

Abstract
In resistance arteries pressure-induced (myogenic) tone (MT) and flow (shear stress)-induced dilation (FD) are potent determinant of vascular resistance. We investigated the role of angiotensin II and endothelin-1 in FD and MT in resistance arteries and their potential change in hypertension.
Flow–diameter–pressure relationship was established in situ, under anaesthesia, in two daughter branches of a mesenteric resistance artery (180 μM, n=7 per group) from spontaneously hypertensive (SHR) or normotensive (WKY) rats. One artery was ligated distally, so that it was submitted to pressure only, while the other was submitted to pressure and flow. Drugs were added to the preparation and external diameter, pressure and flow measured continuously.
External diameter (with flow) ranged from 150±3 to 191±7 μM in WKY (n=28) rats and from 168±6 to 186±6 μM in SHR (n=28). Flow induced a dilation of the non-ligated arteries which was lower in SHR (13±5–31±4 μM vs WKY: 5±5–44±4 μM). In the ligated artery, the diameter did not significantly change, due to MT.
In the vessels submitted to flow angiotensin converting enzyme inhibition (perindopril, 10 μmol L−1) increased the diameter in SHR (+11±2 μM) significantly more than in WKY (+2±1 μM). Angiotensin type 1 receptor (AT1R) blockade (losartan, 10 μmol L−1) increased the diameter in the vessels with flow in SHR only (+6±1 μM). Angiotensin type 2 receptor (AT2R) blockade (PD 123319, 1 μmol L−1) decreased arterial diameter in WKY only (9±2). Endothelin-1 type A receptor (ETAR) blockade (LU135252, 0.1 μmol L−1) increased the diameter only in SHR in the artery submitted to flow (by 6±1 μM).
Thus FD was counteracted by a flow-dependent AT1 and ETA receptors-activation in SHR whereas in WKY FD AT2-dependent dilation is involved.
Keywords: Blood vessels, myogenic tone, angiotensin converting enzyme, flow-induced dilation
Introduction
In resistance arteries pressure induces an active vasoconstrictor (myogenic) tone (Johnson, 1989; Bevan & Laher, 1991; Kuo et al., 1991; D'Angelo & Meininger, 1994; Davis & Hill, 1999) which is opposed by flow-induced dilation, in vitro as well as in vivo (Bevan & Laher, 1991; Kuo et al., 1991; Friebel et al., 1995). Whereas myogenic tone is mainly independent of endothelial factors (Bevan & Laher, 1991; D'Angelo & Meininger, 1994; Friebel et al., 1995; Davis & Hill, 1999), flow (shear stress) depends mainly on the release of endothelium-derived relaxing factors (Friebel et al., 1995; Koller & Huang, 1994; Qiu et al., 1994; Matrougui et al., 1997; 1999). Besides these two mechanical factors, local tissue renin-angiotensin (Caputo et al., 1995a; Vicaut, 1995; Henrion et al., 1997) and endothelin (Schiffrin, 1995; Iglarz et al., 1998) systems are two main hormonal regulators of vascular tone which may interact with flow-induced dilation and myogenic tone. Acute shear stress induces the release of endothelin-1 (Yoshizumi, 1989), whereas long-term changes in flow decrease its production (Kuchan & Frangos, 1993; Malek et al., 1993; Morita et al., 1993; 1994). Locally produced angiotensin II is a potent amplifier of vascular tone (Henrion et al., 1997) and Angiotensin II is also able to stimulate NO release from the endothelium (Caputo et al., 1995b).
In resistance arteries from normotensive rats, flow induces the activation of endothelin-1 type A receptors (ETAR), which counteracts the dilation (Iglarz et al., 1998) and the activation of angiotensin II type 2 receptors (AT2R), which participates to flow-induced dilation through the release of NO by the endothelium (Matrougui et al., 1999). Nevertheless, these two peptides (angiotensin II and endothelin-1) might play a different role in the functional response of resistance arteries to pressure and flow in hypertension. We have previously shown that a chronic ETAR blockade improves flow-induced dilation in SHR's mesenteric resistance arteries (Iglarz et al., 1998). Thus we hypothesized that locally produced angiotensin II and/or endothelin-1 might both counteract the response (change in diameter) of resistance arteries to flow in spontaneously hypertensive rats (SHR) and thus explain, at least in part, the poor dilation induced by flow in hypertensive rats (Koller & Huang, 1994; Matrougui et al., 1997; Qiu et al., 1994). In SHR's mesenteric resistance arteries, perfused in situ, the external diameter was measured after a bifurcation on two distal branches equivalent in size. One branch was perfused while the other was ligated distally so that it was submitted to the same pressure but not to flow. Thus one artery developed myogenic tone only while in the other myogenic tone was counteracted by flow-induced dilation (Matrougui et al., 1999).
