

Abstract
The pharmacological activity of phenylacetyl-Phe-Ser-Arg-N-(2,4-dinitrophenyl)-ethylenediamine (TKI), a tissue kallikrein specific inhibitor, was assessed using models of nociception and inflammation in mice.
Injection of TKI (13.6–136 μmol kg−1, i.p. or 41–410 μmol kg−1, s.c.) produced a dose-related inhibition of the acetic acid-induced writhes (by 37 to 85% or 34 to 80%, respectively). The antinociceptive activity of TKI (41 μmol kg−1, i.p.) was maximal after 30 min injection and lasted for 120 min. The effect was unaltered by pretreatment with naloxone (8.2 μmol kg−1, s.c.) or bilateral adrenalectomy.
TKI (41 and 136 μmol kg−1, i.p.) produced a dose-related decrease of the late phase of formalin-induced nociception by 79 and 98%, respectively. At 136 μmol kg−1, i.p., TKI also shortened the duration of paw licking in the early phase by 69%. TKI (41 and 136 μmol kg−1, i.p.) also reduced the capsaicin-induced nociceptive response (by 51 to 79%).
TKI (41 μmol kg−1, i.p. or 410 μmol kg−1, s.c.) reduced the oedematogenic response, from the second to the fifth hour after carrageenin injection by 36 to 30% or by 47 to 39%, respectively.
Pretreatment with TKI (41 μmol kg−1, i.p.) reduced the capsaicin-induced neurogenic inflammation in the mouse ear by 54%.
It is concluded that TKI presents antinociceptive and antiinflammatory activities mediated by inhibition of kinin formation by tissue kallikrein in mice. The results also indicate that the tissue kallikrein-dependent pathway contributes to kinin generation in nociceptive and inflammatory processes in mice.
Keywords: Tissue kallikrein inhibitor, kinins, nociception, inflammation
Introduction
Activation of the kallikrein-kinin cascade in injured tissues causes the release of the nonapeptide bradykinin (BK) in the plasma, and the closely related decapeptide kallidin (Lys-BK) in tissues. The endogenous precursor of BK is the high molecular weight kininogen (HMWK), while the tissue precursor for Lys-BK is the low molecular weight kininogen (LMWK). Activation of prekallikrein bound to either precursor originates two kininogen-processing serine proteases (kallikreins) that are physicochemically and immunologically distinct (Bhoola et al., 1992a). In the blood, plasma kallikrein releases BK and the active tissue kallikrein releases kallidin, except in rats where BK rather than kallidin is produced. (Bhoola et al. 1992a; Sharma 1993; Walker et al., 1995). Once kinins are generated, they exert their pharmacological effects through interaction with two main specific receptors, B1 and B2 (Sharma, 1993). Local actions of BK and Lys-BK produce, among others, vasodilatation, increased vascular permeability, stimulation of nociceptive nerve terminals causing hyperalgesia and pain, and spasmogenic effect on smooth muscles (Bhoola et al., 1992a; Sharma, 1993; Perkins et al., 1995; Walker et al., 1995; Poole et al., 1999; Ahluwalia & Perretti, 1999). For this reason, kinins have been involved in the pathogenesis of inflammatory processes like rheumatoid arthritis and asthma (Bhoola et al. 1992b; Polosa, 1993; Bond et al., 1997; Deshandra & Bhoola, 1998).
Because LMWK and tissue kallikrein are localized in tissues and plasma kallikrein does not release kinin from LMWK, kallidin is expected to be the predominant kinin formed in tissues in inflammatory responses (Bhoola et al., 1992a; Walker et al., 1995). In addition, the tissue kallikrein-dependent pathway has been involved in rheumathoid arthritis (Selwyn et al., 1989; Williams et al., 1997), allergic diseases (Polosa, 1993) and experimental pancreatitis (Blackberg & Ohlsson, 1994). Burton & Benetos (1989) and Okunishi et al. (1989) reported the effects of tissue kallikrein inhibitors in the blood pressure of the rat. Szelke et al. (1994) described the effects of synthetic inhibitors of tissue kallikrein in a model of allergic inflammation in guinea-pigs. The effects of tissue kallikrein specific inhibitors, however, were not tested in nociceptive transmission and inflammatory response in mice.
