

Abstract
Betaxolol, a β1-adrenoceptor antagonist used for the treatment of glaucoma, is known to be neuroprotective in paradigms of ischaemia/excitotoxicity. In this study, we examined whether betaxolol and other β-adrenoceptor antagonists interact directly with neurotoxin binding to sites 1 and 2 of the voltage-sensitive sodium channel (Na+ channel) in rat cerebrocortical synaptosomes.
Betaxolol inhibited specific [3H]-batrachotoxinin-A 20-α-benzoate ([3H]-BTX-B) binding to neurotoxin site 2 in a concentration-dependent manner with an IC50 value of 9.8 μM. Comparison of all the β-adrenoceptor antagonists tested revealed a potency order of propranolol>betaxolol≈levobetaxolol>levobunolol≈carteolol⩾timolol>atenolol.
None of the drugs caused a significant inhibition of [3H]-saxitoxin binding to neurotoxin receptor site 1, even at concentrations as high as 250 μM.
Saturation experiments showed that betaxolol increased the KD of [3H]-BTX-B binding but had no effect on the Bmax. The association kinetics of [3H]-BTX-B were unaffected by betaxolol, but the drug significantly accelerated the dissociation rate of the radioligand. These findings argue for a competitive, indirect, allosteric mode of inhibition of [3H]-BTX-B binding by betaxolol.
Betaxolol inhibited veratridine-stimulated Na+ influx in rat cortical synaptosomes with an IC50 value of 28.3 μM. Carteolol, levobunolol, timolol and atenolol were significantly less effective than betaxolol at reducing veratridine-evoked Na+ influx.
The ability of betaxolol to interact with neurotoxin site 2 of the Na+ channel and inhibit Na+ influx may have a role in its neuroprotective action in paradigms of excitotoxicity/ischaemia and in its therapeutic effect in glaucoma.
Keywords: betaxolol, Na+ influx, voltage-sensitive sodium channel, batrachotoxin, beta-adrenoceptor antagonist, glaucoma, neuroprotection
Introduction
β-Adrenoceptor antagonists are the most commonly used class of antiglaucoma agents. Their mechanism of action, like that of all antiglaucoma drugs, involves lowering intraocular pressure. However, while raised intraocular pressure is clearly a significant risk factor for glaucoma, much evidence has accumulated which suggests that targeting intraocular pressure alone is not sufficient to prevent death of retinal neurons and consequent loss of visual field. Accordingly, a consensus is evolving which proposes that treatment strategies may be better focused on neuroprotection. In glaucoma, death of retinal ganglion cells, and their axons that form the optic nerve, occurs over a long period of time via a cascade of events which is little understood. Nevertheless, evidence for an ischaemic/excitotoxic-like pathway of ganglion cell death in glaucoma, with some similarities to that occurring in the brain after a stroke or heart attack, is accumulating (Osborne et al., 1999c; Vorwerk et al., 1999).
Recent work indicates that the β1-adrenoceptor antagonist betaxolol is a retinal neuroprotective agent in animals, since topical or i.p. administration of the drug attenuates excitotoxicity-induced damage to ganglion cells and the effects of ischaemia to the retina (Osborne et al., 1997; 1999b). No similar data have been reported for other β-blockers, such as timolol, levobunolol and carteolol, commonly prescribed for glaucoma. These data correlate well with clinical observations, which suggest that glaucoma patients treated with betaxolol fare often better than, and at least as well as, patients treated with timolol with regard to their visual fields (Messmer et al., 1991; Kaiser et al., 1992; 1994; Collignon Brach, 1994; Drance, 1998). Since timolol reduces intraocular pressure more effectively than betaxolol in humans (Sorensen & Abel, 1996), the combined data support the argument that factors other than elevated pressure contribute to glaucomatous optic neuropathy. Various studies have shown that betaxolol, in common with many β-blockers, acts as a calcium channel blocker (Setoguchi et al., 1995). Importantly, of all the β-blockers used in the treatment of glaucoma, betaxolol is the most effective calcium channel blocker (Yu et al., 1999). Moreover, recent work has shown that betaxolol interacts directly with L-type calcium channels (Melena et al., 1999). Since various studies have shown that calcium channel blockers can attenuate NMDA- or ischaemia-induced insults to neurons (Kobayashi & Mori, 1998), it follows that the neuroprotective properties of betaxolol may well be associated with its calcium channel blocking activity (Osborne et al., 1999b). However, the potency of betaxolol in reducing calcium influx into neurons may not be sufficient to account for its complete neuroprotective action, and furthermore there is little evidence that L-type voltage-gated calcium channel blockers are effective neuroprotectants in the retina. For this reason, it was postulated that additional mechanisms of neuroprotection for betaxolol must be considered.
Voltage-sensitive sodium channels (Na+ channels) are responsible for the generation and propagation of action potentials in axons. They comprise three subunits, but the physiological function is primarily subserved by the α-subunit. Interestingly, a considerable structural similarity exists between calcium channel α1-subunits and Na+ channel α-subunits and many calcium channel blockers have affinity for Na+ channels. There are three main types of Na+ channel α-subunits in the CNS, namely types I, II and III, each of which has been identified in rat retinal ganglion cells (Fjell et al., 1997). Several neurotoxins are known to interact with specific, allosterically coupled binding sites on the Na+ channel and alter channel function. At least six classes of toxin receptor sites are currently recognized, the most important of which are sites 1 and 2. Site 1 is thought to be located in the vestibule of the channel and binding of water soluble toxins such as saxitoxin and tetrodotoxin directly leads to inhibition of ion conductance. Site 2 is found in the transmembrane region and is involved in the gating of the channel. Lipophilic toxins including veratridine and batrachotoxin bind at site 2 causing a persistent activation of the channel, which can be reversed by tetrodotoxin in a non-competitive fashion. A number of local anaesthetics, antiarrhythmics and anticonvulsants reduce neuronal excitability through an indirect negative allosteric interaction with receptor site 2 of the Na+ channel (Catterall, 1987). Moreover, some β-adrenoceptor antagonists possess local anaesthetic activity and these compounds are known to interact with the batrachotoxin binding site (Ijzerman et al., 1987).
A variety of sodium channel blockers have been shown to offer protection to neurons in experimental models of cerebral ischaemia and in models of white matter damage (see Narahashi et al., 1997; Carter, 1998; Obrenovitch, 1998). More pertinent with respect to the retina, anoxic injury to the rat optic nerve can be ameliorated by exposure to the sodium channel blocker tetrodotoxin (Stys et al., 1992b; Waxman et al., 1994) and by local anaesthetics (Stys et al., 1992a), which, as mentioned previously, inhibit toxin binding to site 2.