Methods
Twelve-week-old spontaneously hypertensive (SHR, n=28) and normotensive (WKY, n=28) rats (C.E.R. Janvier, St-Berthevin, France) were anaesthetized with sodium pentobarbital (50 mg kg−1, IP). A medial laparotomy was then performed and the last loop of the small intestine was exteriorized and superfused with a physiological salt solution of the following composition (in mmol L−1): NaCl 130, NaHCO3 14.9, KCl 3.7, CaCl2(2H2O) 1.6, KH2PO4 1.2, MgSO4(7H2O) 1.2, Glucose 11 N-2-hydroxy-ethylpiperazine-N-2-ethylsulphonic acid (HEPES) 10 mM, and albumin (4%). The temperature was 37–38°C, the pH 7.4, the pO2 160 mmHg and the pCO2 37 mmHg. Two daughter arteries (second order mesenteric arteries), equivalent in size were dissected free of fat and connective tissues. One artery was distally ligated (178±7.1 μM external diameter, before ligation) while the other was left open (178±4.7 μM external diameter). As previously described (Matrougui et al., 1999) the diameter of the two arteries was measured by video-microscopy and flow (solution as described above) was generated through a first generation branch of the mesenteric artery, upstream located. Pressure was recorded from an arterial branch located upstream of the observed arteries and flow was measured in the perfused arterial segment (Transonic Systems Inc, Ithaca, NY, U.S.A).
Flow–pressure–diameter relationships were established by imposing step increases in flow from 200 to 2000 μl min−1, corresponding to a flow ranging from 71 to 310 μl min−1 (n=56) as measured in the non-ligated artery. Step increases in flow and measurements of diameter and pressure were conducted, in the same vessels, under control conditions and then after the addition of one of the following drugs: Losartan (10 μmol L−1) as an AT1 receptor blocker, PD 123319 (1 μmol L−1), as an AT2 receptor blocker, perindopril (10 μmol L−1) as angiotensin-converting enzyme inhibitors (ACEI) or LU 135252 (0.1 μmol L−1) as an endothelin type A receptor blocker (Iglarz et al., 1998). Specificity and concentrations of the drugs have been previously determined in conditions similar to the present conditions (Matrougui et al., 1999).
At the end of each experiment step increases in flow were repeated in the presence of sodium nitroprusside (1 mmol L−1), EGTA (2 mmol L−1) and in the absence of extracellular calcium to determine the passive diameter (Matrougui et al., 1999).
In another series of experiments (n=4–5), angiotensin II (10 nmol L−1) was added to the physiological solution after pretreatment of the vessels with the AT1 receptor blocker losartan (10 μmol L−1). Shear stress was calculated for each individual segment of artery as previously described (Matrougui et al., 1997):
where ζ is viscosity (poise=dyn.s.cm−2), Q is flow (ml s−1), and r is radius (cm).
Drugs
Losartan was provided by Dupont-Merck Pharmaceutical, (Paris, France) PD 123319 by Parke-Davis (Paris, France), perindopril by Servier (Courbevoie, France), LU135252 by Knoll AG (Ludwigshafen, Germany). All the other reagents were purchased from Sigma Chemical (St Louis, MO, U.S.A.).
Statistical analysis
Changes in diameter due to each drug was calculated as the difference between the diameter values obtained in response to step increases in flow before and then after the addition of the drug. Results are expressed as mean±standard error of the mean (s.e.mean). Significance of the differences between groups was determined by 2-way ANOVA for consecutive measurements or by 1-way ANOVA followed by a Bonferroni test when appropriated. P values less than 0.05 were considered to be significant.
Results
When flow was raised by steps in the mesenteric arterial bed flow increased in the artery observed (non-ligated segment), pressure increased in the two arterial segments observed (ligated and non-ligated) and the external diameter of the two arteries changed as described below. Flow rates measured in the arterial segment left open corresponded to a shear stress ranging from 25±3 to 46±5 dynes cm−2 (n=28) in SHR and from 20±1 to 37±3 dynes cm−2 (n=28, P<0.001 versus SHR). These values were corresponding to mesenteric arterial pressures ranging from 53±4 to 82.6±2 mmHg in SHR and from 45±3 to 66±4 mmHg in WKY rats (P<0.001, WKY versus SHR). Under these flow and pressure conditions diameter (Figure 1) ranged from 144±4 to 150±5 μM in ligated arteries in WKY rats (n=28), from 150±3 to 191±7 μM in the arteries with flow in WKY rats, from 151±6 to 155±7 μM in the ligated arteries in SHR and from 168±6 to 186±6 μM in the arteries with flow in SHR (n=28). Passive arterial diameter was 166±4, 179±5, 185±5 and 192±4 μM in WKY rats and 172±6, 181±6, 190±5 and 197±4 μM in SHR (n=28 per group) in the vessels submitted to flow. Passive diameters in the absence of flow were not significantly different from those in the presence of flow (data not shown).