This study aimed to investigate the pharmacological activity of a novel tissue kallikrein specific inhibitor, phenylacetyl-Phe-Ser-Arg-EDDnp where EDDnp is N-(2,4-dinitrophenyl)-ethylenediamine (TKI) (Juliano et al., 1995; Bizeto et al., 1996; Portaro et al., 1997), in experimental models of chemical and thermal nociception and inflammation in mice. The results indicated that TKI is endowed with antinociceptive and anti-inflammatory properties mediated in part by the tissue kallikrein-dependent pathway.
Methods
Adult albino mice (25–35 g) of either sex, kept under controlled 12/12 h light/dark cycle and temperature (23±2°C) were used. Food and water were provided ad libitum. Animals used in the nociception studies were acclimatized to the observation chamber during 15 min prior to testing. All experiments were conducted according to the ethical guidelines for investigation of experimental pain in conscious animals (Zimmerman, 1983).
Antinociceptive activity
Abdominal constriction models
The abdominal constriction response was induced as previously described with minor modifications (Emim et al., 1994). Mice were treated intraperitoneally (i.p.) or subcutaneously (s.c) with either the vehicle (saline+tween 80, 40 : 1 v v−1, 0.1 ml 10 g−1) or TKI (4.1–136 μmol kg−1, i.p. or 13.6–410 μmol kg−1, s.c.) After 30 min all animals were injected with either 1.2% acetic acid (0.1 ml 10 g−1, i.p.) (Koster et al., 1959) or kaolin (200 mg kg−1, i.p.) (Fujioshi et al., 1989), placed individually in a glass cylinder (18 cm diameter) and the number of abdominal constrictions was counted cumulatively over 20 or 30 min, respectively. Mice treated with HOE-140 (2.7 μmol kg−1, s.c.), a bradykinin B2 receptor antagonist, with PKSI-527 (22.9, 68.6 and 229 μmol kg−1, i.p) or with soybean trypsin inhibitor (SBTI: 0.48. 1.43 and 4.8 μmol kg−1, i.p.), a specific and nonspecific plasma kallikrein inhibitors, respectively, were used for positive controls.
To evaluate the influence of endogenous corticosteroids release, the same doses of TKI were tested in the acetic acid-induced writhing after 1 week bilateral adrenalectomy in mice. Possible mediation by the opioid system was evaluated after injection with naloxone (8.2 μmol kg−1, s.c.) 15 min prior to TKI (41 μmol kg−1, i.p.) administration.
Formalin test
The formalin test was performed in mice using an acrylic box (12×12×12 cm) mounted with a mirror at a 45° angle beneath the floor (Hunskaar et al., 1985). Following the acclimatization period, mice were treated with either the vehicle or TKI (13.6–136 μmol kg−1, i.p.), 30 min before injection of 50 μl 3% formalin (1.2% formaldehyde in PBS, s.c.) into the plantar surface of the right hindpaw. Each animal was then returned to the observation chamber and the amount of time spent by the animals licking or biting the injected paw was taken as the index of nociception. The effects of TKI was investigated during the early phase of the nociceptive response designated as neurogenic pain (0–5 min after formalin injection), and the late phase related to the inflammatory pain (15–30 min after formalin injection) (Hunskaar & Hole, 1987). Mice treated orally with indomethacin (28 μmol kg−1, p.o.) were used for positive control.
A similar procedure was used to assess the TKI effect on the capsaicin-induced neurogenic pain (Sakurada et al., 1992). Following 15 min adaptation to the observation chamber, mice were treated with either the vehicle, TKI (13.6–136 μmol kg−1, i.p.) or fentanyl (19–190 nmol kg−1, s.c.) for positive control. After 30 min all animals were injected with 50 μl capsaicin (32 μg ml−1, in saline, s.c.) into the plantar surface of the right hindpaw and the index of nociception was quantitated as described above.