Given the known affinity of some β-adrenoceptor antagonists for Na+ channels, the affinity of betaxolol for L-type calcium channels and the neuroprotective action of the drug, the aim of this study was to examine whether betaxolol, and other β-adrenoceptor antagonists currently used as antiglaucoma agents, interact directly with Na+ channels. This was achieved by examining their effects on the binding of [3H]-batrachotoxinin ([3H]-BTX-B) and [3H]-saxitoxin ([3H]-STX) and on Na+ influx in rat cortex.
Methods
[3H]-BTX-B binding assays
Preparation of cerebrocortical membranes
Adult Wistar rats (250–350 g) were killed by decapitation and the cerebral cortex separated from other brain regions over ice. The tissue was roughly chopped and homogenized in 10 volumes of ice-cold 0.32 M sucrose, 5 mM K2HPO4, pH 7.4 using a motor-driven polytetrafluoroethylene-glass homogenizer. The homogenate was centrifuged at 1000×g for 10 min at 4°C and the resulting supernatant centrifuged at 39,000×g for 20 min at 4°C. The pellet was resuspended in Na+ free buffer (in mM: choline chloride 130, KCl 5.4, MgSO4 0.8, D-glucose 5.5, HEPES-Tris 50, pH 7.4) and recentrifuged at 39,000×g for 20 min at 4°C. The pellet was resuspended in Na+ free buffer at an approximate protein concentration of 3 mg ml−1, snap frozen in liquid N2 and stored at −80°C until required. Protein concentration was determined using a Bicinchoninic acid protein assay kit (Sigma).
Equilibrium binding assays
[3H]-BTX-B binding was determined essentially as described by Shimidzu et al. (1997). Briefly, aliquots of cortical membranes (200–400 μg protein) were incubated for 60 min at 37°C in Na+ free buffer containing 10 nM (inhibition experiments) or 2.5–80 nM (saturation experiments) [3H]-BTX-B, 1 μM tetrodotoxin, 30 μg scorpion venom (Leiurus quinquestriatus) and 1 mg ml−1 BSA, with or without the drug to be tested. Nonspecific binding was measured in the presence of 300 μM veratridine. The binding reactions were terminated by the addition of 3 ml of ice-cold washing buffer (in mM: choline chloride 163, MgCl2 0.8, CaCl2 1.8, HEPES-Tris 5, pH 7.4). Following rapid vacuum filtration through Whatman GF/B glass fibre filters, the samples were washed a further three times with the same buffer. Bound radioactivity was measured by liquid scintillation spectrometry in 5 ml of Insta-gel Plus.
Kinetic binding assays
The rates of dissociation and association of [3H]-BTX-B from cerebrocortical Na+ channel binding sites were determined essentially as described by Ratnakumari & Hemmings (1996). For dissociation experiments, aliquots of cortical membranes were incubated for 60 min at 37°C with 10 nM [3H]-BTX-B, 30 μg scorpion venom, 1 μM tetrodotoxin and 1 mg ml−1 BSA as in the equilibrium assays. Dissociation was initiated by addition of 300 μM veratridine±betaxolol (10 or 100 μM) at time zero. Reactions were stopped at 5, 10, 20, 30 and 45 min as for equilibrium experiments.
The rate of association of [3H]-BTX-B was measured by incubating aliquots of cortical membranes in Na+ free buffer containing 1 μM tetrodotoxin, 1 mg ml−1 BSA and 30 μg scorpion venom for 15 min at 37°C in the absence or presence of 100 μM betaxolol. [3H]-BTX-B (10 nM) was then added and the incubations terminated after 5, 10, 20, 30 and 60 min as for equilibrium experiments. Non-specific binding was determined in parallel at each time point.
[3H]-STX binding assays
Cortical membranes were prepared as for the [3H]-BTX-B binding assay (see above) and the [3H]-STX assay performed essentially as described by Shimidzu et al. (1997). To each tube was added 50 μl of [3H]-STX (2.5 nM final concentration), 50 μl of Na+ free buffer or drug to be tested and, to initiate the assay, 100 μl of membrane suspension (200–400 μg protein). Incubation occurred at 37°C for 30 min and binding reactions were terminated as described for [3H]-BTX-B assays. Specific binding was defined as the difference between total binding and binding obtained in the presence of 1 μM tetrodotoxin.
Measurement of Na+ influx
Preparation of synaptoneurosomes
Synaptosomes were prepared according to the method of Dunkley et al. (1988). Briefly, adult male Wistar rats were killed by decapitation. Brains were rapidly removed and rinsed in ice-cold gradient buffer consisting of 0.32 M sucrose, 1 mM EDTA and 0.25 mM dithiothreitol, pH 7.4. The cerebral cortex was separated and homogenized in 10 volumes of gradient buffer using a motor-driven polytetrafluoroethylene-glass homogenizer. The homogenate was centrifuged at 1000×g for 10 min at 4°C and the resultant supernatant collected and diluted with gradient buffer to yield a protein concentration of approximately 5 mg ml−1. Aliquots (2 ml) of this fraction were layered onto discontinuous Percoll gradients consisting of four 2.5 ml layers of 3, 10, 15 and 23% (v v−1) filtered (0.45 μm) Percoll gradient buffer. The gradients were centrifuged at 30,000×g for 4.5 min at 4°C. The synaptosomal fraction was collected from the 23/15% interface and diluted approximately 5 fold in ice-cold low Na+ buffer (in mM: choline chloride 130, KCl 5.4, MgSO4 0.8, D-glucose 5.5, NaCl 5, HEPES-Tris 50, pH 7.4), which was previously bubbled for 1 h with 95% O2/5% CO2. The synaptosomes were centrifuged at 25,000×g for 10 min at 4°C and resuspended in low Na+ buffer to give a final protein concentration of 3.5–4.5 mg ml−1.
Na+ influx
Na+ uptake was determined by a modification of the method described by Tamkun & Catterall (1981). Aliquots of freshly prepared synaptosomes containing approximately 350–450 μg of proteins were preincubated at 37°C for 10 min with or without test agents. Following preincubation, 0.5 μCi of 22Na+ in low Na+ buffer was added and the samples were incubated for 10 min at 37°C. Uptake was initiated by the addition of 100 μM veratridine and terminated after 30 s by the addition of 3 ml of ice-cold washing. Samples were rapidly vacuum filtered through Whatman GF/B filters presoaked for 2 h in 0.1% polyethylenimine and washed three times with 3 ml of ice-cold washing buffer. Trapped radioactivity was measured by liquid scintillation spectrometry in 5 ml of Insta-gel Plus. Non-specific uptake of Na+ was determined in the presence of 1 μM tetrodotoxin.
