

Abstract
A newly synthesized benzothiazepine derivative, JTV-519 (JT) has been reported to be cardioprotective. However, the precise mechanism underlying the cardioprotective effect of this drug is unknown.
Coronary-perfused guinea-pig ventricular muscles were subjected to 20-min no-flow ischaemia followed by 60-min reperfusion (I/R). I/R significantly decreased the contraction in untreated preparations (control group, 34±4% of baseline value, n=6). Brief administration of JT (1.0 μM) prior to ischaemia significantly improved the postischaemic contractile recovery (63±5% of baseline value, n=4), as compared to the control group.
JT (1.0 μM) slightly prolonged action potential duration before ischaemia and induced conduction disturbance (2 : 1 block) after the initiation of ischaemia.
The cardioprotective effect of JT was antagonized by chelerythrine (CH, 5.0 μM), an inhibitor of protein kinase C (PKC) or by 5-hydroxydecanoic acid (5-HD, 400 μM), an inhibitor of mitochondrial ATP-sensitive K+ (KATP) channels.
These results suggest that the protective effect of JT is due to the opening of mitochondrial KATP channels, which, in turn, is linked to PKC activation.
Keywords: Pharmacological preconditioning, protein kinase C, mitochondria, ATP-sensitive K+ channels
Introduction
Numerous studies have reported that diltiazem, a benzothiazepine-derivative calcium antagonist, exerts cardioprotective actions against ischaemia-reperfusion (I/R) injuries (Bush et al., 1982; Nasa et al., 1990). Recently, a newly synthesized benzothiazepine derivative, JTV-519 (JT) (4-[3-(4-benzylpiperidin - 1 - yl)propionyl]- 7-methoxy- 2,3,4,5- tetrahydro- 1,4- benzothiazepine monohydrochloride) has been shown to prevent cardiac cells from injury caused by experimental calcium overload (Kaneko, 1994). In the rat Langendorff heart model, this drug is useful for protecting the myocardium from damage caused by 6-h ischaemia (Hachida et al., 1997). However, the mechanisms underlying the protective action of JT are not clear and may not be explained solely by the possible blockade of Ca2+ channels. In our preliminary experiment, JT significantly improved postischaemic contractile recovery even when this drug was applied briefly just before ischaemia. In contrast, the administration of diltiazem applied just before ischaemia did not show such cardioprotective action (Ito et al., 1998). Therefore it was assumed that the cardioprotective effects of JT are not solely due to the Ca2+ channel blocking action. There is a number of evidence that ischaemic preconditioning provides marked cardioprotection. This phenomenon was originally reported by Murry et al. (1986), who found that a brief period of ischaemia markedly decreased the myocardial-infarct size after subsequent prolonged ischaemia. Ischaemic preconditioning can be reproduced even if the brief period of ischaemia is replaced by certain drugs or bioactive substances (pharmacological preconditioning), e.g., adenosine (Liu et al., 1991), noradrenaline (Kitakaze et al., 1994), or bradykinin (Goto et al., 1995). Although the mechanism of preconditioning is still under debate, the agonist-mediated activation of protein kinase C (PKC) seems to play a pivotal role in this protection (Ytrehus et al., 1994) and a downstream cascade may be related to the activation of ATP-sensitive K+ (KATP) channels (Liu et al., 1996; Hu et al., 1996). Recently, it has been suggested that the end effector of ischaemic preconditioning is mitochondrial KATP channels rather than sarcolemmal KATP channels (Garlid et al., 1997; Liu et al., 1998). Since a brief application of JT prior to ischaemia improved the postischaemic recovery of contraction, we hypothesized that the cardioprotective effect of JT may be similar to that seen in pharmacological preconditioning. The purpose of the present study is to investigate the cardioprotective effects of JT in an isolated heart model. The mechanisms underlying the potentially beneficial effects of JT are also examined with the use of chelerythrine, a potent PKC inhibitor (Herbert et al., 1990), and 5-hydroxydecanoic acid (5-HD), a selective mitochondrial KATP channel blocker (Sato et al., 1998; Hu et al., 1999).
Methods
The present investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1985). All procedures were in accordance with the guidelines stipulated by the Physiological Society of Japan and the Animal Ethics Committee of Oita Medical University.
Preparation
The coronary-perfused, isolated right-ventriclar muscle preparations were prepared from guinea-pig hearts, as described previously (Shigematsu et al., 1995). The preparation, in which the coronary artery was connected to a cannula by the aortic root, was mounted in the recording chamber and pinned at the base of the ventricle, the coronary perfusate was then delivered through a roller pump (MP-3, Tokyo Rikakikai Co., Tokyo). The flow rate was maintained at 1.0±0.2 ml min−1 g wet wt−1, with an intra-aortic perfusion pressure ranging from 40–50 mmHg. The preparation was superfused with substrate-free hypoxic Tyrode solution (10 ml min−1) gassed with nitrogen to minimize direct oxygen diffusion into the muscles from the epicardial and endocardial surfaces of the preparation. The temperature of these solutions was maintained at 37°C. These procedures ensured a sufficient supply of oxygen and substrates to the tissue, because normal action potentials could be recorded even from very marginal portions of the preparation, and a constant amplitude of twitch contractions was recorded for more than 2 h after 90-min equilibration period.