Figure 1.
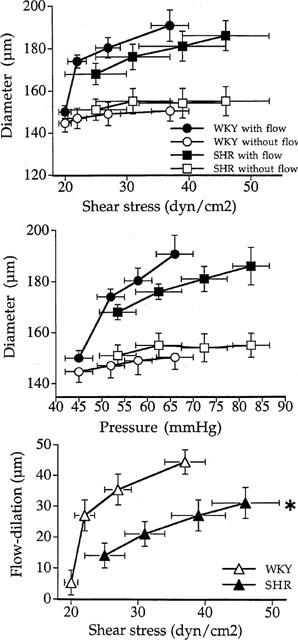
Changes in diameter in responses to increases in flow and pressure in mesenteric resistance artery isolated from SHR (n=28) and WKY rats (n=28). The arterial diameter was measured in arteries segments with or without flow (ligated arteries). Changes in diameter are represented as a function of flow rate (upper panel) or pressure (middle panel). Flow-induced dilation (lower panel) was estimated as the difference in diameter between arteries submitted to flow and pressure and arteries submitted to pressure only in SHR and WKY rats. Mean±s.e.mean are represented. *P<0.001, ANOVA for repeated measures.
When flow was increased by steps the external diameter did not significantly change in the arterial segments distally ligated (submitted to pressure but not to flow), in both SHR and WKY rats (Figure 1). In the non-ligated artery increasing flow induced a dilation (Figure 1) which was significantly less pronounced in SHR (13±5 to 31±4 μM) than in WKY rats (5±5 to 44±4 μM).
Perindopril (10 μmol L−1, n=7 per group) induced a significant increase in diameter in both the ligated and the non-ligated artery, in both strains of rats (Figure 2). There was no difference between the ligated and the non-ligated artery in WKY rats, whereas in SHR perindopril (Figure 2) induced a higher dilation in the non-ligated artery than in the ligated vessel (+11±2 μM in SHR, versus +2±1 μM in WKY rats).
Figure 2.
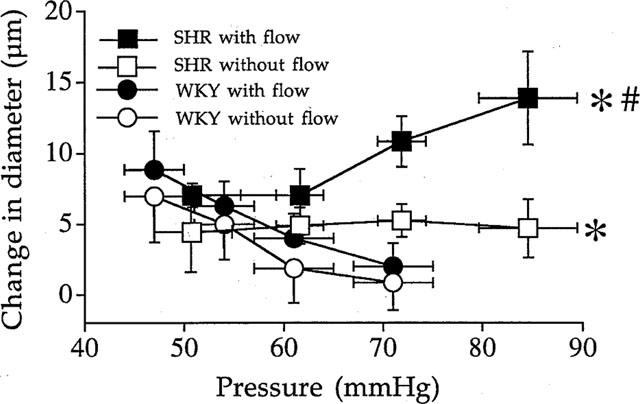
Effect of angiotensin I converting enzyme inhibition with perindoprinol (10 μmol L−1) on the arterial diameter measured in resistance mesenteric artery segments with or without flow (ligated arteries) isolated from SHR or WKY rats. Results are expressed as changes in diameter (μM) due to perindopril. (n=7). Mean±s.e.mean are represented. *P<0.01, 2-way ANOVA for repeated measures, SHR vs WKY. #P<0.01, 2-way ANOVA for repeated measures, vessels with versus vessels without flow.
Angiotensin II type 1 receptor blockade (losartan, 10 μmol L−1, n=7 per group) induced no significant change in arterial diameter in both vessels in WKY rats (Figure 3). In SHR, losartan induced a significant increase in diameter (+6±1 μM) in the artery submitted to flow and pressure and produced no significant change in the ligated artery (Figure 3). The increase in diameter due to losartan in the artery submitted to flow and pressure in SHR corresponded to 23, 25, 21 and 20% of flow-induced dilation in SHR (Figure 1, lower panel).
Figure 3.
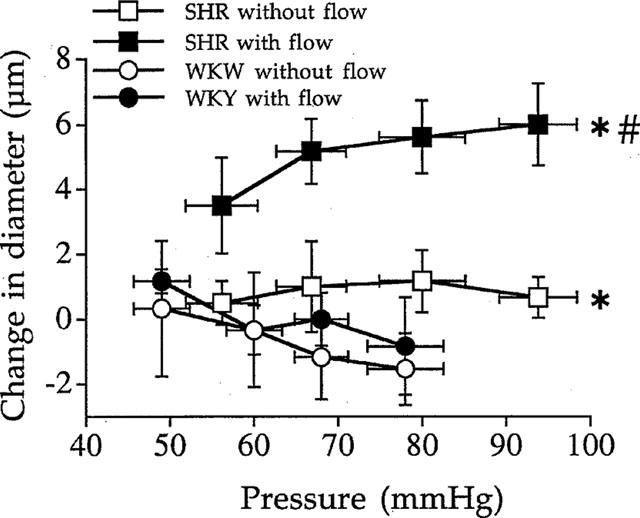
Effect of the angiotensin II type 1 receptor blocker losartan (10 μmol L−1) on the arterial diameter measured in rat resistance mesenteric arteries with or without flow (ligated arteries) isolated from SHR or WKY rats. Results are expressed as changes in diameter due to losartan (μM). Mean±s.e.mean are represented (n=7 per group). *P<0.001, 2-way ANOVA for repeated measures. #P<0.01, 2-way ANOVA for repeated measures, vessels with versus vessels without flow.