Analysis of data
The Hill coefficients and IC50 values for competition binding data were obtained using a nonlinear method (GraphPad Prism 1.0). The dissociation rate constant (K−1) was calculated from linear regression analysis of ln(Bt/B0) versus t, where Bt and B0 are the amount of radioligand bound at t and t zero, respectively. The association rate constant (K+1) was determined using the equation K+1=(Kobs–K−1)/[L]. The observed rate constant (Kobs) was calculated from linear regression analysis of ln(Beq/Beq–Bt) versus t, where Bt and Beq are the amount of radioligand bound at t and equilibrium, respectively, and [L] is the concentration of [3H]-BTX-B. The slope of the plot (Kobs)=[L]K+1+K−1, from which K+1 could be calculated. Statistical analysis was performed by analysis of variance (ANOVA) with the Bonferroni post-test or by Student's t-test.
Materials
[3H]-batrachotoxinin-A 20-α-benzoate (34 Ci mmol−1) and [22NaCl] (1 mCi ml−1) were obtained from NEN Research Products (Stevenage, U.K.) and [11-3H]-saxitoxin diacetate (14.9 Ci mmol−1) from Amersham (Amersham, U.K.). Betaxolol and levobetaxolol hydrochlorides were supplied by Alcon Research Laboratories (Fort Worth, TX, U.S.A.) and levobunolol hydrochloride by Allergan Pharmaceuticals Ltd. (Westport, Ireland). Teoptic® (CIBA Vision Ophthalmics, Hedge End, U.K.) was used as a source of carteolol hydrochloride. All other drugs and reagents were purchased from Sigma (Poole, U.K.) except for tetrodotoxin and Insta-gel Plus which were from Semat Technical Ltd. (St Albans, U.K.) and Packard (Groningen, The Netherlands), respectively.
Results
Effect of beta-adrenoceptor antagonists on [3H]-BTX-B binding
The effects of various β-adrenoceptor antagonists on specific binding of [3H]-BTX-B are shown in Figure 1. All of the drugs tested inhibited binding completely in a concentration-dependent fashion with the exception of atenolol, which even at 1 mM was only weakly active, while none of the drugs displaced non-specific binding. A potency order of propranolol>racemic betaxolol≈levobetaxolol>levobunolol≈carteolol⩾timolol>atenolol was obtained. No stereoselectivity was evident with regard to the effect of betaxolol. The IC50 values for the inhibition of [3H]-BTX-B binding are shown in Table 1. Since the concentration of [3H]-BTX-B used is substantially less than its KD, the IC50 values obtained are effectively equal to the Ki values. Hill coefficients are all close to unity indicating the likelihood of competitive inhibition. The relatively high affinity of propranolol for [3H]-BTX-B binding is well-documented (Postma & Catterall, 1984; Grima et al., 1987) and explained by its action as a local anaesthetic. However, the potency of betaxolol was considerably higher than for the other β-antagonists and accordingly its effect on [3H]-BTX-B binding was examined in more detail.
Figure 1.
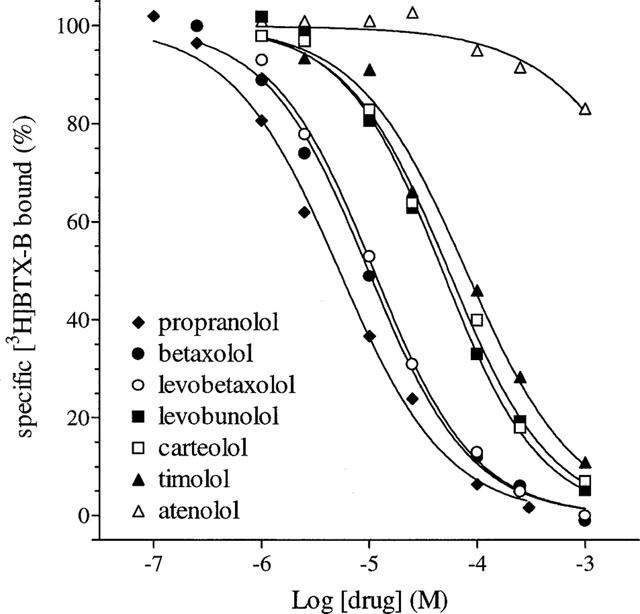
Concentration-response curves for the inhibition of specific binding of 10 nM [3H]-BTX-B to rat cortical membranes by propranolol, betaxolol, levobetaxolol, levobunolol, carteolol, timolol and atenolol. Each curve is the mean of 3–4 independent experiments performed in duplicate as described in Methods. Error bars have been removed for the sake of clarity. The s.e.means ranged from 0.2–8.1.
Table 1.
Affinity values for the inhibitory actions of veratridine and various beta-adrenoceptor antagonists on the binding of [3H]-BTX-B to rat cerebrocortical membranes
Effect of betaxolol on saturation and kinetic properties of [3H]-BTX-B binding
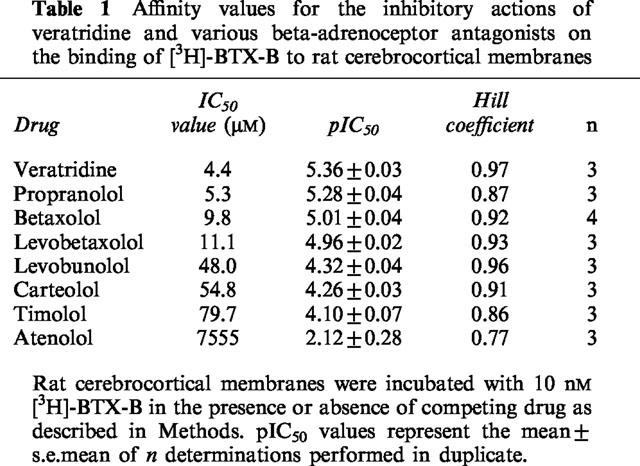
In order to explore further the mechanism of the interaction of betaxolol with neurotoxin site 2, the effect of the drug on saturation and kinetic properties of [3H]-BTX-B binding to cortical membranes was assessed. Saturation analysis of [3H]-BTX-B binding revealed a single class of binding sites with a KD of 29.5±1.24 nM (mean±s.e.mean) and a Bmax of 1.43±0.05 pmol mg−1 protein (Figure 2). Inclusion of 10 μM betaxolol in the assay resulted in a significant increase in the KD 55.7±8.4 nM (n=3; P<0.05) but little change in the Bmax (1.34±0.08 pmol mg−1 protein), suggesting that the inhibition was likely to be competitive in nature.