Electromechanical recordings
The heart was stimulated at 3 Hz throughout the experiment with the use of a pair of platinum electrodes, attached to the basal portion of the preparation and connected to the isolation unit of an electronic stimulator (SEN-3201, SS-302J, Nihon Kohden, Tokyo). Square pulses of 5-ms duration, with a pulse strength 1.5 fold greater than threshold, were used to drive the preparation. Action potentials were recorded from a fibre that was located deep in the subepicardial surface by a flexibly mounted microelectrode. Microelectrodes (tip resistance, 20–30 MΩ) were pulled from filamented borosilicate glass and filled with 3 M KCl. A DC preamplifier (MEZ-7101, Nihon Kohden) with capacitance compensation was used to record the transmembrane potential. The developed tension (dTension) was recorded using a force transducer (TB-612T, Nihon Kohden) connected to the apical end of the preparation. The resting tension (rTension) was adjusted to obtain the optimal dTension. The membrane potential and the dTension were monitored on a multibeam oscilloscope (VC-9A, Nihon Kohden) and recorded on a multi-channel thermal arraycorder (WT-645G, Nihon Kohden).
Solutions and drugs
The composition of oxygenated Tyrode solution was (in mM): NaCl 136.7, NaHCO3 11.9, KCl 5.4, NaH2PO4 0.42, MgCl2 0.5, CaCl2 1.8, and glucose 11 with a pH of 7.35–7.40 when gassed with 97% O2 and 3% CO2. PO2 of the solution was measured by an O2 monitor (POG-200BA, Unique Medical, Tokyo) and found to be >400 mmHg. The hypoxic Tyrode solution (PO2 <10 mmHg) had the same composition as above, except that it contained no glucose and was gassed with 97% N2 and 3% CO2 (pH 7.35–7.40).
JTV-519 (JT, c.f. Figure 1A) (a kind gift from Japan Tobacco Inc., Takatsuki, Japan), and diltiazem (DIL) (Sigma Chemical Co., St. Louis, MO, U.S.A.) were dissolved in 5% dimethyl sulphoxide (DMSO) as an 1-mM stock solution. Chelerythrine (CH) (Sigma Chemical Co.) and 5-hydroxydecanoic acid (5-HD) (ICN Pharmaceuticals Inc., Costa Mesa, CA, U.S.A.) were dissolved in distilled water as 1-mM and 10-mM stock solutions, respectively. An appropriate volume of the stock solution was added to the perfusing solution immediately before use for coronary perfusion, as required. Control experiments were performed with 0.1% of DMSO, which was the maximal concentration used and had no effect on the action potentials and contraction.
Figure 1.
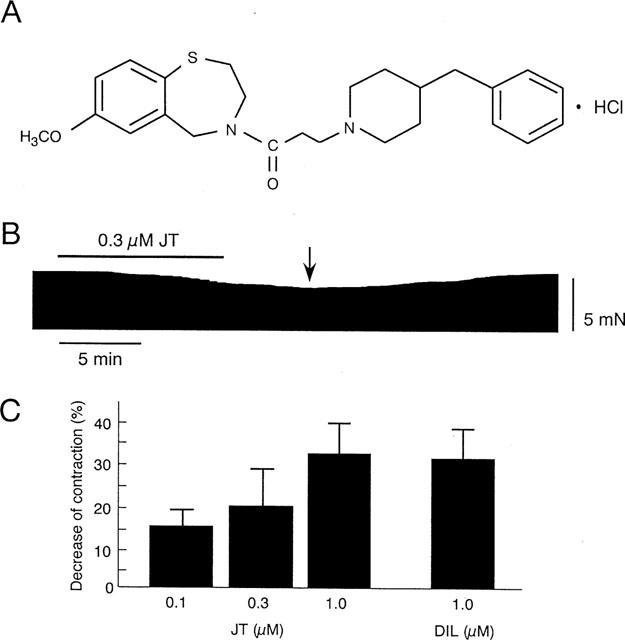
(A) Chemical structure of JTV-519 (JT). (B) Representative recording of contractile tension during 10 min of JT (0.3 μM) application, followed by washout. The arrow indicates the point where the maximal decrease of contraction was observed (approximately 5 min after washout of JT). (C) Summarized data for the per cent decrease of dTension by JT and diltiazem (DIL).
Experimental protocol of I/R
After equilibration (>90 min), the preparations were subjected to 20 min of global ischaemia followed by 60 min of reperfusion (I/R). In the control group, the preparations were subjected to I/R without drugs. In the JT group, the preparations were treated with JT (0.1, 0.3, or 1.0 μM) for 10 min, followed by a 5-min washout period prior to ischaemia. In some experiments (c.f. Figure 2C), the drug was washed out for 20 min, before the onset of I/R. In the DIL group, the preparations were treated with DIL (1.0 μM) for 10 min, followed by a 5-min washout period prior to I/R. In the CH or 5-HD group, the preparations were treated with CH (5.0 μM) or 5-HD (400 μM) for 20 min, followed by a 5-min washout period prior to I/R. In the JT+CH or JT+5-HD group, the treatment of CH (5.0 μM) or 5-HD (400 μM) was started 10 min before the application of JT (1.0 μM) and was continued until the end of the JT perfusion. All the drugs were administered via the coronary artery.
Figure 2.
