

Abstract
The nitric oxide (NO)-donating nitroxybutyl ester of flurbiprofen (NO-flurbiprofen), shows reduced gastro-intestinal toxicity relative to flurbiprofen. NO may exert either pro- or anti-apoptotic effects, while non-steroidal anti-inflammatory drugs may induce apoptosis. The aim of the present work was therefore to compare the effects of flurbiprofen and NO-flurbiprofen on apoptosis in guinea-pig gastric mucous cells.
Apoptotic activity was assessed by assay of caspase activity and from the fragmentation and condensation of nuclei.
Incubation with flurbiprofen for 24 h produced a concentration-dependent induction of apoptosis in cells attached to the culture plate (caspase 3-like activity increased by 257% at 500 μM), while NO-flurbiprofen inhibited basal apoptosis (caspase 3-like activity decreased by 71% at 500 μM).
Caspase activity and nuclear fragmentation were substantially increased in cells that had spontaneously detached from the culture plate. NO-flurbiprofen inhibited caspase activity (55% at 500 μM) but not nuclear fragmentation in these detached cells.
NO flurbiprofen inhibited the activation of apoptosis by 25 μM C6-ceramide in cells attached to the culture plate.
Inhibition of caspase activity by NO-flurbiprofen was detectable after 6 h of incubation with intact cells, but by contrast with the NO-donor S-nitrosyl-N-acetyl-penicillamine, was not demonstrable with cell homogenates.
Activation of caspase 3-like activity by flurbiprofen was slow (>6 h incubation needed) and was inhibited by cycloheximide.
The presence of a nitroxybutyl ester moiety on flurbiprofen prevents the pro-apoptotic activity of the parent compound and may contribute to the reduced gastro-intestinal toxicity of NO-flurbiprofen.
Keywords: Nitric oxide, apoptosis, gastric epithelial cell, non-steroidal anti-inflammatory drug, flurbiprofen, nitroxybutyl flurbiprofen, stomach
Introduction
Apoptosis in the gastro-intestinal tract occurs close to the proliferative zone and at the surface of the epithelium (Hall et al., 1994), and plays an important role under normal circumstances in regulating mucosal cell number. Under pathological conditions apoptosis in the stomach is stimulated by infection with Helicobacter pylori (Mannick et al., 1996; Moss et al., 1996), and by administration of non-steroidal anti-inflammatory drugs (NSAIDs) (Zhu et al., 1998), which could contribute to the gastric atrophy seen with these agents (Taha et al., 1992).
NSAIDs damage the gastric mucosa: possible mechanisms being a direct topical action on epithelial cells, inhibition of prostaglandin biosynthesis and a reduction of gastric blood flow associated with a tumour necrosis factor α (TNFα)-mediated recruitment of neutrophils to the capillary endothelium (Santucci et al., 1994; 1995). One approach to reducing the gastro-intestinal toxicity of NSAIDS has been the development of NO-donating NSAIDs by the addition of a nitroxybutyl moiety. These agents show a significantly reduced gastro-intestinal toxicity without reduction of their anti-inflammatory effect (Wallace et al., 1994). Release of NO may inhibit neutrophil adherence, promote mucosal blood flow and counteract the pro-apoptotic effects of TNFα on endothelial cells by inhibition of caspase activity (Wallace et al., 1994; Fiorucci et al., 1999a,1999b).
The effects of NO on apoptosis vary with cell-type. Thus NO stimulates apoptosis in macrophages, pancreatic islets, and thymocytes (Brune et al., 1998), and in a wide variety of cell lines including one, RGM1, of rat gastric mucosal origin (Honda et al., 1999). Furthermore, recent work with transformed cell lines (Lu et al., 1995; Chan et al., 1998; Simmons et al., 1999; Zhu et al., 1999) has indicated that NSAIDs can induce apoptosis, but effects on gastric epithelial cells, with which they have topical contact, and in which they may accumulate (Wallace, 1997), have not been determined. The purpose of this investigation was therefore to use a primary culture of guinea-pig gastric mucosal cells, which consists of at least 90% of gastric mucous epithelial cells (Byrne & Hanson, 1998; Teshima et al., 1998), to determine whether the NO-donating NSAID nitroxybutyl-flurbiprofen (NO-flurbiprofen) would affect apoptotic activity and to compare the results with those obtained with the parent compound flurbiprofen. Two late manifestations of apoptosis namely nuclear fragmentation and condensation and increased activity of the effector proteolytic enzyme, caspase 3 (Raff, 1998), were used as indicators of programmed cell death.
Methods
Animals
Male Dunkin-Hartley guinea-pigs of 200–300 g body weight were obtained from Charles River, Margate, Kent U.K. and were fed on SDS Economy guinea-pig diet supplied by Lillico, Betchworth, Surrey, U.K.
Materials
RPMI 1640 medium, foetal calf serum, antibiotics and amphotericin B were from Life Technologies, Paisley U.K. N-Hexanoyl-D-sphingosine (C6-ceramide) and caspase substrates and inhibitors were obtained from Alexis, Nottingham, U.K. Pronase E (70,000 PU units per g) was purchased from Merck, Lutterworth, U.K. NO-flurbiprofen was from NicOx SA, Sophia-Antipolis, France. Other reagents were from Sigma, Poole, U.K.