Figure 2.
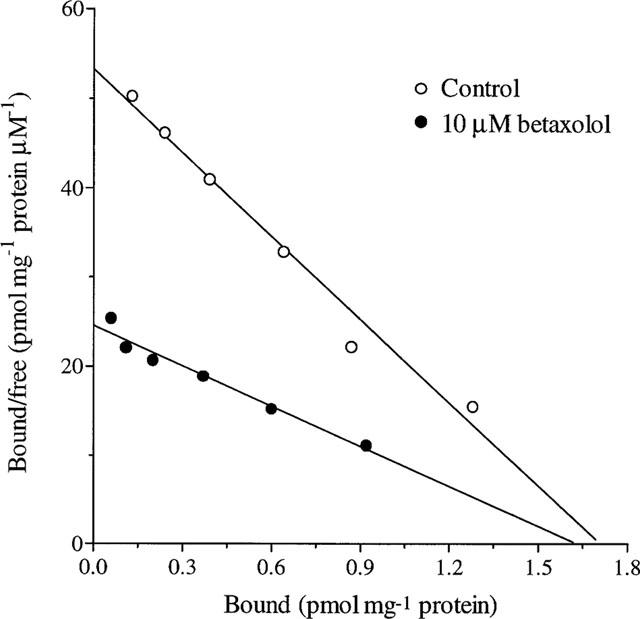
Scatchard analysis of the effect of betaxolol on the specific binding of [3H]-BTX-B to rat cortical membranes. Membranes were incubated in the presence or absence of 10 μM betaxolol as described in Methods. The results represent data from a single typical experiment performed in duplicate. Values for Bmax were 1.46 pmol mg−1 protein and 1.24 pmol mg−1 protein in the absence and presence of betaxolol, respectively. The KD values were 29.2 and 55.8 nM, respectively. Two additional experiments yielded similar results.
In kinetic studies, the dissociation of [3H]-BTX-B from cortical membranes in the presence of a saturating concentration of competitive ligand veratridine was relatively slow with K−1=0.0088±0.0007 min−1 (n=4; Figure 3). Co-incubation with betaxolol (10 μM) caused a slight but insignificant increase in the dissociation rate constant K−1=0.0113±0.0010 (n=4) and a 10 fold higher concentration of betaxolol (100 μM) increased the dissociation rate constant dramatically K−1=0.0241±0.0010 (n=4; P<0.01; Figure 3). In contrast to its effect on the rate of dissociation of [3H]-BTX-B from cortical membranes, betaxolol (up to 100 μM) had no effect on the rate of association of [3H]-BTX-B (K+1=0.0026±0.0006 and 0.0023±0.0020 min−1 nM−1 in the absence and presence of 100 μM betaxolol, respectively).
Figure 3.
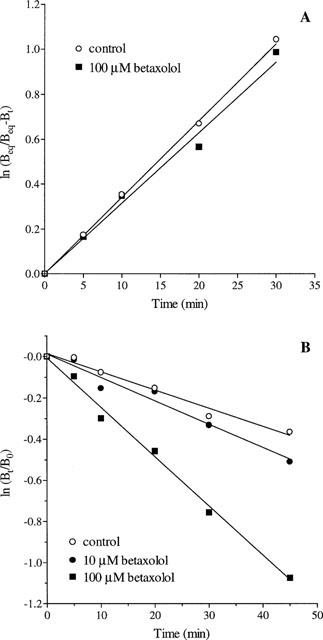
(A) Effect of betaxolol on the association rate of [3H]-BTX-B to rat cortical membranes. Membranes were incubated with 10 nM [3H]-BTX-B in the presence or absence of 100 μM betaxolol as described in Methods. (B) Effect of betaxolol on the dissociation rate of [3H]-BTX-B to rat cortical membranes. Membranes were incubated with 10 nM [3H]-BTX-B plus 300 μM veratridine in the absence or presence of 10 or 100 μM betaxolol as described in Methods. In (A) and (B) the results shown represent the mean of 3–4 independent experiments performed in duplicate. Error bars have been removed for the sake of clarity.
Effect of beta-adrenoceptor antagonists on [3H]-STX binding
As shown in Table 2, none of the β-adrenoceptor antagonists caused a significant inhibition of [3H]-STX binding in rat cortical membranes, even at concentrations as high as 250 μM. In contrast, tetrodotoxin potently displaced [3H]-STX binding with an IC50 value of 37 nM (data not shown). However, it should be noted that propranolol did reduce specific binding by 15%, a level of inhibition which may have reached significance with repeated measurement.
Table 2.
Effect of beta-adrenoceptor antagonists (250 μM) on the binding of [3H]-STX to rat cerebrocortical membranes

Effect of beta-adrenoceptor antagonists on 22Na+ influx
Veratridine (100 μM) caused a 3 fold increase in the basal uptake of 22Na+ into rat cortical synaptosomes from 35720±3020 c.p.m. min−1 mg protein−1 to 108060±7060 c.p.m. min−1 mg protein−1 (mean±s.e.mean; n=10) which was blocked by inclusion of tetrodotoxin (1 μM) in the assay (data not shown). Betaxolol had no effect on basal Na+ influx (data not shown) but inhibited veratridine-stimulated Na+ influx in a concentration-dependent manner (Figure 4) with an EC50 value of 28.3 μM (pIC50 4.55±0.05) and a Hill slope of 0.80. Comparison of the efficacy of various β-adrenoceptor antagonists at 100 μM on veratridine-stimulated Na+ influx into rat cortical synaptosomes revealed that atenolol, timolol, levobunolol and carteolol were significantly less active than betaxolol, whereas no statistically significant differences were observed between the effects of propranolol and levobetaxolol and that of betaxolol (see Table 3).
Figure 4.
