOESS F.G., FREDERICK J.M., Jr, SLEIGHT A.J. Identification of residues in transmembrane regions III and VI that contribute to the ligand binding site of the serotonin 5-HT6 receptor. J. Neurochem. 1998;5:2169–2177. doi: 10.1046/j.1471-4159.1998.71052169.x. [DOI] [PubMed] [Google Scholar]
- BOUSSIF O., LEZOUALC'H F., ZANTA M.A., MERGNY M.D., SCHERMAN D., DEMENEIX B., BEHR J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CAVALLI A., FANELLI F., TADDEI C., DE BENEDETTI P.G., COTECCHIA S. Amino acids of the α1B-adrenergic receptor involved in agonist binding: differences in docking catecholamines to receptor subtypes. FEBS Lett. 1996;399:9–13. doi: 10.1016/s0014-5793(96)01286-0. [DOI] [PubMed] [Google Scholar]
- CLAEYSEN S., SEBBEN M., BECAMEL C., BOCKAERT J., DUMUIS A. Novel brain-specific 5-HT4 receptor splice variants show marked constitutive activity: role of the C-terminal intracellular domain. Mol. Pharmacol. 1999;55:910–920. [PubMed] [Google Scholar]
- EGLEN R.M., WONG E.H.F., DUMUIS A., BOCKAERT J. Central 5-HT4 receptors. Trends Pharmacol. Sci. 1995;16:391–397. doi: 10.1016/s0165-6147(00)89081-1. [DOI] [PubMed] [Google Scholar]
- HEDGE S., EGLEN R.M. Peripheral 5-HT4 receptors. FASEB J. 1996;10:1398–1407. doi: 10.1096/fasebj.10.12.8903510. [DOI] [PubMed] [Google Scholar]
- HIBERT M.F., TRUMPP-KALLMEYER A., BRUINVELS A., HOFLACK J. Three dimensional models of neurotransmitter G-binding protein coupled receptors. Mol. Pharmacol. 1991;40:8–15. [PubMed] [Google Scholar]
- HO Y.B., KARSCHIN A., BRANCHEK T., DAVIDSON N., LESTER H.A. The role of conserved aspartate and serine residues in ligand binding and in function of the 5-HT1A receptor: a site-directed mutation study. FEBS Lett. 1992;312:259–262. doi: 10.1016/0014-5793(92)80948-g. [DOI] [PubMed] [Google Scholar]
- KAUMANN A.J. Do human atrial 5-HT4 receptors mediate arrhythmias. Trends Pharmacol. Sci. 1994;15:451–455. doi: 10.1016/0165-6147(94)90058-2. [DOI] [PubMed] [Google Scholar]
- LEBESGUE D., WALLUKAT G., MIJARES A., GRANIER C., ARGIBAY J., HOEBEKE J. An agonist-like monoclonal antibody against the human β2-adrenoreceptor. Eur. J. Pharmacol. 1998;348:123–133. doi: 10.1016/s0014-2999(98)00136-8. [DOI] [PubMed] [Google Scholar]
- NEIMARK J., BRIAND J.P. Development of a fully automated multichannel peptide synthesizer with integrated TFA cleavage capability. Peptide Res. 1993;6:219–228. [PubMed] [Google Scholar]
- NORDVALL G., HACKSELL U. Binding-site modeling of the muscarinic ml receptor: a combination of homology-based and indirect approaches. J. Med. Chem. 1993;36:967–976. doi: 10.1021/jm00060a003. [DOI] [PubMed] [Google Scholar]
- PERSSON A., JERGIL B. Purification of plasma membranes by aqueous two-phase affinity partitioning. Anal. Biochem. 1992;204:131–136. doi: 10.1016/0003-2697(92)90151-v. [DOI] [PubMed] [Google Scholar]
- REYNOLDS G.P., MASON L., MELDRUM A., DE KECZER S., PARNES H., WONG E.H.F. 5-Hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: distribution, pharmacology and affects of neurodegenerative diseases. Br. J. Pharmacol. 1995;114:993–998. doi: 10.1111/j.1476-5381.1995.tb13303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RI Y., BALLESTEROS J.A., ABRAMS C.K., OH S., VERSELIS V.K., WEINSTEIN H., BARGIELLO T.A. The role of a conservated proline residue in mediating conformational changes associated with voltage gating of CX 32 Gap junctions. Biophys. J. 1999;76:2887–2898. doi: 10.1016/S0006-3495(99)77444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTH B.L., SHOHAM M., CHOUDHARY M.S., KHAN N. Identification of conserved aromatic residues essential for agonist binding and second messenger production at 5-hydroxytryptamine2A receptors. Mol. Pharmacol. 1997;52:259–266. doi: 10.1124/mol.52.2.259. [DOI] [PubMed] [Google Scholar]
- SANKARARAMAKRISHNAN R., VISHVESHWARA S. Conformational studies on peptides with proline in the right-handed alpha-helical region. Biopolymers. 1990;30:287–298. doi: 10.1002/bip.360300307. [DOI] [PubMed] [Google Scholar]
- SAVARESE T.M., FRASER C.M. In vitro mutagenesis and the search for structure-function relationships among G protein-coupled receptors. Biochem, J. 1992;283:1–19. doi: 10.1042/bj2830001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAXENA P.R. Serotonin receptors: subtypes, functional responses and therapeutic relevance. Pharmacol. Ther. 1995;66:339–368. doi: 10.1016/0163-7258(94)00005-n. [DOI] [PubMed] [Google Scholar]
- SEALFON S.C., CHI L., EBERSOLE B.J., RODIC W., ZHANG D., BALLESTEROS J.A., WEINSTEIN H. Related contribution of specific helix 2 and 7 residues to conformational activation of the serotonin 5-HT2A receptor. J. Biol. Chem. 1995;270:16683–16688. doi: 10.1074/jbc.270.28.16683. [DOI] [PubMed] [Google Scholar]
- TRUMPP-KALLMEYER S., HOFLACK J., BRUINVELS A., HIBERT M. Modeling of G-protein-coupled receptors: application to dopamine, adrenaline, serotonin, acetylcholine, and mammalian opsin receptors. J. Med. Chem. 1992;35:3448–3462. doi: 10.1021/jm00097a002. [DOI] [PubMed] [Google Scholar]
- VAN RHEE A.H., JACOBSON K.A. Molecular architecture of G protein-coupled receptors. Drug. Dev. Des. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG C.-D., GALLAHER T.K., SHIH J.C. Site-directed mutagenesis of the serotonin 5-hydroxytryptamine2 receptor: identification of amino acids necessary for ligand binding and receptor activation. Mol. Pharmacol. 1993;43:931–940. [PubMed] [Google Scholar]
- WONG E.H., REYNOLDS G.P., BONHAUS D.W., HSU S., EGLEN R.M. Characterization of [3H]- GR113808 binding to 5-HT4 receptors in brain tissues from patients with neurodegenerative disorders. Behav. Brain. Res. 1996;73:249–252. doi: 10.1016/0166-4328(96)00106-4. [DOI] [PubMed] [Google Scholar]
- YANG D., SOULIER J.-L., SICSIC S., MATHÉ-ALLAINMAT M., BRÉMONT B., CROCI T., CARDAMONE R., AUREGGI G., LANGLOIS M. New esters of 4-amino-5-chloro-2-methoxybenzoic acid as potent agonists and antagonists for 5-HT4 receptors. J. Med. Chem. 1997;40:608–621. doi: 10.1021/jm960320m. [DOI] [PubMed] [Google Scholar]