Isolation and culture of gastric mucous cells
The method has been described in detail previously (Byrne & Hanson, 1998). Gastric mucosa was minced with fine scissors, incubated with 45 ml of RPMI 1640 containing 2 g l−1 bovine serum albumin (isolation medium) and 0.5 mg ml−1 pronase for 20 min at 37°C, and then, after centrifugation, with 45 ml isolation medium containing 0.4 mg ml−1 of collagenase for 20 min. Cells were filtered through 150 μm nylon mesh, washed in culture medium (RPMI 1640 containing 10% foetal calf serum, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin and 2.5 μg ml−1 of amphotericin B) and cultured on six-well culture plates. The culture medium was renewed after 24 h, and again at 48 h, at which time the experiments were initiated by the inclusion of agents in the culture medium. All measurements were made after a further 24 h. Agents were dissolved in dimethylsulphoxide, the final concentration of which was 0.25% (v v−1) in all wells, including control wells.
Detection of apoptosis by staining with Hoechst 33258
Cells in suspension, and those removed from the plate by exposure to trypsin/EDTA, were subjected to centrifugation for 20 s at 12,000 ×g. Cells were resuspended in 4% (w v−1) paraformaldehyde in 138 mM NaCl, 2.7 mM KCl, 10 mM phosphate (PBS) for 10 min at room temperature, subjected to centrifugation at 12,000 ×g for 10 s, resuspended in ethanol: water (4 :1) and stored at 4°C. Cells were transferred to glass slides by use of a Shandon cytocentrifuge, and nuclei were stained with 8 μg ml−1 of Hoechst 33258. Slides were coded, and then the proportion of apoptotic nuclei was determined by counting using a fluorescent microscope with apoptosis defined as the presence of two or more condensed bodies per nucleus.
Preparation of homogenates
The incubation medium was removed from the culture plate into 2 ml microfuge tubes. The plate was placed on ice and 1 ml of PBS was added to each well. The cells were removed from the plate with a cell-scraper and transferred to microfuge tubes. All microfuge tubes were subjected to centrifugation for 20 s at 12,000 ×g. Pellets were rinsed with 1.0 ml of ice cold PBS and then were resuspended, by pipetting up and down 10 times, in 110 μl of homogenization buffer (mM): HEPES 100, NaCl 140, EDTA 1, (pH 7.4), containing 0.5 mM phenylmethylsulphonyl fluoride, 5 μg ml−1 aprotinin, 5 μg ml−1 pepstatin and 10 μg ml−1 leupeptin. Extracts were then frozen in liquid nitrogen and stored at −70°C. Before assay homogenates were thawed and refrozen twice, before a final thaw and centrifugation at 12,000 ×g for 20 min at 4°C.
Caspase assay
Twenty five μl of supernatant was incubated at 37°C with 175 μl of assay buffer consisting of 100 mM HEPES (pH 7.4) containing protease inhibitors at the same concentration as above, and caspase substrate at a final concentration of 15 μM. The substrates Ac-Tyr-Val-Ala-Asp-7-amido-4-methylcoumarin and Ac-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin were used for caspase 1-like and caspase 3-like activity respectively. Formation of fluorescent product was measured on a Victor multilabel counter.
Acid phosphatase activity
Enzyme activity was determined by the breakdown of p-nitrophenyl phosphate at pH 4.8 as described by Wong & Tepperman (1994).
Protein concentration determination
Protein was measured by using bicinchoninic acid reagent with bovine serum albumin as standard (Redinbaugh & Turley, 1986).
Statistical analysis
Results are presented as means±s.e.mean, with n equal to the number of cell preparations, and unless stated otherwise were subjected to analysis of variance, to remove variation between cell cultures, followed by either a Newman-Keuls or a Dunnett's multiple comparison test. Data on percentage of apoptotic cells were arcsine-transformed before analysis.
Results
Effect of detachment from the culture plate and of C6-ceramide on apoptotic activity in gastric epithelial cells
Caspase 3-like activity was increased in cells which had detached from the culture plate, and was also increased by incubation for 24 h with C6-ceramide but only in cells which remained attached (Figure 1a). Analogous findings on the effects of C6-ceramide and detachment were obtained if nuclear fragmentation and condensation was used as the measure of apoptotic activity (Figure 1b).
Figure 1.
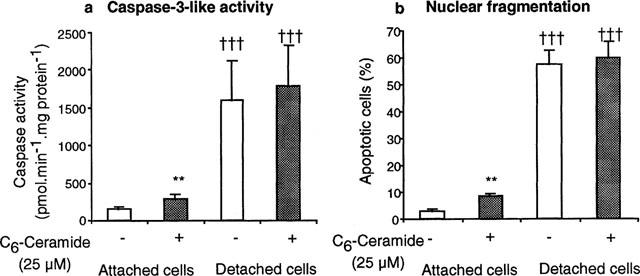
Incubation of gastric epithelial cells for 24 h with C6-ceramide, or detachment of cells from the culture plate increase (a) caspase 3-like activity and (b) nuclear condensation and fragmentation. Results are means±s.e.mean from six separate cell preparations, and have been analysed by analysis of variance followed by a Newman-Keuls multiple comparison test. **P<0.01 for difference from appropriate control without ceramide, and †††P<0.001 for effect of detachment from the plate.