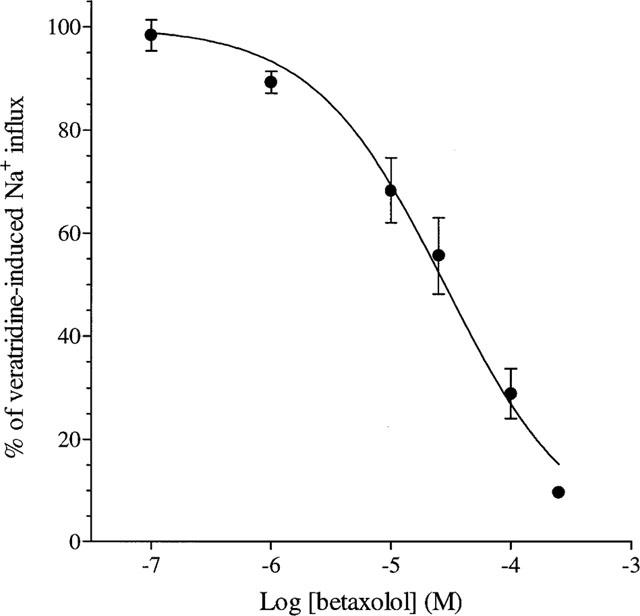
Concentration-response curve for the inhibition of specific veratridine-induced 22Na+ uptake into rat cortical synaptosomes by betaxolol. The results shown represent the mean±s.e.mean of four independent experiments performed in duplicate as described in Methods.
Table 3.
Effect of beta-adrenoceptor antagonists (100 μM) on the specific veratridine-induced 22Na+ uptake into rat cerebrocortical synaptosomes

Discussion
It has recently been demonstrated that betaxolol can interact directly with L-type Ca2+ channels in rat cortex (Melena et al., 1999). The data presented in this study show that betaxolol can interact with voltage-sensitive sodium channels on cortical synaptosomes and reduce Na+ influx. The affinity and manner by which betaxolol displaced [3H]-BTX-B binding in rat cortex (IC50=9.8 μM; nH=0.92) correlated well with the inhibitory action of the drug on veratridine-stimulated Na+ influx in the same tissue (IC50=28.3 μM; nH=0.80), suggesting that these effects occur at a single class of sites. However, the ligand had no effect on the binding of saxitoxin to receptor site 1. It is clear, therefore, that betaxolol does not interfere with ion conductance directly but rather modulates the gating mechanism of the Na+ channel. As very similar data were generated for racemic betaxolol and levobetaxolol, we can conclude that stereoselectivity is not important with regard to the affinity of betaxolol for the batrachotoxin site. Stereospecificity is discernible for some local anaesthetics, but like betaxolol, the d- and l-isomers of propranolol are equipotent at [3H]-BTX-B sites (Postma & Catterall, 1984).
The results of Scatchard analysis indicate that the inhibition of [3H]-BTX-B binding by betaxolol is competitive, since in the presence of betaxolol an increase in KD was recorded without a reduction in total binding capacity. Kinetic analysis showed that the association rate of the radioligand was unaffected by betaxolol, but the dissociation rate of [3H]-BTX-B was accelerated by increasing concentrations of the drug. These findings argue for an indirect allosteric mode of inhibition by betaxolol. In fact, the results we obtained with betaxolol are very similar to those reported by Ratnakumari & Hemmings (1996) for propofol. Our data thus support the view that allosteric inhibition of the interaction between activating neurotoxins and receptor site 2 by various drugs is best accounted for by selective high affinity binding to a hydrophobic site distinct from, yet coupled to, neurotoxin site 2. This binding site is able to interact with a number of chemically diverse drugs, including local anaesthetics, anti-convulsants and antiarrhythmics (Catterall, 1987), the general anaesthetic propofol (Ratnakumari & Hemmings, 1996), β-adrenoceptor antagonists (Ijzerman et al., 1987), calcium channel blockers (Grima et al., 1988) and neuroprotective compounds (Shimidzu et al., 1997). These drugs are thought to inhibit Na+ channel activity by preferential interaction with the inactivated state of the channel (Catterall, 1987).
The potency of betaxolol at cortical [3H]-BTX-B sites was similar to, and higher than, many drugs whose function is mediated by blockade of Na+ channels (Postma & Catterall, 1984). However, significant pharmacological differences are apparent between cardiac and neuronal isoforms of Na+ channels (Fozzard & Hanck, 1996). For example, local anaesthetics such as lidocaine and antiarrhythmics have a higher efficacy on heart rather than nerve Na+ channels, permitting drugs to be used to treat cardiac conditions without interfering with neuronal function. Conversely, the novel neuroprotective compound NS-7 inhibited [3H]-BTX-B binding in cardiac cells 13 fold less potently than in brain tissue (Shimidzu et al., 1997). Given the lack of antiarrhythmic activity of betaxolol, it would be expected that the drug is likewise significantly more active at brain rather than cardiac Na+ channels.
Interestingly, betaxolol displayed at least 5 fold higher potency for Na+ channels than the other commonly prescribed β-adrenoceptor antagonists used for the treatment of glaucoma (carteolol, timolol, levobunolol). The most logical explanation for the greater affinity of betaxolol is provided by the lipophilic nature of the compound. Figure 5 reveals that, with the exception of carteolol, a good correlation exists between the partition co-efficients of β-adrenoceptor antagonists and their potencies at rat cortical [3H]-BTX-B binding sites. Propranolol and betaxolol are highly lipophilic and potent displacers of [3H]-BTX-B binding, levobunolol and timolol are moderately lipophilic and are less potent inhibitors, while atenolol is hydrophilic and possesses little affinity for neurotoxin site 2. Lipophilicity is of prime importance in determining the affinity of various ligands for the [3H]-BTX-B binding site, which is understandable given its hydrophobic location within the membrane. Ijzerman et al. (1987) showed that lipophilicity is the key determinant as to whether β-adrenoceptor antagonists inhibit [3H]-BTX-B binding. However, in Ijzerman's study, lipophilicity did not account for all of the observed variance in binding affinities between different β-adrenoceptor antagonists. Similarly, in this study, propranolol displays higher potency for cortical [3H]-BTX-B binding sites than betaxolol, yet the compounds are equally lipophilic, while atenolol and carteolol are both hydrophilic and yet carteolol shows much greater affinity for Na+ channels. An explanation for this anomaly was provided by Ijzerman who showed that steric factors play a part in determining affinity of β-adrenoceptor antagonists for [3H]-BTX-B sites. Substitution para to the aliphatic side chain introduces not only β1-selectivity but also, through steric hindrance, a reduced affinity for [3H]-BTX-B binding sites. Betaxolol and atenolol have bulky para substituents and consequently are less effective Na+ channel blockers than propranolol and carteolol, respectively, which have no para substitution. In fact, the additional potency of propranolol over betaxolol at Na+ channels is presumably a contributory factor in the potent local anaesthetic properties displayed by propranolol. Local anaesthetic activity, also known as membrane stabilizing activity, is an undesirable side-effect of β-adrenoceptor antagonists used in glaucoma therapy and is manifested as a reduction in corneal sensitivity. Unlike propranolol, which rapidly induces corneal desensitization, betaxolol possesses only minor local anaesthetic properties, which do not prevent its use as a therapeutic agent (Buckley et al., 1990; Zimmerman, 1993).