Tail flick test
The nociceptive stimuli were induced in mice by a constant focused heat stimulus on the tail delivered by an analgesiometer apparatus (Ugo Basile, Italy) as previously described (D'Amour & Smith, 1941; Guillén et al., 1997). Animals used in this test were selected according to their responses to the nociceptive stimuli and by eliminating those mice with reaction time up to 7 s. A cut-off time of 20 s was maintained throughout the procedure to prevent tissue damage. Each mouse response was elicited every 30 min from 1 h up to 2 h after treatment with either the vehicle. TKI (41 and 136 μmol kg−1, i.p.) or the opioid analgesic fentanyl (567 nmol kg−1, i.p.) for positive control. The animal's reaction to the heat was quantitated as the latency of the tail flick response.
Antiinflammatory activity
Mouse paw oedema induced by carrageenin, bradykinin, histamine and serotonin
Mice were treated with either the vehicle or TKI (13.6 and 41 μmol kg−1, i.p., or 136 and 410 μmol kg−1, s.c.) 30 min before injection of 50 μl 1% carrageenin, s.c., into the plantar surface of the right hind paw. Positive control animals were given indomethacin (28 μmol kg−1, p.o.) 60 min before. The contralateral paw was injected with equal volume of saline. The paw volumes were determined hourly for 5 h in a hydroplethysmometer (Ugo Basile) and the swelling, expressed in μl, was calculated as the difference between the two paws (Henriques et al., 1987).
The effect of TKI (41 μmol kg−1, i.p.) was also assessed on the mouse paw oedema induced by intraplantar injection of either bradykinin, histamine or serotonin (50 μg each, s.c.) after 30 min treatment. The respective positive control animals were given HOE-140 (0.5 μmol kg−1), diphenhydramine (206 μmol kg−1) or cyproheptadine (1.5 μmol kg−1), i.p.
Carrageenin-induced peritonitis
The acute inflammatory response was induced by injection of 1% carrageenin (0.25 ml in saline, i.p.) after 30 min treatment with either the vehicle or TKI (13.6–136 μmol kg−1, i.p.). The positive control group was pretreated with dexamethasone (1.3 μmol kg−1, i.p.). After 4 h carrageenin injection, all animals were killed under ether anaesthesia and 2 ml of PBS containing heparin (10 iu ml−1) was injected into the peritoneal cavity. Following a gentle massage, peritoneal fluids were collected and the number of leukocytes that had migrated to the peritoneal cavity was counted in a Neubauer chamber (Ferrándiz & Alcaraz, 1992).
Ear oedema-induced by croton oil and capsaicin
Thirty minutes after treatment of mice with the vehicle or TKI (41 μmol kg−1, i.p.) 10 μl of croton oil solution (2.5% in acetone) were applied topically to the inner surface of the right ear. The left ear was treated with the same volume of acetone for control. After 4 h, all animals were killed by cervical dislocation under ether anaesthesia and a 6 mm diameter disk removed from each ear lobe. The difference between the ear disk weights was taken as the oedema induced by croton oil (Schianterelli et al., 1982).
Neurogenic inflammation was induced in mice by topical application of 20 μl capsaicin solution (5 mg ml−1 in acetone) to the inner surface of the right ear (Inoue et al., 1993) 30 min after treatment with either the vehicle or TKI (41 μmol kg−1, i.p.). The left ear was treated with 20 μl of acetone for control. After 30 min, all animals were killed by cervical dislocation under ether anaesthesia and the ear oedema quantitated as described above.