Effect of flurbiprofen and NO-flurbiprofen on apoptotic activity in gastric epithelial cells
Incubation with flurbiprofen (250 and 500 μM) for 24 h increased caspase 3-like activity in cells attached to the culture plate, but did not further enhance the already elevated activity in detached cells (Figure 2a). NO-flurbiprofen exerted a dose-related inhibition of caspase 3-like activity in both attached cells, and in those that had detached from the culture plate (Figure 2b). Inhibition of caspase 3-like activity by 100 μM NO-flurbiprofen in attached cells was 58±9 and 69±11% in the presence and absence of the guanylate cyclase inhibitor 1H-[1,2,4] Oxadiazole [4,3-a] quinoxaline-1-one (ODQ, 10 μM) (n=3, no significant difference by paired t-test). For attached cells, the effects of flurbiprofen and NO-flurbiprofen (500 μM) on the percentage of cells showing condensation and fragmentation of DNA were qualitatively similar to those on caspase activity (Figure 3a). In detached cells flurbiprofen (500 μM) increased the proportion of apoptotic cells despite the absence of any change in caspase activity which was however already substantially elevated (Figure 3b). NO-flurbiprofen did not decrease the proportion of detached cells which were apoptotic (Figure 3b) despite decreasing caspase activity (Figure 2), which, however, at 545±147 pmol min−1 mg protein−1 was still above (P<0.05, paired t-test, n=5) the control value for attached cells of 182±26 pmol min−1 mg protein−1.
Figure 2.
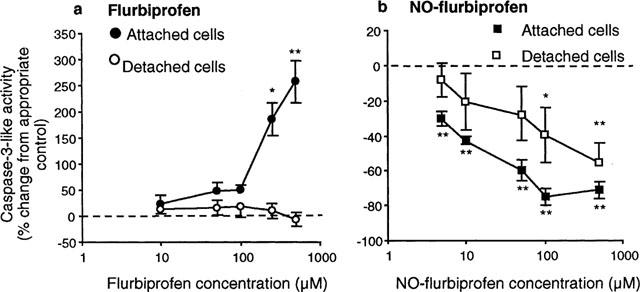
Flurbiprofen activates caspase 3-like activity while NO-flurbiprofen inhibits it. Gastric epithelial cells were incubated for 24 h with different concentrations of (a) flurbiprofen and (b) NO-flurbiprofen. Results in (a) are means±s.e.mean from six separate cell cultures and are expressed as the per cent change from activities in the absence of agent which were: 147±36 and 1779±775 pmol min−1 mg protein−1 for attached and detached cells respectively. Results in (b) are means±s.e.mean from five separate cell cultures and are expressed as the per cent change from activities in the absence of agent which were: 182±26 and 1439±353 pmol min−1 mg protein−1 for attached and detached cells respectively. *P<0.05, **P<0.01 for difference from appropriate control by analysis of variance and Dunnett's test.
Figure 3.
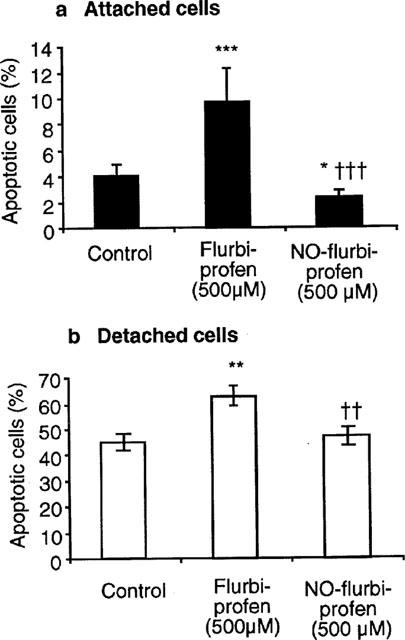
Effect of incubation with flurbiprofen or NO-flurbiprofen on nuclear condensation and fragmentation in gastric epithelial cells, which after 24 h were attached (a) or detached (b) from the culture plate. Results are means±s.e.mean from five separate cell cultures and have been analysed by analysis of variance followed by a Newman-Keuls multiple comparison test. *P<0.05, **P<0.01, ***P<0.001 for difference from control, and ††P<0.01, †††P<0.001 for difference between flurbiprofen and NO-flurbiprofen.
Effect of flurbiprofen and NO-flurbiprofen on cellular protein and release of enzyme activity from gastric epithelial cells
Attached cellular protein (μg well−1, n=4) was: 216±28, 236±24 and 249±20 for control cultures and those incubated for 24 h with flurbiprofen (500 μM) and NO-flurbiprofen (500 μM) respectively; there being no significant effect of the added agents by analysis of variance. The cellular protein present in suspension was (per cent of total cellular protein per well, n=4): 10±3, 17±6 and 12±3 for control cultures and those incubated for 24 h with flurbiprofen (500 μM) and NO-flurbiprofen (500 μM) respectively. Again there was no significant effect of agents when the data were subjected to analysis of variance. Release of acid phosphatase activity into the culture medium, which was used previously as an indicator of damage to gastric mucosal cells (Wong & Tepperman, 1994), was (per cent of total activity, n=4) 1.33±0.24, 1.35±0.60 and 1.67±0.44 for control, flurbiprofen (500 μM) and NO-flurbiprofen (500 μM) respectively (no significant effect of treatments by analysis of variance).