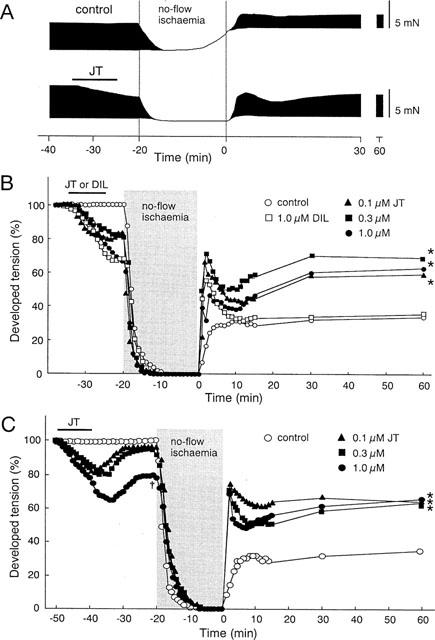
Effects of JT on dTension before and during I/R. (A) Representative recordings of contractile tension in the control group and JT (1.0 μM) group before and during I/R. (B) Per cent change of dTension before and during I/R in the control group, JT group, and DIL group. In the control, the dTension measured after 60 min of reperfusion was decreased to 34±4% of the baseline value (n=6). Brief treatment with JT significantly improved the dTension up to 60±4% (0.1 μM, n=5), 70±5% (0.3 μM, n=5), and 63±5% (1.0 μM, n=4) when compared with the pre-drug values; treatment with DIL produced no significant improvement (36±4%, n=4). (C) Per cent change of dTension before and during I/R with a prolonged washout period. JT was washed out for 20 min prior to ischaemia in these experiments. The data of the control group are the same as shown in Figure 2B. By prolonging the washout period, the dTension was restored to 95±2% (0.1 μM, n=4), 91±6% (0.3 μM, n=4), and 78±4% (1.0 μM, n=4) of the pre-drug values at the start of ischaemia. The JT treatment improved the recovery of the dTension measured at 60 min of reperfusion to 62±5% (0.1 μM), 64±5% (0.3 μM), and 66±6% (1.0 μM) of the pre-drug values. *P<0.01: significant difference vs control. †P<0.01: significant difference vs 0.1 μM-JT.
Data acquisition and analysis
All data were stored on magnetic tapes using a PCM data recording system (RP-880, NF Electronic Instruments, Tokyo) and reproduced by using a computer (Macintosh 8100, Apple Japan Inc., Tokyo) equipped with an analogue-to-digital converter (MacLab 2e, AD Instruments, Japan, Tokyo). The preparations that developed severe arrhythmias (sustained ventricular tachycardia or ventricular fibrillation persisting for more than 30 s) were excluded from the electrical and mechanical analyses. In all series of experiments, the developed tension (dTension) and the action potential duration at 90% repolarization (APD90) were expressed as a per cent of the baseline value (before drug application). Statistical analyses were performed at 10, 15, 30, and 60 min of reperfusion. All data were expressed as mean±s.e.mean. The significance of difference was determined by use of ANOVA combined with Fisher post hoc test. A P value <0.05 was regarded as significant.
Results
Effects of JT on contraction during preischaemic normal perfusion
We first examined the effects of JT on contraction during preischaemic normal perfusion. As shown in Figure 1B, the administration of JT (0.3 μM) for 10 min gradually decreasing the dTension. The decrease continued approximately 5 min after the washout of JT and then recovered gradually. The per cent decreases of the dTension measured 5 min after the washout of JT were summarized in Figure 1C. The application of JT concentration-dependently decreased the contraction; the per cent decreases are 16±2% (0.1 μM, n=5), 19±4% (0.3 μM, n=5), and 33±4% (1.0 μM, n=4). DIL at a concentration of 1.0 μM decreased the dTension by 32±3% (n=4); the degree of reduction was comparable to that achieved by 1.0 μM JT.
Effects of JT on contraction during I/R
To examine whether the brief administration of JT prior to ischaemia (10-min administration and 5-min washout) improves the dTension after I/R, we tested three concentrations (0.1, 0.3, or 1.0 μM) of JT. Figure 2A shows the representative recordings of the dTension before and during I/R in the control group and in the JT (1.0 μM) group. In the control group, the dTension was rapidly decreased after the onset of ischaemia, associated with an elevation of the rTension. The dTension was restored after reperfusion, but, the extent of recovery was only partial and the elevation of the rTension persisted. In the JT group, the dTension also decreased after ischaemia, but an elevation of the rTension was not observed. After reperfusion, the dTension rapidly and transiently increased in the very early phase of reperfusion, which was followed by a greater recovery of the dTension (compared to that in the control group after 60 min of reperfusion). The per cent changes of the dTension in the control and in the JT groups given different concentrations of the drug are shown in Figure 2B; the DIL group is also shown in this figure for comparison. In the control group, the recovery of the dTension after reperfusion was only partial (34±8% of the preischaemic value after 60 min of reperfusion). In contrast, the dTension in the JT groups was rapidly and transiently restored in the early phase of reperfusion, which was followed by a subsequent and gradual increase. The per cent recovery of the dTension in the JT groups (0.1, 0.3 and 1.0 μM) was greater than that in the control group. To test whether the improvement of contractile recovery in the JT groups was mediated by the possible Ca2+ channel blocking action of JT, we examined the effect of brief treatment of DIL, which is known to have a protective action on ischaemic hearts (Bush et al., 1982; Nasa et al., 1990). The treatment by DIL (1.0 μM) decreased the contractions during preischaemia to an extent similar to that caused by JT (1.0 μM). However, this brief treatment with DIL showed no ameliorative effect on postischaemic contractile depression; i.e., the recovery of the dTension was almost the same as that seen in the control group (Figure 2B).