Drugs and chemicals
Drugs used were: bradykinin, capsaicin, histamine hydrochloride, naloxone hydrochloride, serotonin hydrochloride, cyproheptadine hydrochloride, diphenhydramine, soybean trypsin inhibitor (Sigma, U.S.A.), κ-carrageenin (Cialgas, Brazil), Croton Oil (Veafarm, Brazil), Fentanyl (Janssen Farmacêutica, Brazil), and Heparine (Roche®, Brazil). HOE-140 (D-Arg-[Hyp3-Thi5-D-Tic7-Oic8]-bradykinin) (Hoechst, Germany). Phenylacetyl-FSR-EDDnp was obtained by peptide synthesis in solution using anhydride procedures and tert-butyloxycarbonyl-amino acids, purified in silica gel and characterized by HPLC as previously described (Juliano & Juliano, 1985; Chagas et al., 1991). PKSI-527 was provided by Dr Yoshio Okada from the Faculty of Pharmacy Sciences, Kobe-Gakuin University, Kobe, Japan. For pharmacological tests, the drug was dissolved in physiological saline (0.9%) using tween 80 (40 : 1). All other reagents were of analytical grades. Stock drug solutions were prepared just before use in 0.9% w v−1 of NaCl, except indomethacin and capsaicin which were dissolved in NaHCO3 and ethanol, respectively. Formalin solution was prepared in phosphate buffer saline (PBS: NaCl 135 mM, KCl 2.7 mM and phosphate buffer 10 mM).
Statistical analysis
All data were expressed as means±s.e.mean. ID50 (doses to reduce the number of responses to 50% of control) values were calculated by linear regression from the data against logarithm of doses. Statistical significance of the results was determined using one-way analysis of variance followed by the Duncan method (Sokal & Rohlf, 1981). Differences between two means were compared using unpaired Student's t-test. Data were considered different at the level of P<0.05.
Results
Effect of TKI on nociceptive transmission
Acetic acid-induced writhing
Mice treated with TKI (4.1 –136 μmol kg−1, i.p.) did not show signs of toxicity up to 24 h afterwards compared to control animals injected with the vehicle (saline+tween). A few animals presented occasional abdominal constrictions immediately after injection of the highest dose.
Injection of 1.2% acetic acid (0.1 ml 10 g−1, i.p.) in control mice treated i.p. or s.c. with the vehicle produced respectively, 26.2±3.3 (n=9) or 30.5±2.3 writhes (n=10) over 20 min. Previous treatment with TKI (13.6–136 μmol kg−1, i.p.) produced a long lasting and dose-related inhibition of abdominal constrictions by 37 to 85% of control, with a mean effective dose (ID50) of 17.7 μmol kg−1 (Figure 1A). In mice pretreated with 41 μmol kg−1, i.p., the TKI-induced antinociceptive response reached its maximum after 30 min injection, lasted for 120 min and then decayed (Figure 2). At a dose below 13.6 μmol kg−1 TKI did not affect the acetic acid-induced nociceptive response (Figure 1A).
Figure 1.
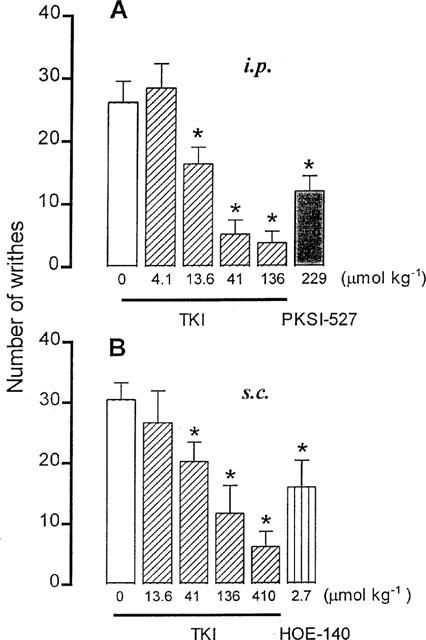
Accumulative number of writhings induced by injection of acetic acid (1.2%, 0.1 ml 10 g−1, i.p.) in mice treated 30 min before with either the vehicle (0), TKI (4.1–136 μmol kg−1) and PKSI (229 μmol kg−1) intraperitoneally (i.p.) in (A) or with TKI (13.6– 410 μmol kg−1) and HOE-140 (2.7 μmol kg−1) subcutaneously (s.c.), in (B) The columns and vertical bars are means±s.e.mean of 6–8 animals. *Different from the vehicle-treated mice (P<0.05).
Figure 2.