Angiotensin II type 2 receptor blockade (PD 123329, 1 μmol L−1, n=7 per group) had no significant effect in arteries (ligated and non-ligated) from SHR (Figure 4). In WKY rats PD 123329 induced a significant contraction (−9±2 μM) in the artery submitted to flow without affecting the diameter of the artery distally ligated (Figure 4). The decrease in diameter due to PD 123329 in the artery submitted to flow and pressure in WKY rats corresponded to 100, 30, 30 and 23% of flow-induced dilation (Figure 1, lower panel).
Figure 4.
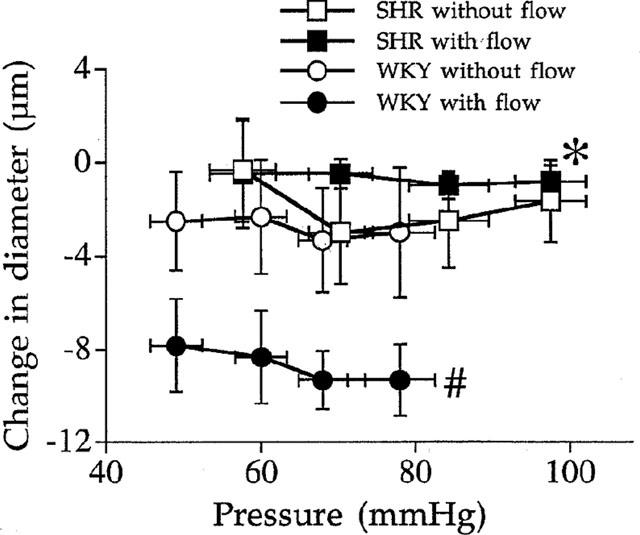
Effect of the angiotensin II type 2 receptor blocker PD 123319 (1 μmol L−1) on the arterial diameter measured in rat resistance mesenteric arteries with or without flow (ligated arteries) isolated from SHR or WKY rats. Results are expressed as changes in diameter due to PD 123319 (μM). Mean±s.e.mean are represented (n=7 per group). *P<0.001, 2-way ANOVA for repeated measures. #P<0.01, 2-way ANOVA for repeated measures, vessels with versus vessels without flow.
In SHR endothelin type A receptor blockade (LU 135252, 0.1 μmol L−1, n=7 per group) induced a significant increase in diameter in the mesenteric arterial branches submitted to flow and pressure (+6±1 μM) but not in the mesenteric arterial branches submitted to pressure only (Figure 5). In WKY rats LU 135252 induced no significant change in both arteries (Figure 5). The increase in diameter due to LU 135252 in the artery submitted to flow and pressure in SHR corresponded to 60, 30, 23 and 17% of flow-induced dilation in SHR (Figure 1, lower panel).
Figure 5.
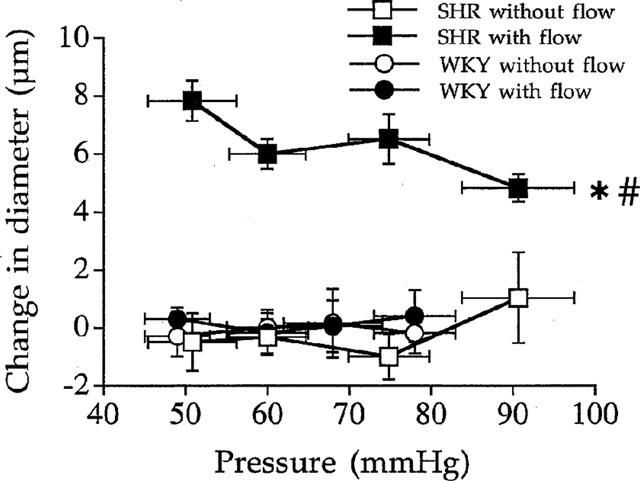
Effect of the endothelin type A receptor blocker LU 13525 (0.1 μmol L−1) on the arterial diameter measured in rat resistance mesenteric arteries with or without flow (ligated arteries) isolated from SHR or WKY rats. Results are expressed as change in diameter (μM). Mean±s.e.mean are represented (n=7 per group). *P<0.01, 2-way ANOVA for repeated measures, vessels with versus vessels without flow.