One may speculate that the improvement of the dTension in the JT group was mediated by the JT-induced negative inotropic effect during preischaemia, since the decrease of the dTension at the onset of ischaemia continued even 5 min after JT washout. Thus, in another protocol, the washout period of JT was prolonged up to 20 min (Figure 2C). Although the decrease of the dTension in the group given 1.0 μM JT still remained at the onset of ischaemia, the JT-induced decrease of the dTension was almost completely restored in the groups given 0.1 and 0.3 μM JT. The recovery of the dTension compared after 60 min of reperfusion was still greater in the JT-treated groups than in the control group. These findings indicated that the protective effects of JT are not secondary to the Ca2+ channel blocking action or the negative inotropic effect of JT.
Relationship between JT and PKC activation
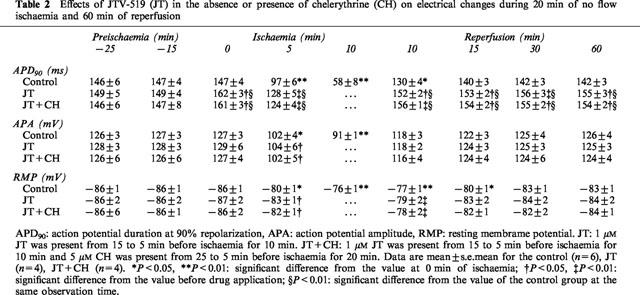
We speculated that the protective effect of JT might be mediated by the activation of protein kinase C (PKC), because our mode of application was similar to that reported in experiments of pharmacological preconditioning. Thus, we tested the effects of chelerythrine (CH) a potent PKC inhibitor, on the protective effect of JT. Figure 3A shows representative recordings of contractile tension before and during I/R in the control group, the JT (1.0 μM) group and the JT+CH (5.0 μM) group. In the presence of CH, JT exerted negative inotropic effects on contractions that were similar to those seen in the control trace. Namely, during ischaemia the dTension decreased and the elevation of the rTension occurred. The increase of the rTension was significant but was much less than that seen in the control group (the absolute values of the rTension are shown in Table 1). In the JT+CH group, a rapid recovery of the dTension in the very early phase of reperfusion (as seen in the JT group) was not observed and the recovery of the dTension after 60 min of reperfusion was less than that of the JT group. The per cent changes of the dTension in the control, the JT group, the CH group, and the JT+CH group are compared in Figure 3B and the absolute values of the dTension are shown in Table 1. The per cent recovery of the dTension in the JT+CH group measured after 60 min of reperfusion was greater than that in the control group but less than that in the JT group. To exclude the direct action of CH on postischaemic contractile recovery, CH was administered without JT. In this case, the contractile recovery in the CH group was comparable to that in the control group. These data suggest that the cardioprotective effect of JT is mediated, at least in part, by the activation of PKC.
Figure 3.
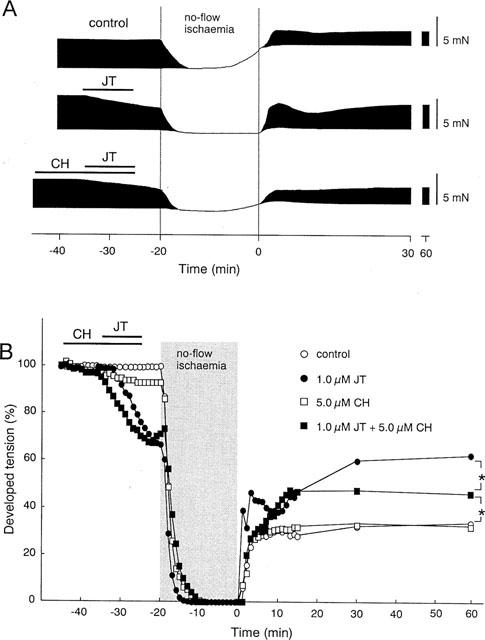
Effects of CH with or without JT on dTension before and during I/R. (A) Representative recordings of contractile tension in the control group, JT (1.0 μM) group, and JT (1.0 μM) plus CH (5.0 μM) group before and during I/R. (B) Per cent change of dTension before and during I/R in the control group, the JT group, the CH group, and the JT+CH group. The data of the control group and the JT group (1.0 μM) are the same as shown in Figure 2B. The per cent recovery of the dTension measured at 60 min of reperfusion was significantly greater in the JT+CH group (46±4%, n=4) than in the control (34±4%, n=6), and was significantly less than that in the JT group (63±5%, n=5), suggesting that the treatment with CH partially antagonized the effect of JT. The per cent recovery of the dTension in the group with CH alone, measured at 60 min of reperfusion (32±8%, n=4), was the same as that in the control group. *P<0.05: significant difference between the data indicated by the brackets.
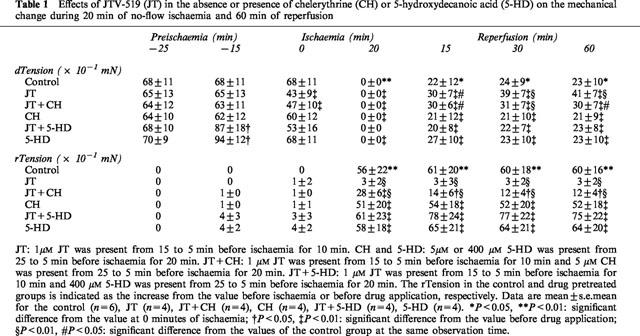
Table 1.