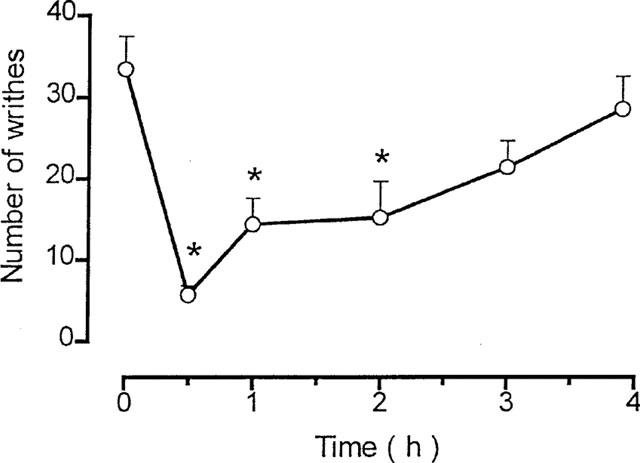
Time course of the antinociceptive effect of TKI (41 μmol kg−1, i.p.) on the acetic acid-induced abdominal constrictions in mice. Symbols and vertical bars are means±s.e.mean of 6–8 animals. *Different from vehicle-treated mice (P<0.05).
A similar effect was obtained after s.c. injection of TKI (41–410 μmol kg−1) which reduced abdominal constrictions by 34 to 84%, with ID50 value of 85.7 μmol kg−1 (Figure 1B).
In positive control animals treated with PKSI-527 (22.9–229 μmol kg−1, i.p.), a specific plasma kallikrein inhibitor (Teno et al., 1993), the acetic acid-induced writhes were reduced by 10 to 60%, with ID50 value of 186.5 μmol kg−1 (Figure 1). Likewise, pretreatment of mice with SBTI (0.48–4.8 μmol kg−1, i.p.), a non specific plasma kallikrein inhibitor (Katori et al., 1989), reduced the acetic acid-induced abdominal constrictions by 37 to 58%, with ID50 value of 3.1 μmol kg−1. Comparatively, injection of mice with HOE-140 (2.7 μmol kg−1, s.c.), a bradykinin B2 receptor antagonist (Wirth et al., 1991), reduced the acetic acid-induced writhing by 48%.
Effect of naloxone and adrenalectomy
The inhibitory effect of TKI on the acetic-acid induced writhing was unaffected by a dose of naloxone (8.2 μmol kg−1, s.c.) that completely reversed the analgesic activity of the opioid fentanyl (190 nmol kg−1, s.c.) (Table 1). Likewise, adrenalectomy (ADX) of mice did not alter the TKI antinociceptive activity as compared to the observed in sham-operated (S) animals (S: 36.5±6.6 and TKI-treated: 9.9±2.0 writhes, ADX: 26.6±4.6 and TKI-treated: 4.2±3.1 writhes, n=6 each).
Table 1.
Effect of naloxone (NAL) on antinociception induced by phenylacetyl-Phe-Ser-Arg-EDDnp (TKI) or fentanyl (FEN) in the acetic acid-induced abdominal constrictions in mice

Kaolin-induced writhing
Injection of kaolin (200 mg kg−1, i.p.) to control vehicle-treated mice 30 min beforehand induced 8.8±1.6 writhes (n=6). Treatment with the kininase II inhibitor enalapril (20.3 μmol kg−1, s.c.), 120 min before kaolin, increased the number of abdominal constrictions by 153% (22.3±2.6 writhes in 30 min, n=6). Injected at a dose that inhibited the acetic acid-induced writhing, TKI (136 μmol kg−1, s.c.) reduced the number of abdominal constrictions in enalapril-treated animals by 47% (11.8±2.8 writhes in 30 min). Similar effect was observed in animals pretreated with SBTI (1.4 μmol kg−1, i.p.) which inhibited writhing by 72% of control (17.7±2.0, writhes in 30 min, n=8).