Exogenous angiotensin II, in the presence of losartan (10 μmol L−1) induced a significant dilation (125±8 to 139±7 μM) in the arteries submitted to flow in WKY (Figure 6). Exogenous angiotensin II, in the presence of losartan, did not significantly affect the diameter of the arteries submitted to flow in SHR (138±7 before and 136±8 μM after angiotensin II; and Figure 6).
Figure 6.
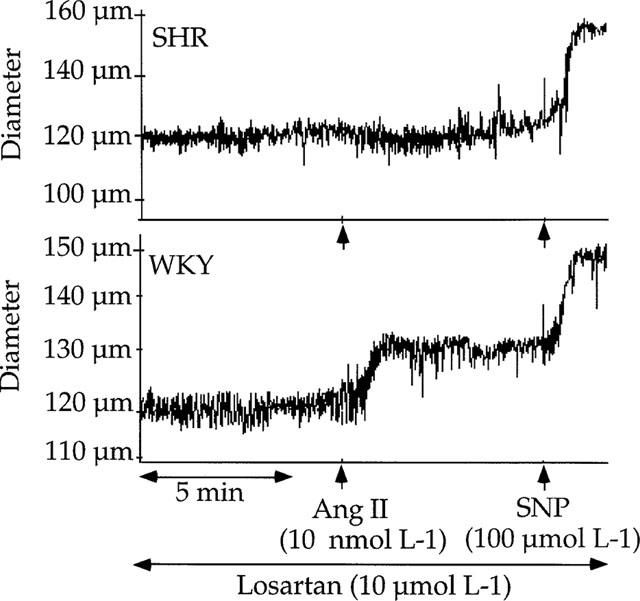
Effect of exogenous angiotensin II (Ang II, 10 nmol L−1) on the internal diameter of mesenteric resistance arteries from SHR or WKY rats submitted to pressure and flow. Vessels were pretreated with losartan (10 μmol L−1). Sodium nitroprusside (SNP, 100 μmol L−1) was added at the end of the experiment in order to induce a full vessel relaxation.
Discussion
The major new findings of the present study is that angiotensin II and endothelin-1 were produced locally by resistance arteries from SHR and/or WKY rats, mainly in the presence of flow, and that they interacted with flow-induced dilation.
In resistance vessels, flow (shear stress) induces a dilation (Bevan & Laher, 1991; Kuo et al., 1991; Koller & Huang, 1994; Qiu et al., 1994; Friebel et al., 1995; Matrougui et al., 1997), and in some situations a constriction (Bevan & Henrion, 1994), whereas pressure induces myogenic tone (Bevan & Laher, 1991; Kuo et al., 1991; Friebel et al., 1995). As previously shown (Juncos et al., 1995; Matrougui et al., 1999), the design of the experiments described in the present study allowed the in situ measurement of a basal tone in the presence or in the absence of flow in two daughter arterial branches similar in size. In the branch distally ligated, only pressure was acting on the vessel wall, so that myogenic tone developed in the absence of flow-induced dilation. in the arterial branch submitted to both flow and pressure, myogenic tone was opposed by flow-induced dilation. The difference in diameter between the two branches can be considered as flow-induced dilation (Matrougui et al., 1999), although a possible influence of substances released by the artery upstream located, in response to flow, cannot be excluded.
Recent studies have reported the occurrence of a local renin-angiotensin system located in blood vessels (Campbell, 1987; Samani & Swales, 1991; Caputo et al., 1995a; Henrion et al., 1997; Matrougui et al., 1999), including in the rat mesenteric arteries (Henrion et al., 1997; Matrougui et al., 1999). The relaxing effect of the ACE inhibitor perindopril was effective in both strains, but it was higher in the presence of flow in SHR. In parallel, AT1R blockade (losartan) was effective only in the presence of flow in SHR. These observations suggest that in mesenteric arteries ACE inhibition relaxed the arteries through angiotensin II synthesis inhibition only in SHR's arteries submitted to flow. Indeed, in the other situations (WKY with or without flow and SHR without flow) the inhibition of bradykinin breakdown mediates a great part of the relaxing effect of ACE inhibition, as previously shown in the same model (Matrougui et al., 1999). Thus in SHR, flow-induced dilation was counteracted by AT1R stimulation. This involvement of AT1R stimulation by flow was not seen in WKY, in which flow stimulates the endothelial release of NO in part through the activation of AT2R (present study and Matrougui et al., 1999). By contrast, in SHR (present study), blockade of AT2 receptors had no effect on the diameter of mesenteric arteries whether they were submitted to pressure alone or to pressure and flow. In this type of mesenteric arteries, flow-dilation (Qiu et al., 1994; Matrougui et al., 1997) and AT2R-dependent dilation (Matrougui et al., 1999) involve mainly NO production. NO-production independent of AT2R stimulation is most probably due to the direct stimulation of NO synthase through a transduction signal triggered by shear stress of the endothelial cells surface (Lehoux & Tedgul, 1998). Both AT1R and AT2R are present in mesenteric resistance arteries (Matrougui et al., 1999; Touyz et al., 1999). In mesenteric resistance arteries from SHR both AT1R and AT2R stimulation by exogenous angiotensin II induces a contraction, whereas in WKY rats only AT1R stimulation by exogenous angiotensin II induces a contraction, in no-flow-conditions (Touyz et al., 