Figure 5.
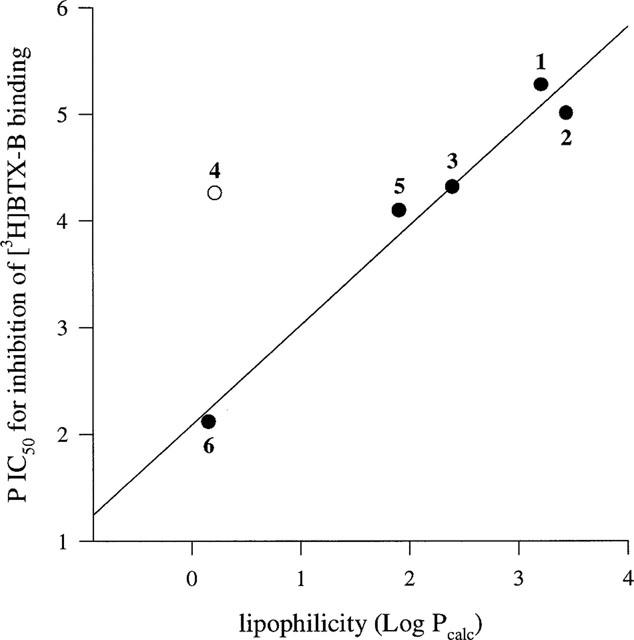
Linear relationship between calculated lipophilicities and experimentally determined pIC50 values for inhibition of [3H]-BTX-B binding to rat cerebrocortical membranes of various β-adrenoceptor antagonists. 1=propranolol, 2=betaxolol, 3=levobunolol, 4=carteolol, 5=timolol, 6=atenolol. Omission of carteolol from the regression analysis revealed a good correlation for the other β-adrenoceptor antagonists (r=0.97). Lipophilicities were obtained from Wang et al. (1991) and Sasaki et al. (1994).
The rationale for examining whether betaxolol can interact with Na+ channels stems from the considerable interest into whether drugs used for the treatment of glaucoma can act as neuroprotectants for the ganglion cells of the retina. In glaucoma, death of neurons occurs gradually over many years, and thus pursuing a strategy of neuroprotection should theoretically be more rewarding for this disease than for severe, acute conditions such as stroke (Osborne et al., 1999a). A wealth of indirect evidence points to a mild yet chronic ischaemic/hypoxic-like pathway being responsible for ganglion cell loss in glaucoma. It follows, therefore, that drugs which can diminish the intensity of such an insult should ameliorate to some extent the damage to ganglion cells and the optic nerve head. Reducing calcium influx is one potential strategy for achieving this aim and much evidence exists that betaxolol can act as a calcium channel blocker (Setoguchi et al., 1995). The finding of this study that betaxolol can inhibit Na+ influx through a direct interaction with the Na+ channel is not unsurprising given firstly, the impressive structural similarity that is evident between the Na+ and Ca2+ α-subunits (Fozzard & Hanck, 1996) and secondly, the known ability of many calcium channel blockers to bind with high affinity to Na+ channels (Velly et al., 1987; Grima et al., 1988). Na+ channel blockade is considered to be an effective means of prolonging the survival of cells during periods of physiological stress caused by limited oxygen and nutrient supply (Goldin et al., 1995; Urenjak & Obrenovitch, 1998). Direct support for the idea that a combination of Na+ and Ca2+ channel blockade is responsible for the neuroprotective action of betaxolol in retinal paradigms of ischaemia/excitotoxicity comes from the study of Gross et al. (1999). They showed that betaxolol reversibly reduced the voltage-gated sodium and calcium currents in retinal ganglion cells and reduced the rate of spontaneous ganglion cell firing induced by glutamate, while timolol, which displays a markedly inferior affinity for voltage-sensitive Na+ and Ca2+ channels relative to betaxolol had no effect on any of these currents. Indirect evidence comes from the knowledge that Na+ channel blockers, acting at either neurotoxin site 1 or site 2, are neuroprotective in the ischaemic rat optic nerve model (Stys et al., 1992a,1992b) and that betaxolol protects against NMDA-induced ganglion cell loss by a mechanism that does not involve either β-adrenoceptor antagonism (Osborne et al., 1997) or induction of basic fibroblast growth factor expression (unpublished observations). Despite the fact that propranolol cannot be used as an antiglaucoma agent due to its undesirable effects on the cornea, it would be interesting to determine whether the drug affords an equivalent degree of neuroprotection in paradigms of excitotoxicity/ischaemia to betaxolol, since the drugs displayed similar profiles in this study.
Finally it should be stated that betaxolol is known to reach the retina in significant amounts after topical administration (Osborne et al., 1999b) and owing to its highly lipophilic nature may well accumulate in cell membranes at sufficient quantities to interact with ion channels. As a consequence, it is not unlikely that the reason why glaucoma patients treated topically with betaxolol have better preserved visual fields than patients treated with other β-blockers is due to the action of betaxolol on Na+ and Ca2+ channels. Nevertheless, without detailed knowledge of the exact concentration of betaxolol which accumulates in the immediate vicinity of the ganglion cells or their axons in patients after topical administration this theory will remain unproved.
In conclusion, this study demonstrates that betaxolol displaces [3H]-BTX-B binding and reduces Na+ influx in rat cortex. The compound is more effective than other β-adrenoceptor antagonists currently used in glaucoma therapy but less potent than the combined local anaesthetic/β-adrenoceptor antagonist propranolol. Future work is needed to examine the effect of betaxolol and other β-adrenoceptor antagonists on Na+ channels within the retina.
Acknowledgments
The authors are grateful to the British Council for the Prevention of Blindness for providing financial support, to Dr L. DeSantis of Alcon Research Laboratories (Fort Worth, TX, U.S.A.) for his scholarly and financial assistance and to Allergan Pharmaceuticals Ltd. (Westport, Ireland) for a supply of levobunolol hydrochloride. Dr J. Melena is supported by a post-doctoral Marie Curie grant (TMR programme, European Commission).