Selectivity of caspase activation
Caspase 1-like activity, measured with the substrate Ac-Tyr-Val-Ala-Asp-7-amido-4-methylcoumarin, was much lower (P<0.05) than caspase 3-like activity under control conditions (respectively 10.4±4.7 (5) and 147±37 (6) pmol min−1 mg protein−1. Furthermore treatment for 24 h with respectively flurbiprofen (500 μM), NO-flurbiprofen (100 μM) or C6-ceramide (25 μM), or detachment from the culture plate, gave the following results for caspase 1-like activity: 7.1±2 (6), 12.2±2.3 (4), 14.7±2.5 (6) and 5.2±1.5 (6). These treatments did not therefore produce changes in caspase 1-like activity of the magnitude seen with the caspase 3 substrate. The effect of preincubating homogenates for 10 min with the inhibitor Ac-Asp-Glu-Val-Asp-CHO (10 nM) on subsequent caspase 3-like activity was investigated. Inhibition of activity (%) on samples obtained after incubation for 24 h under control conditions, with flurbiprofen (250 μM) or after detachment from the culture plate was 90.5±1.4 (3), 96.3±1.3 (5) and 96.2±1.2 (5) respectively.
Effect of NO-flurbiprofen on apoptotic activity in gastric epithelial cells incubated with C6-ceramide
Both caspase 3-like activity and the proportion of apoptotic cells was reduced by 500 μM NO-flurbiprofen (Figure 4) in attached cells incubated with C6-ceramide (25 μM). NO-flurbiprofen did not affect either the proportion of apoptotic cells, or caspase 3-like activity, in cells which became detached in the presence of 25 μM C6-ceramide (Figure 4).
Figure 4.
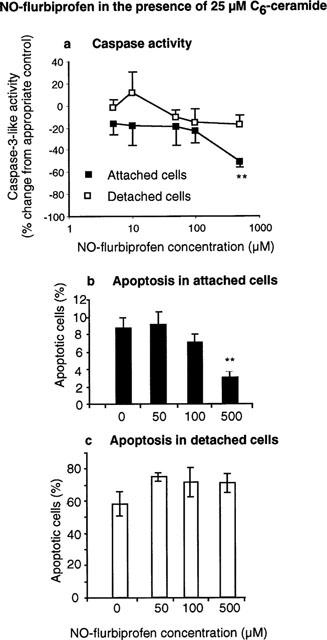
Incubation for 24 h with NO-flurbiprofen inhibits (a) caspase activity and (b, c) nuclear condensation and fragmentation in attached gastric epithelial cells in the presence of 25 μM C6-ceramide. Results in (a) are means±s.e.mean from five separate cell cultures and are expressed as the per cent change from activities in the absence of agent which were: 311±67 and 2165±725 pmol min−1 mg protein−1 for attached and detached cells respectively. Results in (b) and (c) are means±s.e.mean from three separate cell cultures. **P<0.01 for difference from appropriate control by analysis of variance and Dunnett's test.
Time course of changes in caspase 3-like activity
All treatments decreased caspase 3-like activity in attached cells after 1 h relative to control. The reason for this is unclear, but it should be emphasized that the vehicle, dimethyl sulphoxide, was present in all wells. After 6 h NO-flurbiprofen (500 μM) decreased activity relative to control while C6-ceramide increased it whether or not flurbiprofen (500 μM) was present. Flurbiprofen (500 μM) alone had no effect. The same situation pertained after 24 h of incubation except that there was now a marked stimulation of caspase activity with flurbiprofen (500 μM) alone. There were insufficient detached cells for measurement to be made at either 1 or 6 h (Figure 5).
Figure 5.
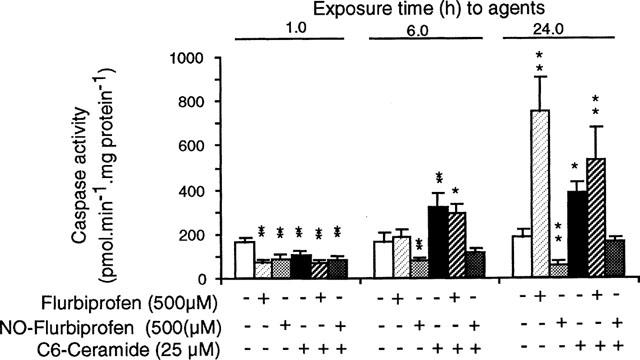
Effects of C6-ceramide and NO-flurbiprofen precede those of flurbiprofen on caspase activity in gastric epithelial cells attached to the culture plate. Results are means±s.e.mean from five separate cell cultures. *P<0.05, and **P<0.01 for difference from the control result for each time period by analysis of variance and a Newman-Keuls multiple comparison test.
Effect of cycloheximide on the stimulation of caspase activity by flurbiprofen
Treatment for 24 h with cycloheximide (1 μM) reduced basal caspase activity in attached cells, while the higher concentration (5 μM) was without effect (Figure 6). One hundred μM cycloheximide produced variable, and in some instances highly elevated caspase activity 1100±522 pmol min−1 mg protein−1 (4). The stimulatory effect of flurbiprofen (500 μM) was reduced by cycloheximide (1 μM) below that found under control conditions or with cycloheximide (5 μM).