1999). Nevertheless the amount of AT2R mRNA is similar in WKY rats and SHR (Touyz et al., 1999). In the present study and in a previous one (Matrougui et al., 1999), we found a different effect of AT1R and AT2R blocked in the presence or in the absence of flow. Flow or shear stress might stimulate the angiotensin I converting enzyme, as shown in bovine pulmonary artery endothelial cells (Rieder et al., 1997). An alternative explanation could be that flow changes the mass transport of angiotensin II to its receptors, as previously shown for adenosine receptors (Dull & Davies, 1991; Mo et al., 1991). The involvement of AT2R stimulation in flow-induced dilation (8–10 μM dilation) represented 100–23% of the total flow-dependent dilation for low and high flow rates, respectively. This, and the observation that the AT2R-dependent dilation due to a supra-maximal concentration of angiotensin II is limited to a 10 μM dilation (Figure 6), suggest that flow-stimulation of AT2R-dependent dilation is more sensitive than the direct stimulation by flow of NO-dependent dilation. Thus for higher flow rates AT2R-dependent dilation is representing a lower percentage of the total flow-dilation. This observation does not apply to the AT1R-dependent contraction stimulated by flow in SHR (Figure 3) which seems to increase with flow, but in this type of artery AT1R-dependent contraction is not limited to a low level as is AT2R-dependent dilation. In mesenteric resistance arteries similar to those used in the present study, we have found that the AT1R-dependent contraction represents a 50–100 μM decrease in diameter (Loufrani et al., 1999).
Flow-induced dilation, in SHR's arteries, was also improved by endothelin-1 type A receptor blockade. Endothelin-1 type A receptors are expressed by vascular smooth muscle cells and cause vasoconstriction (Haynes et al., 1995). In the present study, the blockade of ETAR improved the dilation due to flow. This was not observed in the ligated artery. Thus endothelin-1 release was stimulated by flow and opposed flow-induced dilation in SHR (ETAR blockade was not effective in WKY). This is in agreement with our previous work showing that flow-induced dilation in SHR's mesenteric arteries was improved after chronic ETAR blockade (Iglarz et al., 1998). This is also in agreement with previous studies showing that acute changes in flow induce the release of endothelin-1 from cultured endothelial cells (Malek et al., 1995; Wang et al., 1995). The absence of effect of the ETAR blockade in the artery submitted only to pressure suggest that in this type of artery (mesenteric), in WKY as well as in SHR, endothelin-1 might not be involved in myogenic tone, whereas in rat skeletal muscle arterioles both endothelin-1 and prostaglandin H2 enhance myogenic tone in SHR, not in normotensive rats (Huang & Koller, 1997). As for the ETAR-dependent dilation, the effect of ETAR blockade on flow-dilation was proportionally decreasing with increasing flow rates (60–17% of total flow-dilation). This could be due to the biphasic effect of flow on ET-1 production as described by Malek et al. (1995).
The participation of both endothelin-1 and angiotensin II to the functional response to flow in mesenteric resistance arteries from SHR as contractile agents might explain, at least in part, the difference in the amplitude of flow-induced dilation between resistance arteries from normotensive and hypertensive rats (present study, Koller & Huang, 1994; Qiu et al., 1994; Matrougui et al., 1997; 1999; Iglarz et al., 1998). The main difference between WKY rats and SHR (present study) is that angiotensin II was involved in response to flow as a contractile agent in SHR (through AT1 receptor stimulation) and as a dilator agent in WKY rats (through AT2 receptor stimulation).
In conclusion, the present study suggests that in mesenteric resistance arteries from SHR flow-induced dilation was counteracted by locally produced angiotensin II and endothelin-1. Such an altered endothelial response to flow in resistance arteries might be an important factor in the increase in peripheral resistance in hypertension.
Acknowledgments
Khalid Matrougui was a fellow of the ‘Recherche et Partage' foundation, Paris, France. This work was supported in part by a grant from IRIS (Servier, Paris, France).