Effects of JTV-519 (JT) in the absence or presence of chelerythrine (CH) or 5-hydroxydecanoic acid (5-HD) on the mechanical change during 20 min of no-flow ischaemia and 60 min of reperfusion
Effects of JT on action potentials during I/R
To investigate the mechanism underlying the protective effects of JT, we investigated the change in the APD before and during I/R. Figure 4A shows the per cent change of the APD90 during I/R in the control group, the JT (1.0 μM) group, and the JT+CH group. The absolute values of APD90, action potential amplitude, and resting membrane potential are also shown in Table 2. In the control group, the APD90 was gradually shortened after introduction of ischaemia (C1, C2, C3 in Figure 4A,B). Upon reperfusion, the APD90 was rapidly restored but yet remained shortened until about 20 min of reperfusion. In the JT group, the application of JT slightly prolonged the APD90 during preischaemic perfusion (J0, J1 in Figure 4A,B). After introduction of ischaemia, the APD90 gradually shortened (J2 in Figure 4A,B) and a conduction disturbance (2 : 1 block) developed in all preparations tested within 8 min (J2′ in Figure 4A,B) and continued throughout ischaemia. This block was not preventable even when the pacing output was increased 10 fold. On reperfusion, the APD90 was rapidly restored and persistently prolonged until the end of the observation. The APD90 estimated at 10, 15, 30 and 60 min of reperfusion in the JT group was significantly longer than that in the control group. On the other hand, the action potential amplitude and resting membrane potential in the JT group were almost the same as those in the control group (Table 2). In the JT+CH group, the changes of the APD90 before and during I/R were almost the same as seen in the JT group. The conduction block caused by JT may be involved in the protective effect of JT; however, this possibility is unlikely because the presence of CH did not affect the JT-induced conduction block, but significantly inhibited the cardioprotective effects of JT (Figure 3). Furthermore, to test the effects of conduction block on the contractile recovery, the stimulation rate was reduced from 3–1.5 Hz during 8–20 min of ischaemia in three other preparations. However, the recovery of the dTension after 60 min of reperfusion (36±4%) was not different from that encountered in the control group (data not shown).
Figure 4.
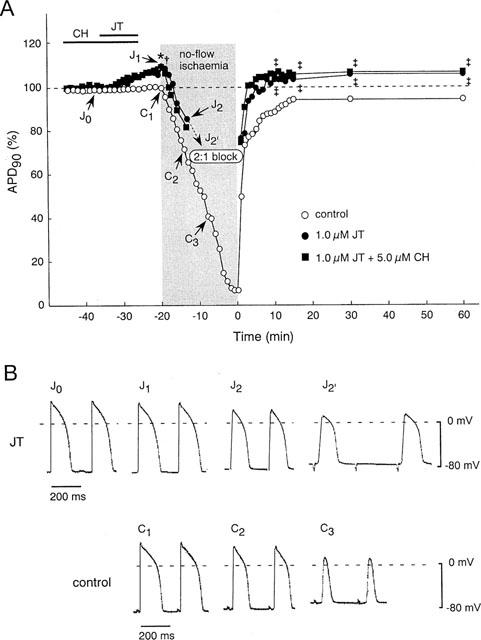
Effects of JT with or without CH on transmembrane action potentials before and during I/R. (A) Time course of changes in the action-potential duration at 90% repolarization (APD90) in the control group (n=4), the JT group (n=4) and the JT+CH group (n=4). (B) Representative recordings of action potentials in the control group and the JT group. In the control group, the APD90 (C1) was shortened by no-flow ischaemia and reached 66±5% at 5 min of ischaemia (C2) and 39±10% at 10 min of ischaemia (C3). On reperfusion, the APD90 was rapidly restored to 74±2% at 2 min of reperfusion, and remained shortened at 96±3% of the baseline value after 60 min of reperfusion. In the JT group, the APD90 was prolonged after the application of JT and reached 109±3% just before the onset of ischaemia (J1). During the ischaemia, APD90 was shortened to 86±9% at 5 min of ischaemia (J2) followed by the evolution of the 2 : 1 block (J2). On reperfusion, the APD90 was rapidly restored and reached 109±4%, at 60 min of reperfusion. In the JT+CH group, the changes of the APD90 before and during I/R were similar to those of the JT group.
Table 2.
Effects of JTV-519 (JT) in the absence or presence of chelerythrine (CH) on electrical changes during 20 min of no flow ischaemia and 60 min of reperfusion
Effects of JT on mitochondrial KATP channels
In the final series of experiments, we tested whether the protective effects of JT were mediated via the activation of KATP channels. It is unlikely that the protective effects of JT are mediated via the activation of sarcolemmal KATP channels, because JT treatment did not shorten the APD but rather prolonged it. Thus, we tested the possible involvement of mitochondrial KATP channels in the protective action of JT. The effects of 5-hydroxydecanoic acid (5-HD), a selective mitochondrial KATP channel blocker, were examined against the anti-ischaemic effects of JT using similar experimental conditions. Figure 5A shows representative recordings of contractile tension before and during I/R in the control group, the JT (1.0 μM) group and the JT+5-HD group. In the presence of 5-HD, the effect afforded by JT, namely, the prevention of the rise of the rTension during ischaemia was completely abolished (the absolute values of the rTension are shown in Table 1). The rapid recovery of the dTension occurring immediately after reperfusion (seen in the JT group) was not inhibited by 5-HD; however, the JT-induced enhancement in the recovery of the dTension seen after 60 min of reperfusion were abolished. The per cent changes of the dTension in the control group, the JT group, the 5-HD group, and the JT+5-HD group are compared in Figure 5B and the absolute values of the dTension are shown in Table 1. The treatment with 5-HD alone increased the dTension up to 120% of the baseline value. However, such increases returned to the baseline level shortly thereafter. The treatment with 5-HD alone did not affect the recovery of the dTension, but the beneficial effects of JT on contractile recovery were completely abolished in the presence of 5-HD.