Figure 6.
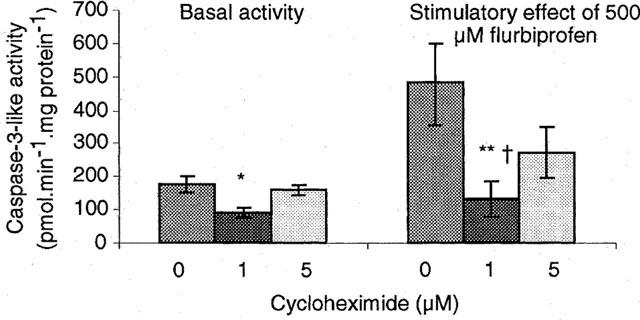
Cycloheximide inhibits the stimulation of caspase activity by flurbiprofen. Results are means±s.e.mean from four separate cell preparations and have been analysed by analysis of variance and a Newman-Keuls test. Left-hand set: *P<0.05 for a significant difference of basal activity at 1 μM cycloheximide from activity at 0 or 5 mM. Right-hand set: **P<0.01 for difference in stimulation of caspase activity by 500 μM flurbiprofen at 0 and 1 μM cycloheximide, and †P<0.05 for difference between 1 and 5 μM cycloheximide.
Effect of S-nitroso-N-acetyl-penicillamine (SNAP) on the stimulation of caspase activity by flurbiprofen
Flurbiprofen (500 μM) did not stimulate caspase activity in attached cells in the presence of the NO-donor SNAP. Results (pmol min−1 mg protein−1, n=3) were: control, 236±33; SNAP (1 mM), 45±7.0; and SNAP (1 mM) plus flurbiprofen (500 μM), 58±4.8, with the two results in the presence of SNAP being not different from one another and lower (P<0.01) than the control (Newman-Keuls test).
Action of S-nitroso-N-acetyl-penicillamine (SNAP) and NO-flurbiprofen on caspase activity in homogenates and in intact cells: effects of dithiothreitol in the assay
Homogenates from control attached cells were preincubated at room temperature for 0, 1, or 3 h before being assayed over the subsequent 40 min for caspase 3-like activity. Enzyme activity was relatively stable under control conditions and this situation was unaltered if NO-flurbiprofen (100 μM) was included during the preincubation period (Figure 7). However, incubations with SNAP (100 μM) produced a progressive decline in caspase activity, which was largely reversed by the inclusion of dithiothreitol (20 mM) during the assay period. If homogenates were taken from cells (n=3 separate experiments), which had been preincubated with 1 mM SNAP for 24 h, then the inhibitory effect of SNAP on caspase 3-like activity was respectively 76±4% and 75±5% with and without inclusion of dithiothreitol (20 mM) in the assay. Similarly the inhibitory effect of incubation with NO-flurbiprofen (100 μM) for 24 h on caspase activity was (n=4 separate experiments): respectively 64±9% and 68±9% assayed in the presence and absence of dithiothreitol. Inclusion of dithiothreitol (20 mM) in the assay did not affect basal caspase 3-like activity in the above experiments.
Figure 7.
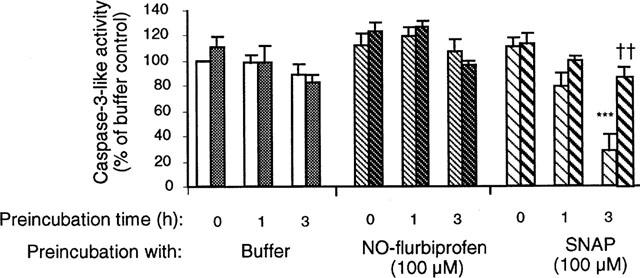
Pre-incubation of 12,000 ×g cell supernatants of gastric epithelial cells with S-nitroso-N-acetyl-penicillamine (SNAP), but not NO-flurbiprofen, causes a time-dependent inhibition of caspase activity. Results are means±s.e.mean from four separate cell cultures. The left-hand and right-hand bars of each pair represent activity measured in the absence and presence of 20 mM dithiothreitol respectively. Results have been analysed by analysis of variance followed by a Newman-Keuls multiple comparison test. The difference highlighted *** is P<0.001 for the comparison between samples incubated with buffer and SNAP for 3 h, and †† is P<0.01 for the effect of dithiothreitol in samples preincubated with SNAP for 3 h.
Discussion
The main finding of this work is that NO-flurbiprofen inhibited apoptosis in guinea-pig gastric epithelial cells, while the parent compound, flurbiprofen promoted apoptosis. Apoptosis was assessed by the condensation and fragmentation of nuclei and by changes in caspase activity. Two pieces of evidence suggest that the hydrolysis of the substrate Ac-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin represented the activity of caspase 3 and similar proteases and not some non-selective peptidase. Firstly, activity was profoundly inhibited by a low concentration of the caspase 3 inhibitor Ac-Asp-Glu-Val-Asp-CHO, and secondly substitution of three amino acids to generate a substrate for caspase 1 gave very much lower enzyme activities. Generally there was good agreement between the two indices of apoptotic activity. Thus both caspase activity and nuclear fragmentation were elevated by flurbiprofen and C6-ceramide in attached cells and by detachment of cells from the culture plate under control conditions, while NO-flurbiprofen decreased both parameters in attached cells. However, detached cells treated with flurbiprofen exhibited an increase in apoptosis without a significant change in an already elevated caspase activity, and NO-flurbiprofen did not inhibit apoptosis in detached cells despite decreasing caspase activity although not to the basal value. In these two instances caspase activity would not seem to have been limiting apoptotic activity.