Abbreviations
- MT
myogenic tone
- FD
flow-induced dilation
- SHR
spontaneously hypertensive rats
- WKY
Wistar-Kyoto
- AT1R
angiotensin type 1 receptor
- AT2R
angiotensin type 2 receptor
- NO
nitric oxide
- ETAR
endothelin-1 type A receptors
- ACEI
angiotensin converting enzyme inhibition
References
- BEVAN J.A., HENRION D. Pharmacological implications of the flow-dependence of vascular smooth muscle tone. Ann. Rev. Pharmacol. Toxicol. 1994;34:173–190. doi: 10.1146/annurev.pa.34.040194.001133. [DOI] [PubMed] [Google Scholar]
- BEVAN J.A., LAHER I. Pressure and flow-dependent vascular tone. FASEB J. 1991;5:2267–2273. doi: 10.1096/fasebj.5.9.1860618. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.J. Circulating and tissue angiotensin system. J. Clin. Invest. 1987;79:1–6. doi: 10.1172/JCI112768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAPUTO L., BENESSIANO J., BOULANGER C.M., LEVY B.I. Angiotensin II increases cGMP content via endothelial angiotensin II AT1 subtype receptors in the rat carotid artery. Arterioscler. Thromb. Vasc. Biol. 1995a;15:1646–1651. doi: 10.1161/01.atv.15.10.1646. [DOI] [PubMed] [Google Scholar]
- CAPUTO L., TEDGUI A., LÉVY B.I. Control of carotid vasomotor tone by local renin-angiotensin system in normotensive and spontaneously hypertensive rats. Role of endothelium and flow. Circ. Res. 1995b;77:303–309. doi: 10.1161/01.res.77.2.303. [DOI] [PubMed] [Google Scholar]
- D'ANGELO G., MEININGER G.A. Transduction mechanisms involved in the regulation of myogenic activity. Hypertension. 1994;23:1096–1105. doi: 10.1161/01.hyp.23.6.1096. [DOI] [PubMed] [Google Scholar]
- DAVIS M.J., HILL M.A. Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- DULL R.O., DAVIES P.F. Flow modulation of agonist (ATP) response (Ca2+) coupling in vascular endothelial cells. Am. J. Physiol. 1991;261:H149–H156. doi: 10.1152/ajpheart.1991.261.1.H149. [DOI] [PubMed] [Google Scholar]
- FRIEBEL M., KLOTZ K.F., LEY K., GAEHTGENS P., PRIES A. Flow-dependent regulation of arteriole diameter in rat skeletal muscle in situ: role of endothelium-derived relaxing factor and prostanoids. J. Physiol. 1995;483:715–726. doi: 10.1113/jphysiol.1995.sp020616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYNES W.G., STRACHAN F.E., WEBB D.J. Endothelin ETA and ETB receptors cause vasoconstriction of human resistance and capacitance vessels in vivo. Circulation. 1995;92:357–363. doi: 10.1161/01.cir.92.3.357. [DOI] [PubMed] [Google Scholar]
- HENRION D., BENESSIANO J., LÉVY B.I. In vitro modulation of a resistance artery diameter by the tissue renin-angiotensin system of a large donor artery. Circ. Res. 1997;80:189–195. doi: 10.1161/01.res.80.2.189. [DOI] [PubMed] [Google Scholar]
- HUANG A., KOLLER A. Endothelin and prostaglandin H2 enhance arteriolar myogenic tone in hypertension. Hypertension. 1997;30:1210–1215. doi: 10.1161/01.hyp.30.5.1210. [DOI] [PubMed] [Google Scholar]
- IGLARZ M., MATROUGUI K., LÉVY B.I., HENRION D. Chronic blockade of endothelin ETA receptors improves flow-dependent dilation in resistance arteries of hypertensive rats. Cardiovasc. Res. 1998;39:657–664. doi: 10.1016/s0008-6363(98)00151-5. [DOI] [PubMed] [Google Scholar]
- JOHNSON P.C. The myogenic response in the microcirculation and its interaction with other control systems. J. Hypertens. 1989;4:S33–S40. [PubMed] [Google Scholar]
- JUNCOS L.A., GARVIN J., CARRETERO O.A., SADAYOSHI I. Flow modulates myogenic responses in isolated microperfused rabbit afferent arterioles via endothelium-derived nitric oxide. J. Clin. Invest. 1995;95:2741–2748. doi: 10.1172/JCI117977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLLER A., HUANG A. Impaired nitric oxide-mediated flow-induced dilatation in arterioles of spontaneously hypertensive rats. Circ. Res. 1994;74:416–421. doi: 10.1161/01.res.74.3.416. [DOI] [PubMed] [Google Scholar]
- KUCHAN M.J., FRANGOS J.A. Shear stress regulates endothelin-1 release via protein kinase C and cGMP in cultured endothelial cells. Am. J. Physiol. 1993;264:H150–H156. doi: 10.1152/ajpheart.1993.264.1.H150. [DOI] [PubMed] [Google Scholar]