Abbreviations
- [3H]-BTX-B
[3H]-batrachotoxinin-A 20-α-benzoate
- [3H]-STX
[11-3H]-saxitoxin diacetate
References
- BUCKLEY M.M., GOA K.L. , CLISSOLD S.P. Ocular betaxolol. A review of its pharmacological properties, and therapeutic efficacy in glaucoma and ocular hypertension. Drugs. 1990;40:75–90. doi: 10.2165/00003495-199040010-00005. [DOI] [PubMed] [Google Scholar]
- CARTER A.J. The importance of voltage-dependent sodium channels in cerebral ischaemia. Amino Acids. 1998;14:159–169. doi: 10.1007/BF01345257. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Common modes of drug action on Na(+) channels: Local anesthetics, antiarrhythmics and anticonvulsants. Trends. Pharmacol. Sci. 1987;8:57–65. [Google Scholar]
- COLLIGNON BRACH J. Longterm effect of topical beta-blockers on intraocular pressure and visual field sensitivity in ocular hypertension and chronic open-angle glaucoma. Surv. Ophthalmol. 1994;38 Suppl:S149–S155. doi: 10.1016/0039-6257(94)90059-0. [DOI] [PubMed] [Google Scholar]
- DRANCE S.M. A comparison of the effects of betaxolol, timolol, and pilocarpine on visual function in patients with open-angle glaucoma. J. Glaucoma. 1998;7:247–252. doi: 10.1097/00061198-199808000-00006. [DOI] [PubMed] [Google Scholar]
- DUNKLEY P.R., HEATH J.W., HARRISON S.M., JARVIE P.E., GLENFIELD P.J. , ROSTAS J.A. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: homogeneity and morphology of subcellular fractions. Brain Res. 1988;441:59–71. doi: 10.1016/0006-8993(88)91383-2. [DOI] [PubMed] [Google Scholar]
- FJELL J., DIB HAJJ S., FRIED K., BLACK J.A. , WAXMAN S.G. Differential expression of sodium channel genes in retinal ganglion cells. Brain Res. Mol. Brain Res. 1997;50:197–204. doi: 10.1016/s0169-328x(97)00187-3. [DOI] [PubMed] [Google Scholar]
- FOZZARD H.A. , HANCK D.A. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol. Rev. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- GOLDIN S.M., SUBBARAO K., SHARMA R., KNAPP A.G., FISCHER J.B., DALY D., DURANT G.J., REDDY N.L., HU L.Y., MAGAR S., PERIMAN M.E., CHEN J., GRAHAM S.H., HOLT W.F., BERLOVE D. , MARGOLIN L.D. Neuroprotective use-dependent blockers of Na+ and Ca2+ channels controlling presynaptic release of glutamate. Ann. N.Y. Acad. Sci. 1995;765:210–229. doi: 10.1111/j.1749-6632.1995.tb16578.x. [DOI] [PubMed] [Google Scholar]
- GRIMA M., FREYSS BEGUIN M., MILLANVOYE VAN BRUSSEL E., DECKER N. , SCHWARTZ J. Effects of various antianginal drugs on sodium influx in rat brain synaptosomes and in rat heart muscle cells in culture. Eur. J. Pharmacol. 1987;138:1–8. doi: 10.1016/0014-2999(87)90330-x. [DOI] [PubMed] [Google Scholar]
- GRIMA M., VELLY J., DECKER N., MARCINIAK G. , SCHWARTZ J. Inhibitory effects of some cyclohexylaralkylamines related to perhexiline on sodium influx, binding of [3H]batrachotoxinin A 20-alpha-benzoate and [3H]nitrendipine and on guinea pig left atria contractions. Eur. J. Pharmacol. 1988;147:173–185. doi: 10.1016/0014-2999(88)90776-5. [DOI] [PubMed] [Google Scholar]
- GROSS R.L., HENSLEY S.H. , WU S.M. Retinal ganglion cell dysfunction induced by hypoxia and glutamate: potential neuroprotective effects of β-blockers. Surv. Ophthalmol. 1999;43:S162–S170. doi: 10.1016/s0039-6257(99)00054-5. [DOI] [PubMed] [Google Scholar]
- IJZERMAN A.P., NAGESSER A. , GARRITSEN A. The membrane stabilizing activity of beta-adrenoceptor ligands. Quantitative evaluation of the interaction of phenoxypropanolamines with the [3H]batrachotoxinin A 20-alpha-benzoate binding site on voltage-sensitive sodium channels in rat brain. Biochem. Pharmacol. 1987;36:4239–4244. doi: 10.1016/0006-2952(87)90664-2. [DOI] [PubMed] [Google Scholar]
- KAISER H.J., FLAMMER J., MESSMER C., STUMPFIG D. , HENDRICKSON P. Thirty-month visual field follow-up of glaucoma patients treated with beta-blockers. J. Glaucoma. 1992;1:153–155. doi: 10.1016/0039-6257(94)90060-4. [DOI] [PubMed] [Google Scholar]
- KAISER H.J., FLAMMER J., STUMPFIG D. , HENDRICKSON P. Longterm visual field follow-up of glaucoma patients treated with beta-blockers. Surv. Ophthalmol. 1994;38 Suppl:S156–S159. doi: 10.1016/0039-6257(94)90060-4. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI T. , MORI Y. Ca2+ channel antagonists and neuroprotection from cerebral ischemia. Eur. J. Pharmacol. 1998;363:1–15. doi: 10.1016/s0014-2999(98)00774-2. [DOI] [PubMed] [Google Scholar]
- MELENA J., WOOD J.P.M. , OSBORNE N.N. Betaxolol, a β1-adrenoceptor antagonist, has an affinity for L-type Ca2+ channels. Eur. J. Pharmacol. 1999;378:317–322. doi: 10.1016/s0014-2999(99)00459-8. [DOI] [PubMed] [Google Scholar]
- MESSMER C., FLAMMER J. , STUMPFIG D. Influence of betaxolol and timolol on the visual fields of patients with glaucoma. Am. J. Ophthalmol. 1991;112:678–681. doi: 10.1016/s0002-9394(14)77274-5. [DOI] [PubMed] [Google Scholar]
- NARAHASHI T., HUANG C.S., SONG J.H. , YEH J.Z. Ion channels as targets for neuroprotective agents. Ann. N.Y. Acad. Sci. 1997;825:380–388. doi: 10.1111/j.1749-6632.1997.tb48448.x. [DOI] [PubMed] [Google Scholar]