Figure 5.
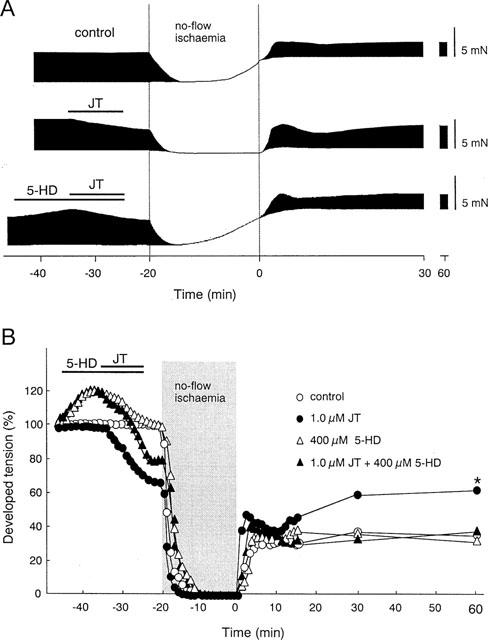
Effects of 5-HD with or without JT on developed tension before and during I/R. (A) Representative recordings of contractile tension in the control group, the JT (1.0 μM) group, and the JT (1.0 μM) plus 5-HD (400 μM) group before and during I/R. (B) Per cent change of the dTension before and during I/R in the control group, the JT group, the 5-HD group, and the JT+5-HD group. The data of the control group and the JT group (1.0 μM) were the same as shown in Figure 2B). The per cent recovery of the dTension in the JT+5-HD group measured at 60 min of reperfusion (34±6%, n=4) was comparable to that in the control group, suggesting that the treatment with 5-HD completely antagonized the effect of JT. The per cent recovery of the dTension in the group with 5-HD alone, measured at 60 min of reperfusion (33±7%, n=4) was the same as that in the control group. *P<0.01: significant difference vs JT (1.0 μM) group.
Discussion
In the present study, we demonstrate that JT attenuated the contractile dysfunction after I/R in isolated guinea-pig hearts. This potentially beneficial effect of JT was partially antagonized with a PKC inhibitor. In addition, treatment with 5-HD completely blocked the effects of JT, thereby suggesting that the activation of mitochondrial KATP channels mediated the cardioprotective effects of JT. The application of JT alone significantly decreased the dTension in a concentration-dependent manner (Figure 1C). Moreover, JT at a concentration of 1.0 μM decreased the dTension to an extent similar to that caused by 1.0 μM DIL (Figure 1C). It has been reported that, in guinea-pig ventricular cells, the potency of L-type Ca2+ channel blocking effect of JT was comparable to that of DIL (Okuyama et al., 1994; Kimura et al., 1999). Therefore, such negative inotropic effects of JT may be produced by the inhibition of L-type Ca2+ channels.
Previous studies have shown that most calcium antagonists attenuated myocardial stunning when the drug was given before ischaemia (Du Toit & Opie, 1992; Ehring et al., 1992). In addition, the reduction in calcium overload, the major mechanism of the cardioprotective effect of calcium antagonists is supposed to be their ability to spare ATP consumption during ischaemia, as a consequence of their negative inotropic properties (Nayler et al., 1980; Lange et al., 1984). However, it is assumed that the cardioprotective effect of JT did not merely depend on the Ca2+ channel blocking property, because the treatment with DIL (1.0 μM) which showed a negative inotropic effect comparable to that of JT (1.0 μM), did not exert a cardioprotective effect in this model (Figure 2B). In other words, the JT-induced negative inotropic effect did not contribute to the protective effect of JT. The latter notion is further based on the finding that the cardioprotective effect of JT (0.1∼1.0 μM) did not depend on the concentration used, although JT dose-dependently decreased the contraction (Figure 2B). In addition, JT showed an effect even when the washout period was prolonged up to 20 min (Figure 2C).
Since the relatively brief (10 min) administration of JT prior to ischaemia significantly improved the postischaemic contractile recovery, we considered that the cardioprotective effects of JT were mediated via mechanisms similar to those encountered in pharmacological preconditioning. Thus, we tested whether the inhibition of PKC by CH interferes with the protective action of JT. The IC50 of CH for PKC inhibition has been reported to be 0.7 μM (Herbert et al., 1990). In the rat heart model, 2.0 μM CH completely abolished the effect of ischaemic preconditioning (Bugge & Ytrehus, 1995). Therefore, the concentration of CH (5.0 μM) that we used in the present study seems sufficient to inhibit PKC. We found that the treatment with 5.0 μM CH attenuated the postischaemic recovery of the dTension afforded by JT, accompanied by moderate elevation of the rTension during ischaemia (Figure 3A,B and Table 1). In addition, treatment with CH not completely but significantly inhibited the effects of JT on the postischaemic recovery of the dTension. These results support to the notion that the cardioprotective effects of JT were mediated at least in part via the activation of PKC. Interestingly, different concentrations of JT showed a comparable degree of cardioprotection (Figure 2B,C). This finding may be explained by the notion that 0.1 μM JT might be enough to exceed the threshold for PKC activation. Recently, it was reported that JT stimulated the translocation of delta PKC from the cytosol to the sarcolemma (Inagaki et al., 2000).