- KUO L., WILLIAM M.C., DAVIS M.J. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am. J. Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- LEHOUX S., TEDGUI A. Signal transduction of mechanical stresses in the vascular wall. Hypertension. 1998;32:338–345. doi: 10.1161/01.hyp.32.2.338. [DOI] [PubMed] [Google Scholar]
- LOUFRANI L., HENRION D., CHANSEL D., ARDAILLOU R., LÉVY B.I. Functional evidence for an angiotensin IV receptor in rat resistance arteries. J. Pharmacol. Exp. Ther. 1999;291:583–588. [PubMed] [Google Scholar]
- MALEK A.M., IZUMO S. Control of endothelial cell gene expression by flow. J. Biomech. 1995;28:1515–1528. doi: 10.1016/0021-9290(95)00099-2. [DOI] [PubMed] [Google Scholar]
- MALEK A.M., GREENE A.L., IZUMO S. Regulation of endothelin 1 gene by fluid shear stress is transcriptionally mediated and independent protein kinase C and cAMP. Proc. Natl. Acad. Sci. USA. 1993;90:5999–6003. doi: 10.1073/pnas.90.13.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATROUGUI K., LOUFRANI L., LÉVY B.I., HENRION D. Role of angiotensin II type 2 receptor stimulation in the response to flow and pressure of rat resistance mesenteric arteries. Hypertension. 1999;34:659–665. doi: 10.1161/01.hyp.34.4.659. [DOI] [PubMed] [Google Scholar]
- MATROUGUI K., MACLOUF J., LEVY B., HENRION D. Impaired nitric oxide- and prostaglandins-mediated responses to flow in resistance mesenteric arteries from spontaneously hypertensive rats. Hypertension. 1997;30:942–947. doi: 10.1161/01.hyp.30.4.942. [DOI] [PubMed] [Google Scholar]
- MO M.S., ESKIN S.G., SCHILLING W.P. Flow-induces changes in Ca2+ signalling of vascular endothelial cells: effects of shear stress and ATP. Am. J. Physiol. 1991;265:H3–H8. doi: 10.1152/ajpheart.1991.260.5.H1698. [DOI] [PubMed] [Google Scholar]
- MORITA T., KURIHARA H., MAEMURA K., YOSHIZUMI M., NAGAI R., YAZAKI Y. Role of Ca2+ and protein kinase C in shear stress-induced actin depolymerisation and endothelin 1 gene expression. Circ. Res. 1994;75:630–636. doi: 10.1161/01.res.75.4.630. [DOI] [PubMed] [Google Scholar]
- MORITA T., KURIHARA H., MAEMURA K., YOSHIZUMI M., YAZAKI Y. Disruption of cytoskeletal structures mediates shear stress-induced endothelin-1 gene expression in culture porcine aortic endothelial cells. J. Clin. Invest. 1993;92:1706–1712. doi: 10.1172/JCI116757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIU H.Y., HENRION D., LÉVY B.I. Alteration in flow-dependent vasomotor tone in spontaneously hypertensive rats. Hypertension. 1994;24:474–479. doi: 10.1161/01.hyp.24.4.474. [DOI] [PubMed] [Google Scholar]
- RIEDER M.J., CARMONA R., KRIEGER J.E., PRITCHARD K.A., Jr, GREENE A.S. Suppression of angiotensin-converting enzyme expression and activity by shear stress. Circ. Res. 1997;80:312–319. doi: 10.1161/01.res.80.3.312. [DOI] [PubMed] [Google Scholar]
- SAMANI N.J., SWALES J.D. Molecular biology of the vascular renin antiotensin system. Blood vessels. 1991;28:210–216. doi: 10.1159/000158864. [DOI] [PubMed] [Google Scholar]
- SCHIFFRIN E.L. Endothelin: Potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25:1135–1143. doi: 10.1161/01.hyp.25.6.1135. [DOI] [PubMed] [Google Scholar]
- TOUYZ R.M., ENDEMANN D., HE G., LI J.S., SCHIFFRIN E.L. Role of AT2 receptors in angiotensin II-stimulated contraction of small mesenteric arteries in young SHR. Hypertension. 1999;33:366–372. doi: 10.1161/01.hyp.33.1.366. [DOI] [PubMed] [Google Scholar]
- VICAUT E., HOU X. Duration of inhibition of local microvascular ACE in sponteneously hypertensive rats. J. Hypertens. 1995;13:1758–1763. [PubMed] [Google Scholar]
- WANG D.L., WUNG B.S., PENG Y.C., WANG J.J. Mechanical strain increase endothelin-1 gene expression via protein kinase C pathway in human endothelial cells. J. Cell. Physiol. 1995;163:400–406. doi: 10.1002/jcp.1041630220. [DOI] [PubMed] [Google Scholar]
- YOSHIZUMI M., KURIHARA H., SUGIYAMA T., TAKAKU F., YANAGISAWA M., MASAKI T., YASAKO Y. Haemodynamic shear stress stimulates endothelin production by cultured endothelial cells. Biochem. Biophys. Res. Commun. 1989;161:859–864. doi: 10.1016/0006-291x(89)92679-x. [DOI] [PubMed] [Google Scholar]