Formalin-induced nociception
In control vehicle-treated mice the duration of paw licking during the first (neurogenic pain) and second phase (inflammatory pain) of the formalin-induced nociception was 78.4±8.9 and 120.7±17.3 s (n=8), respectively. Previous administration of TKI (41 and 136 μmol kg−1, i.p.) shortened the second phase of the formalin-induced nociception by 79 and 98%, respectively (Figure 3A). A low dose of TKI (13.6 μmol kg−1, i.p.) did not affect the second phase, while a high dose (136 μmol kg−1, i.p.) shortened the first phase by 69% (Figure 3A). The ID50 values calculated in this assay were 88.4 and 20.4 μmol kg−1 in the first and second phase, respectively.
Figure 3.
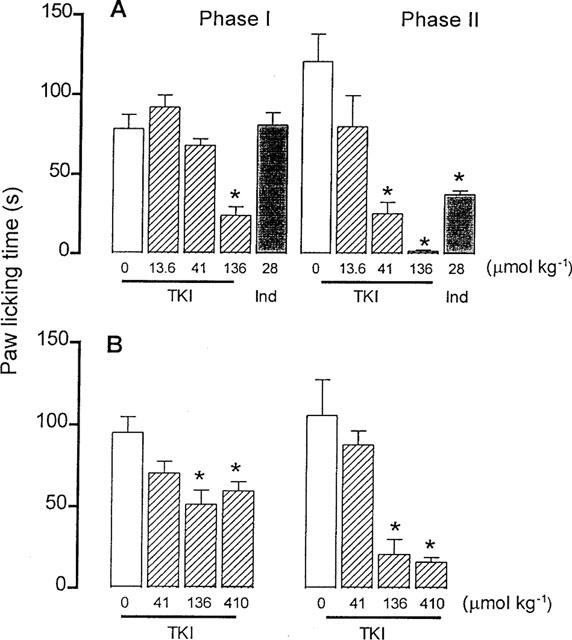
Effect of the vehicle (0) and TKI injected intraperitoneally (13.6–136 μmol kg−1), in (A) or subcutaneously (41–410 μmol kg−1), in (B) on the formalin-induced (50 μl 1.2% formaldehyde in PBS) first (neurogenic pain) and second (inflammatory pain) phases of nociception in mice. Positive control mice were treated orally with indomethacin (Ind, 28 μmol kg−1, A). The columns and vertical bars are means±s.e.mean of 8–9 animals. *Different from vehicle-treated mice (P<0.05).
When injected s.c. TKI (136 and 410 μmol kg−1) shortened the licking time during the first phase, by 46 and 37% of control (95.1±9.5 s), as well as that related to the second phase, by 80 and 85% (control : 105.8±25.0 s) (Figure 3B). Pretreatment of mice with the non steroidal anti-inflammatory agent indomethacin (28 μmol kg−1, p.o.) did not affect the licking time during the first phase, but it shortened the licking time observed during the second phase of the formalin-induced pain by 69% of control (81.0±7.7 s) (Figure 3B).
Capsaicin-induced nociception
Plantar injection of capsaicin (0.016 to 6.4 μg) into the mouse right paw produced a short lasting paw licking related to the dose that was maximal at 1.6 μg (43.5±7.8 s, n=8). Pretreatment of mice with TKI up to 13.6 μmol kg−1, i.p., did not influence the capsaicin-induced nociceptive response. At 41 and 136 μmol kg−1, however, TKI reduced the capsaicin-induced neurogenic pain by 51 and 79%, respectively (Figure 4A). The calculated ID50 of TKI in this assay was 23 μmol kg−1, similar to that determined in the late phase of the formalin test.
Figure 4.