The brief administration of JT prolonged the APD both during preischaemia and after reperfusion (Figure 4A,B). These findings indicate that the cardioprotection rendered by JT is not secondary to the opening of sarcolemmal KATP channels. In a patch-clamp study, we identified that JT inhibits the rapid component of delayed rectifying K+ current (IKr) in guinea-pig ventricular myocytes (our unpublished data). Thus, such JT-induced APD prolongation may be due to the inhibition of IKr. On the other hand, JT produced a conduction disturbance (i.e., 2 : 1 block) after approximately 8 min of ischaemia. This change was not affected by the CH treatment and was probably caused by the blockade of sodium channels (Kimura et al., 1999). We do not think this conduction block is the major mechanism for the cardioprotective action of JT, because the preparation, which was stimulated at a much lower rate of 1.5 Hz (usually 3 Hz) beginning after 8 min of ischaemia, failed to improve the contractile recovery (data not shown).
Recent work disclosed that ischaemic preconditioning opens the KATP channels not of the sarcolemma but of the mitochondria (Garlid et al., 1997; Liu et al., 1998). To investigate whether JT exhibits its cardioprotective effect via the opening of mitochondrial KATP channels, we tested 5-HD, a selective mitochondrial KATP channel blocker. Treatment with 400 μM 5-HD abolished the beneficial effects of JT, i.e., the improvement of the dTension during reperfusion and the attenuation of the rTension elevation during ischaemia (Figure 5A,B). Incidentally, 5-HD temporarily increased the dTension to ∼120% of the control value. We did not investigate the mechanism of this increase, but it is assumed that this phenomenon does not relate to the 5-HD inhibition of cardioprotection afforded by JT, because 100 and 200 μM 5-HD, which increased the dTension to an extent similar to that seen in the case of 400 μM 5HD, did not abolish the protective effect of JT (data not shown). JT also inhibited the rise of the rTension during ischaemia, which may also be related to the mitochondrial KATP channel opening. Sato et al. (1998) have reported that the mitochondrial KATP channel opening is enhanced by the activation of PKC; suggesting mitochondrial KATP channels must exist downstream to PKC. However, the cardioprotective effect of JT was not completely inhibited by CH. It is thought that there are two possibilities here: first, the CH concentration (5.0 μM) might not be enough to inhibit mitochondrial KATP channels activated by PKC, and second, mitochondrial KATP channels might also be activated by another pathway. Further study is required to clarify this question.
In recent years, thanks to the development of interventional recanalizatiion, an increasingly large number of patients are subjected to coronary reperfusion in an effort to preserve cardiac function. Meanwhile, the occurrence of contractile dysfunction due to I/R injury in these patients could significantly delay the benefits of reperfusion therapy. Accordingly, the development of a potent cardioprotective agent has been desired and JT is thought to be such a candidate. In conclusion, JT is a potent cardioprotective agent against I/R injury and its potentially beneficial effect is probably induced by the activation of mitochondrial KATP channels mediated by PKC activation.
Acknowledgments
We thank Japan Tobacco Inc. (Takatsuki, Japan) for their kind supply of JTV-519. This study was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
Abbreviations
- 5-HD
5-hydroxydecanoic acid
- APD
action potential duration
- APD90
APD at 90% repolarization
- CH
chelerythrine
- DIL
diltiazem
- dTension
developed tension
- I/R
ischaemia-reperfusion
- JT
JTV-519
- KATP
ATP-sensitive K+
- PKC
protein kinase C
- rTension
resting tension
References
- BUGGE E. , YTREHUS K. Ischaemic preconditioning is protein kinase C dependent but not through stimulation of alpha adrenergic or adenosine receptors in the isolated rat heart. Cardiovasc. Res. 1995;29:401–406. [PubMed] [Google Scholar]
- BUSH L.R., ROMSON J.L., ASH J.L. , LUCCHESI B.R. Effects of diltiazem on extent of ultimate myocardial injury resulting from temporary coronary artery occlusion in dogs. J. Cardiovasc. Pharmacol. 1982;4:285–296. doi: 10.1097/00005344-198203000-00018. [DOI] [PubMed] [Google Scholar]
- DU TOIT J. , OPIE L.H. Modulation of severity of reperfusion stunning in the isolated rat heart by agents altering calcium flux at onset of reperfusion. Circ. Res. 1992;70:960–967. doi: 10.1161/01.res.70.5.960. [DOI] [PubMed] [Google Scholar]
- EHRING T., BOHM M. , HEUSCH G. The calcium antagonist nisoldipine improves the functional recovery of reperfused myocardium only when given before ischaemia. J. Cardiovasc. Pharmacol. 1992;20:63–74. [PubMed] [Google Scholar]