NO donors have been shown to inhibit caspase activity by S-nitrosylation of the cysteine residue at the active site (Dimmeler et al., 1997; Li et al., 1997) and this is thought to be the mechanism by which such agents inhibit apoptosis in hepatocytes preventing both caspase activity and activation (Kim et al., 1997; Li et al., 1999). Such a mechanism would be compatible with NO-flurbiprofen seeming a more effective inhibitor of caspase activity in control cells compared with those activated by C6-ceramide, or by detachment from the culture plate. Thus the NO, or other reactive species, would be essentially titrated against the caspase activity which would be higher in activated cells. An involvement of cyclic GMP in the inhibitory effect of NO donors on caspase activity has been suggested (Kim et al., 1997; Brune et al., 1998; Fiorucci et al., 1999a). However this mechanism seems unlikely in the present case since the guanylate cyclase inhibitor ODQ did not prevent inhibition of caspase activity by NO-flurbiprofen.
The NO donor SNAP was shown in this work to inhibit caspase activity in homogenates and the reversal by 20 mM dithiothreitol strongly implicates a mechanism involving S-nitrosylation (Dimmeler et al., 1997; Li et al., 1997). However, no such reversal by dithiothreitol was seen if intact cells were pretreated with 1 mM SNAP. A possible explanation is that in intact cells NO prevents the activation of caspase 3 by effects on proximal caspases (Li et al., 1999) rather than inhibiting activated caspase 3. NO-flurbiprofen produced no inhibition of caspase-3-like activity in homogenates, despite inhibiting caspase activity in intact cells within 6 h of addition. Cirino et al. (1996) noted that NO-flurbiprofen released very little NO in vitro, and a possible explanation for the above results is that this compound requires metabolic activation within intact cells. Indeed metabolism of NO-flurbiprofen by an intestinal cytochrome P-450 isoenzyme has been suggested (Bertrand et al., 1998). Alternatively a mercapto group (-SH)-containing compound might be required for release of NO from NO-flurbiprofen (Feelish, 1991). It would therefore be of interest to determine formation of NO, nitrite, nitrate and S-nitrosothiols from NO-flurbiprofen both in the presence and absence of intact cells, but such an investigation is beyond the scope of the present work.
NO-donating NSAIDs inhibit apoptosis induced by TNFα in endothelial cells (Fiorucci et al., 1999b), and this mechanism is probably involved in the mucosal damage and stimulation of apoptosis caused by administration of NSAIDs to rats in vivo not being seen with NO-releasing NSAIDs (Fiorucci et al., 1999a,1999b). We have shown in the present study that a NO-releasing NSAID can also prevent apoptosis in gastric epithelial cells. Furthermore, this mechanism is operative in cells stimulated with C6-ceramide. Ceramide has recently been implicated in the mechanism by which Helicobacter pylori induce apoptosis (Masamune et al., 1999), and there is also evidence that stimulation of apoptosis by TNFα may involve generation of ceramide (Mathias et al., 1998).
Flurbiprofen increased apoptosis in gastric epithelial cells at concentrations above 100 μM. There was no evidence of gross cytotoxicity, such as major loss of cellular protein or increased enzyme leakage, and it can be presumed that the drug was interacting with a specific site. The dependence of protein synthesis could reflect either flurbiprofen directly influencing the synthesis of a particular protein, or that its action on apoptosis depended on a protein which turned over rapidly. The above finding resembles the activation of apoptosis by high concentrations of NSAIDs in transformed cells, which, as here, was slow to take effect and dependent on protein synthesis (Lu et al., 1995; Chan et al., 1998; Simmons et al., 1999; Zhu et al., 1999). This appears to be the first demonstration that such a pathway exists in gastro-intestinal cells in primary culture. However, further investigation will be required to establish whether these cells respond in this way to all NSAIDs. Uncoupling of mitochondrial oxidative phosphorylation by NSAIDs, which is abolished on modification of the carboxyl group as in NO-flurbiprofen, may contribute to their gastro-intestinal toxicity (Mahmud et al., 1996). However such mechanism would not seem to be promoting apoptosis in the gastric epithelial cells since uncoupling by reducing ATP will tip the balance away from apoptosis towards necrosis (Leist et al., 1999), and a mitochondrially-mediated activation of caspase activity should be more rapid and independent of protein synthesis. Metabolism of NO-flurbiprofen within gastric epithelial cells seems likely (see above). The pro-apoptotic effect of any flurbiprofen that is released would however be masked by inhibition of caspase activity by NO or similar reactive species. Indeed the pro-apoptotic effect of flurbiprofen is not seen if cells are also incubated with the NO donor SNAP.