- OBRENOVITCH T.P. Neuroprotective strategies: voltage-gated Na+-channel down-modulation versus presynaptic glutamate release inhibition. Rev. Neurosci. 1998;9:203–211. doi: 10.1515/revneuro.1998.9.3.203. [DOI] [PubMed] [Google Scholar]
- OSBORNE N.N., CAZEVIEILLE C., CARVALHO A.L., LARSEN A.K. , DESANTIS L. In vivo and in vitro experiments show that betaxolol is a retinal neuroprotective agent. Brain Res. 1997;751:113–123. doi: 10.1016/s0006-8993(96)01393-5. [DOI] [PubMed] [Google Scholar]
- OSBORNE N.N., CHIDLOW G., NASH M.S. , WOOD J.P.M. The potential of neuroprotection in glaucoma treatment. Curr. Opin. Ophthalmol. 1999a;10:82–92. doi: 10.1097/00055735-199904000-00002. [DOI] [PubMed] [Google Scholar]
- OSBORNE N.N., DE SANTIS L.M., BAE J.H., UGARTE M., WOOD J.P.M., NASH M.S. , CHIDLOW G. Topically applied betaxolol attenuates NMDA-induced toxicity to ganglion cells and the effects of ischaemia to the retina. Exp. Eye Res. 1999b;69:331–342. doi: 10.1006/exer.1999.0706. [DOI] [PubMed] [Google Scholar]
- OSBORNE N.N., UGARTE M., CHAO M., CHIDLOW G., BAE J.H., WOOD J.P. , NASH M.S. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Surv. Ophthalmol. 1999c;43 Suppl 1:S102–S128. doi: 10.1016/s0039-6257(99)00044-2. [DOI] [PubMed] [Google Scholar]
- POSTMA S.W. , CATTERALL W.A. Inhibition of binding of [3H]batrachotoxinin A 20-alpha-benzoate to sodium channels by local anesthetics. Mol. Pharmacol. 1984;25:219–227. [PubMed] [Google Scholar]
- RATNAKUMARI L. , HEMMINGS H. C., JR Inhibition by propofol of [3H]-batrachotoxinin-A 20-alpha-benzoate binding to voltage-dependent sodium channels in rat cortical synaptosomes. Br. J. Pharmacol. 1996;119:1498–1504. doi: 10.1111/j.1476-5381.1996.tb16064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SASAKI H., IGARASHI Y., NISHIDA K. , NAKAMURA J. Intestinal permeability of ophthalmic beta-blockers for predicting ocular permeability. J. Pharm. Sci. 1994;83:1335–1338. doi: 10.1002/jps.2600830926. [DOI] [PubMed] [Google Scholar]
- SETOGUCHI M., OHYA Y., ABE I. , FUJISHIMA M. Inhibitory action of betaxolol, a beta 1-selective adrenoceptor antagonist, on voltage-dependent calcium channels in guinea-pig artery and vein. Br. J. Pharmacol. 1995;115:198–202. doi: 10.1111/j.1476-5381.1995.tb16339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIDZU T., ITOH Y., TATSUMI S., HAYASHI S., UKAI Y., YOSHIKUNI Y. , KIMURA K. Blockade of voltage-sensitive sodium channels by NS-7, a novel neuroprotective compound, in the rat brain. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;355:601–608. doi: 10.1007/pl00004990. [DOI] [PubMed] [Google Scholar]
- SORENSEN S.J. , ABEL S.R. Comparison of the ocular beta-blockers. Ann. Pharmacother. 1996;30:43–54. doi: 10.1177/106002809603000109. [DOI] [PubMed] [Google Scholar]
- STYS P.K., RANSOM B.R. , WAXMAN S.G. Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. J. Neurophysiol. 1992a;67:236–240. doi: 10.1152/jn.1992.67.1.236. [DOI] [PubMed] [Google Scholar]
- STYS P.K., WAXMAN S.G. , RANSOM B.R. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J. Neurosci. 1992b;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAMKUN M.M. , CATTERALL W.A. Ion flux studies of voltage-sensitive sodium channels in synaptic nerve-ending particles. Mol. Pharmacol. 1981;19:78–86. [PubMed] [Google Scholar]
- URENJAK J. , OBRENOVITCH T.P. Neuroprotection – rationale for pharmacological modulation of Na+-channels. Amino Acids. 1998;14:151–158. doi: 10.1007/BF01345256. [DOI] [PubMed] [Google Scholar]
- VELLY J., GRIMA M., MARCINIAK G., SPACH M.O. , SCHWARTZ J. Effects of some antianginal and vasodilating drugs on sodium influx and on the binding of 3H-batrachotoxinin-A 20-alpha-benzoate and 3H-tetracaine. Naunyn-Schmiedeberg's Arch. Pharmacol. 1987;335:176–182. doi: 10.1007/BF00177720. [DOI] [PubMed] [Google Scholar]
- VORWERK C.K., GORLA M.S.R. , DREYER E.B. An experimental basis for implicating excitotoxicity in glaucomatous optic neuropathy. Surv. Ophthalmol. 1999;43:S142–S150. doi: 10.1016/s0039-6257(99)00017-x. [DOI] [PubMed] [Google Scholar]
- WANG W., SASAKI H., CHIEN D.S. , LEE V.H. Lipophilicity influence on conjunctival drug penetration in the pigmented rabbit: a comparison with corneal penetration. Curr. Eye Res. 1991;10:571–579. doi: 10.3109/02713689109001766. [DOI] [PubMed] [Google Scholar]
- WAXMAN S.G., BLACK J.A., RANSOM B.R. , STYS P.K. Anoxic injury of rat optic nerve: ultrastructural evidence for coupling between Na+ influx and Ca(2+)-mediated injury in myelinated CNS axons. Brain Res. 1994;644:197–204. doi: 10.1016/0006-8993(94)91680-2. [DOI] [PubMed] [Google Scholar]
- YU D.Y., SU E.N., CRINGLE S.J., ALDER V.A., YU P.K. , DESANTIS L. Systemic and ocular vascular roles of the antiglaucoma agents beta-adrenergic antagonists and Ca2+ entry blockers. Surv. Ophthalmol. 1999;43 Suppl 1:S214–S222. doi: 10.1016/s0039-6257(99)00042-9. [DOI] [PubMed] [Google Scholar]
- ZIMMERMAN T.J. Topical ophthalmic beta blockers: a comparative review. J. Ocul. Pharmacol. 1993;9:373–384. doi: 10.1089/jop.1993.9.373. [DOI] [PubMed] [Google Scholar]