- GARLID K.D., PAUCEK P., YAROV-YAROVOY V., MURRAY H.N., DARBENZIO R.B., D'ALONZO A.J., LODGE N.J., SMITH M.A. , GROVER G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circ. Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- GOTO M., LIU Y., YANG X.M., ARDELL J.L., COHEN M.V. , DOWNEY J.M. Role of bradykinin in protection of ischaemic preconditioning in rabbit hearts. Circ. Res. 1995;77:611–621. doi: 10.1161/01.res.77.3.611. [DOI] [PubMed] [Google Scholar]
- HACHIDA M., KANEKO N., OHKADO A., HOSHI H., NONOYAMA M., SAITO S., BONKOHARA Y., HANAYAMA N., MIYAGISHIMA M. , KOYANAGI H. Significant effect of 1,4-benzothiazepine derivative (K201) in improving myocardial preservation. Transplant. Proc. 1997;29:1346–1348. doi: 10.1016/s0041-1345(96)00591-x. [DOI] [PubMed] [Google Scholar]
- HERBERT J.M., AUGEREAU J.M., GLEYE J. , MAFFRAND J.P. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- HU H, , SATO T., SEHARASEYON J., LIU Y., JOHNS D.C., O'ROURKE B. , MARBAN E. Pharmacological and histochemical distinctions between molecularly defined sarcolemmal KATP channels and native cardiac mitochondrial KATP channels. Mol. Pharmacol. 1999;55:1000–1005. [PubMed] [Google Scholar]
- HU K., DURAN D., LI G.R. , NATTEL S. Protein kinase C activates ATP-sensitive K+ current in human and rabbit ventricular myocytes. Circ. Res. 1996;78:492–498. doi: 10.1161/01.res.78.3.492. [DOI] [PubMed] [Google Scholar]
- INAGAKI K., KIHARA Y., HASHIDA W., IZUMI T., YONEDA T., TAKEUCHI Y., INAGAWA Y., KATSUO S., MUSO E. , SASAYAMA S. Anti-ischaemic effect of a novel cardioprotective agent, JTV519, is mediated through specific activation of δ-isoform of protein kinase C in rat ventricular myocardium. Circulation. 2000;101:797–804. doi: 10.1161/01.cir.101.7.797. [DOI] [PubMed] [Google Scholar]
- ITO K., SATO T., ABE T., LI Y. , ARITA M. Anti-stunning effect of JTV-519 in coronary perfused guinea pig ventricular muscles. J. Mol. Cell. Cardiol. 1998;30:A324. [Google Scholar]
- KANEKO N. New benzothiazepine derivative, K201, demonstrates cardioprotective effects against sudden cardiac cell death and intracellular calcium blocking action. Drug. Dev. Res. 1994;33:429–438. [Google Scholar]
- KIMURA J., KAWAHARA M., SAKAI E., YATABE J. , NAKANISHI H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn. J. Pharmacol. 1999;79:275–281. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- KITAKAZE M., HORI M., MORIOKA T., MINAMINO T., TAKASHIMA S., SATO H., SHINOZAKI Y., CHUJO M., MORI H. , INOUE M. Alpha 1-adrenoceptor activation mediates the infarct size-limiting effect of 5′-nucleotidase activity. J. Clin. Invest. 1994;93:2197–2205. doi: 10.1172/JCI117216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGE R., INGWALL J., HALE S.L., ALKER K.J., BRAUNWALD E. , KLONER R.A. Preservation of high-energy phosphates by verapamil in reperfused myocardium. Circulation. 1984;70:734–741. doi: 10.1161/01.cir.70.4.734. [DOI] [PubMed] [Google Scholar]
- LIU G.S., THORNTON J., VAN WINKLE D.M., STANLEY A.W., OLSSON R.A. , DOWNEY J.M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- LIU Y., GAO Y.D., O'ROURKE B. , MARBAN E. Synergistic modulation of ATP-sensitive K+ currents by protein kinase C and adenosine: implications for ischaemic preconditioning. Circ. Res. 1996;78:443–454. doi: 10.1161/01.res.78.3.443. [DOI] [PubMed] [Google Scholar]
- LIU Y., SATO T., O'ROURKE B. , MARBAN E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection. Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- MURRY C.E., JENNINGS R.B. , REIMER K.A. Preconditioning with ischaemia: a delay of lethal cell injury in ischaemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- NASA Y., ICHIHARA K. , ABIKO Y. Both d-cis- and 1-cis-diltiazem have anti-ischaemic action in the isolated, perfused working rat heart. J. Pharmacol. Exp. Ther. 1990;255:680–689. [PubMed] [Google Scholar]
- NAYLER W.G., FERRARI R. , WILLIAMS A. Protective effect of pretreatment with verapamil, nifedipine, and propranolol on mitochondrial function in ischaemic and reperfused myocardium. Am. J. Cardiol. 1980;46:242–248. doi: 10.1016/0002-9149(80)90064-8. [DOI] [PubMed] [Google Scholar]
- OKUYAMA R., ADACHI-AKAHANE S. , NAGAO T. Differential potentiation by depolarization of the effects of calcium antagonists on contraction and Ca2+ current in guinea-pig heart. Br. J. Pharmacol. 1994;113:451–456. doi: 10.1111/j.1476-5381.1994.tb17010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO T., O'ROURKE B. , MARBAN E. Modulation of Mitochondrial ATP-dependent K+ channels by protein kinase C. Circ. Res. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- SHIGEMATSU S., SATO T., ABE T., SAIKAWA T., SAKATA T. , ARITA M. Pharmacological evidence for the persistent activation of ATP-sensitive K+ channels in early phase of reperfusion and its protective role against myocardial stunning. Circulation. 1995;92:2266–2275. doi: 10.1161/01.cir.92.8.2266. [DOI] [PubMed] [Google Scholar]
- YTREHUS K., LIU Y. , DOWNEY J.M. Preconditioning protects ischaemic rabbit heart by protein kinase C activation. Am. J. Physiol. 1994;266:H1145–H1152. doi: 10.1152/ajpheart.1994.266.3.H1145. [DOI] [PubMed] [Google Scholar]