NO-releasing NSAIDs have been shown to have a reduced gastric toxicity compared with the parent compound (Wallace et al., 1994; Wallace, 1997) and in animal models this has been ascribed to maintenance of the gastric microcirculation through the prevention of the effects of TNFα on neutrophil recruitment to endothelial cells and increased apoptosis in these cells (Fiorucci et al., 1999a,1999b). The present work suggests that any apoptotic effect of NSAID, either direct, or mediated by TNFα, on gastric epithelial cells, will not occur with the corresponding NO-NSAID and that such agents would be less likely than the parent compound to promote apoptosis and gastric atrophy in human subjects.
Acknowledgments
This work was supported by a grant from INTAS.
Abbreviations
- C6-ceramide
N-Hexanoyl-D-sphingosine
- NO-flurbiprofen
flurbiprofen nitroxybutyl ester
- NO
nitric oxide
- NSAIDS
non-steroidal anti-inflammatory drugs
- SNAP
S-nitroso-N-acetyl-penicillamine
- TNFα
tumour necrosis factor α
References
- BERTRAND V., GUIMBAUD R., SOGNI P., LAMRANI A., MAUPRIVEZ C., GIROUD J.-P., COUTURIER D., CHAUVELOT-MOACHON L. , CHAUSSADE C. Role of tumour necrosis factor-α and inducible nitric oxide synthase in the prevention of nitro-flurbiprofen small intestinal toxicity. Eur. J. Pharmacol. 1998;356:245–253. doi: 10.1016/s0014-2999(98)00550-0. [DOI] [PubMed] [Google Scholar]
- BRUNE B., VON KNETHEN A. , SANDAU K.B. Nitric oxide and its role in apoptosis. Eur. J. Pharmacol. 1998;351:261–272. doi: 10.1016/s0014-2999(98)00274-x. [DOI] [PubMed] [Google Scholar]
- BYRNE C.R. , HANSON P.J. Induction of heat shock protein 72 by a nitric oxide donor in guinea-pig gastric mucosal cells. Eur. J. Pharmacol. 1998;353:117–122. doi: 10.1016/s0014-2999(98)00388-4. [DOI] [PubMed] [Google Scholar]
- CHAN T.A., MORIN P.J., VOGELSTEIN B. , KINZLER K.W. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1998;95:681–686. doi: 10.1073/pnas.95.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIRINO G., WHEELER-JONES C.P.D., WALLACE J.L., DEL SOLDATO P. , BAYDOUN A.R. Inhibition of inducible nitric oxide synthase expression by novel nonsteroidal anti-inflammatory derivatives with gastrointestinal-sparing properties. Br. J. Pharmacol. 1996;117:1421–1426. doi: 10.1111/j.1476-5381.1996.tb15301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMMELER S., HAENDELER J., NEHLS M. , ZEIHER A.M. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J. Exp. Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEELISCH M. The biochemical pathways of nitric oxide formation from nitrovasodilators: appropriate choice of exogenous NO donors and aspects of preparation and handling of aqueous NO solutions. J. Cardiovasc. Pharm. 1991;17 Suppl.3:S25–S33. [Google Scholar]
- FIORUCCI S., ANTONELLI E., SANTUCCI L., MORELLI O., MIGLIETTI M., FEDERICI B., MANNUCCI R., DEL SOLDATO P. , MORELLI A. Gastrointestinal safety of nitric oxide-derived aspirin is related to inhibition of ICE-like cysteine proteases in rats. Gastroenterology. 1999a;116:1089–1106. doi: 10.1016/s0016-5085(99)70012-0. [DOI] [PubMed] [Google Scholar]
- FIORUCCI S., SANTUCCI L., FEDERICI B., ANTONELLI E., DISTRUTTI E., MORELLI O., DI RENZO G., COATA G., CIRINO G., DEL SOLDATO P. , MORELLI A. Nitric oxide-releasing NSAIDs inhibit interleukin-1β converting enzyme-like cysteine proteases and protect endothelial cells from apoptosis induced by TNFα. Aliment. Pharmacol. Ther. 1999b;13:421–435. doi: 10.1046/j.1365-2036.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- HALL P.A., COATES P.J., ANSARI B. , HOPWOOD D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J. Cell. Sci. 1994;107:3569–3577. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- HONDA K., KATO K., DAIRAKU N., KUBOTA Y., KONNO Y., SUZUKI T., ARIKAWA T., SEKINE H., OHARA S., ASAKI S., SHIMOSEGAWA T. , TOYOTA T. Bax induction and cytochrome c release from mitochondria during nitric oxide induced apoptosis in rat gastric mucosal cell. Gastroenterology. 1999;116:A423. [Google Scholar]
- KIM Y-M., TALANIAN R.V. , BILLIAR T.R. Nitric oxide inhibits apoptosis by preventing increases in caspase 3-like activity via two distinct mechanisms. J. Biol. Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- LEIST M., SINGLE B., NAUMANN H., FAVE E., SIMON B., KUHNLE S. , NICOTERA P. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp. Cell. Res. 1999;249:396–403. doi: 10.1006/excr.1999.4514. [DOI] [PubMed] [Google Scholar]
- LI J., BILLIAR T.R., TALANIAN R.V. , KIM Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- LI J., BOMBECK C.A., YANG S., KIM Y-M. , BILLIAR T.R. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochrondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- LU X., XIE W., REED D., BRADSHAW W.S. , SIMMONS D.L. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7961–7965. doi: 10.1073/pnas.92.17.7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAHMUD T., WRIGGLESWORTH J.M., SCOTT D.L. , BJARNASON I. Mitochondrial function and modification of NSAID carboxyl moiety. Immunopharmacology. 1996;4:189–194. [Google Scholar]
- MANNICK E.E., BRAVO L.E., ZARAMA G., REALPE J.L., ZHANG X-J., RUIZ B., FONTHAM E.T.H., MERA R., MILLER M.J.S. , CORREA P. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in Helicobacter pylori gastritis: effect of antibiotics and antioxidants. Cancer Res. 1996;56:3238–3243. [PubMed] [Google Scholar]
- MASAMUNE A., SHIMOSEGAWA T., MASAMUNE O., MUKAIDA N., KOIZUMI M. , TOYOTA T. Helicobacter pylori-dependent ceramide production may mediate increased interleukin 8 expression in human gastric cancer cell lines. Gastroenterology. 1999;116:1330–1341. doi: 10.1016/s0016-5085(99)70497-x. [DOI] [PubMed] [Google Scholar]
- MATHIAS S., PENA L.A. , KOLESNICK R.N. Signal transduction of stress via ceramide. Biochem. J. 1998;335:465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOSS S.F., CALAM J., AGARWAL B., WANG S. , HOLT P.R. Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut. 1996;38:498–501. doi: 10.1136/gut.38.4.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAFF M. Cell suicide for beginners. Nature. 1998;396:119–122. doi: 10.1038/24055. [DOI] [PubMed] [Google Scholar]
- REDINBAUGH M.G. , TURLEY R.B. Adaptation of the bicinchoninic acid protein assay for use with microtitre plates and sucrose gradient fractions. Anal. Biochem. 1986;153:267–271. doi: 10.1016/0003-2697(86)90091-6. [DOI] [PubMed] [Google Scholar]
- SANTUCCI L., FIORUCCI S., DI MATTEO F.M. , MORELLI A. Role of tumour necrosis factor α release and leukocyte margination in indomethacin-induced gastric injury in rats. Gastroenterology. 1995;108:393–401. doi: 10.1016/0016-5085(95)90065-9. [DOI] [PubMed] [Google Scholar]
- SANTUCCI L., FIORUCCI S., GIANSANTI M., BRUNORI P.M., DI MATTEO F.M. , MORELLI A. Pentoxifylline prevents indomethacin-induced acute gastric mucosal damage in rats: role of tumour necrosis factor alpha. Gut. 1994;35:909–915. doi: 10.1136/gut.35.7.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMMONS D.L., BOTTING R.M,. , ROBERTSON P.M., MADSEN M.L. , VANE J.R. Induction of acetaminophen-sensitive cyclo-oxygenase with reduced sensitivity to nonsteroid antiinflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3275–3280. doi: 10.1073/pnas.96.6.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAHA A.S., NAKSHABENDI I., LEE F.D., STURROCK R.D. , RUSSELL R.I. Chemical gastritis and Helicobacter pylori related gastritis in patients receiving non-steroidal antiinflammatory drugs: Comparison and correlation with peptide ulceration. J. Clin. Pathol. 1992;45:135–139. doi: 10.1136/jcp.45.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESHIMA S., ROKUTAN K., NIKAWA T. , KISHI K. Guinea pig gastric mucosal cells produce abundant superoxide anion through an NADPH oxidase-like system. Gastroenterology. 1998;115:1186–1196. doi: 10.1016/s0016-5085(98)70090-3. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L. Nonsteroidal antiinflammatory drugs and gastroenteropathy: The second hundred years. Gastroenterology. 1997;112:1000–1016. doi: 10.1053/gast.1997.v112.pm9041264. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., REUTER B., CICALA C., MCKNIGHT W., GRISHAM M.B. , CIRINO G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology. 1994;107:173–179. doi: 10.1016/0016-5085(94)90074-4. [DOI] [PubMed] [Google Scholar]
- WONG H.M. , TEPPERMAN B.L. Reduced glutathione modulates Ca2+-mediated damage to rabbit isolated gastric mucosal cells. Am. J. Physiol. 1994;267:G1–G9. doi: 10.1152/ajpgi.1994.267.1.G1. [DOI] [PubMed] [Google Scholar]
- ZHU G.H., WONG B.C.Y., EGGO M.C., CHING C.K., YUEN S.T., CHAN E.Y.T., LAI K.C. , LAM S.K. Non-steroidal anti-inflammatory drug-induced apoptosis in gastric cancer cells is blocked by protein kinase C activation through inhibition of c-myc. Br. J. Cancer. 1999;79:393–400. doi: 10.1038/sj.bjc.6690062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU G.H., YANG X.L., LAI K.C., CHING C.K., WONG B.C.Y., YUEN S.T., HO J. , LAM S.K. Non-steroidal antiinflammatory drugs could reverse Helicobacter pylori-induced apoptosis and proliferation in gastric epithelial cells. Dig. Dis. Sci. 1998;43:1957–1963. doi: 10.1023/a:1018830408397. [DOI] [PubMed] [Google Scholar]