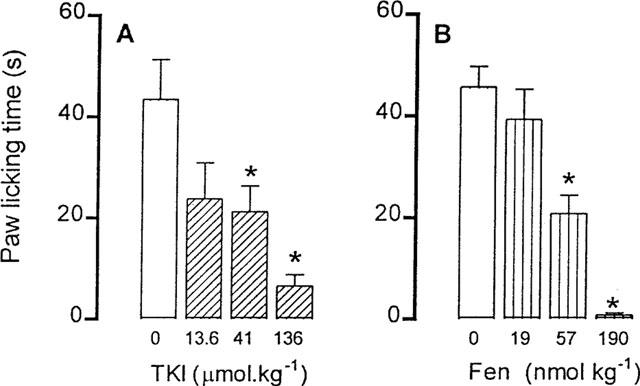
Effect of previous intraperitoneal injection of the vehicle (0) or TKI (13.6–136 μmol kg−1) on capsaicin-induced (1.6 μg paw−1) neurogenic pain in mice. Positive control animals were pretreated with fentanyl (Fen, 19–190 nmol kg−1). The light column represents the values of animals injected with the vehicle. The columns and vertical bars are means±s.e.mean of eight animals. *Different from vehicle treated mice (P<0.05).
Pretreatment with the opioid analgesic fentanyl (56.7 and 190 nmol kg−1, s.c.) was more effective than that with TKI, reducing the capsaicin-induced neurogenic pain by 54 and 98%, respectively (Figure 4B).
Tail flick test
In control mice given the vehicle i.p. or s.c., the basal tail flick latency was 4.7±0.3 or 4.8±0.2 s (n=7 in each group). Treatment with TKI (41 and 136 μmol kg−1, i.p. or 136 μmol kg−1, s.c.) did not alter the animal's nociceptive response over 2 h measurements (i.p.: 4.9±0.5 and s.c.: 4.6±0.3 s, n=7, respectively). Treatment with fentanyl (567 nmol kg−1, s.c.), however, increased the tail flick latency from 4.8±0.2 s (control) to 11.2±1.9 s (n=6 each).
Effect of TKI on the inflammatory responses
Carrageenin-induced paw oedema
Injection of carrageenin in control mice treated with the vehicle, i.p. or s.c., induced a progressive swelling of the paw that reached a maximum volume (i.p.: 121.0±4.8, s.c.: 101.7±8.6 μl, n=8) after 4 h. Pretreatment with TKI (41 μmol kg−1, i.p. or 410 μmol kg−1, s.c.) inhibited the paw oedema by 36 to 30% or by 47 to 39%, respectively, from the second to the fifth hour after carrageenin injection (Figure 6). In positive control animals treated with indomethacin (28 μmol kg−1, p.o.), the maximal carrageenin paw oedema was decreased by 45% of control (Figure 5).
Figure 6.
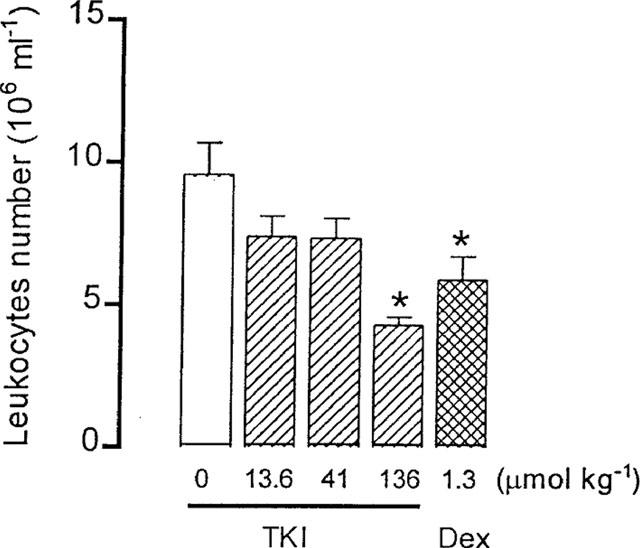
Effect of previous intraperitoneal (i.p.) injection of the vehicle (0) or TKI (13.6–136 μmol kg−1) on cell migration of the carrageenin (0.25 ml 1% in saline) induced peritonitis in mice. Positive control mice were treated with dexamethasone (Dex. 1.3 μmol kg−1, i.p.). The light column represents the values of animals injected with the vehicle. The columns and vertical bars are means±s.e.mean of 6–8 animals. *Different from vehicle treated mice (P<0.05).
Figure 5.
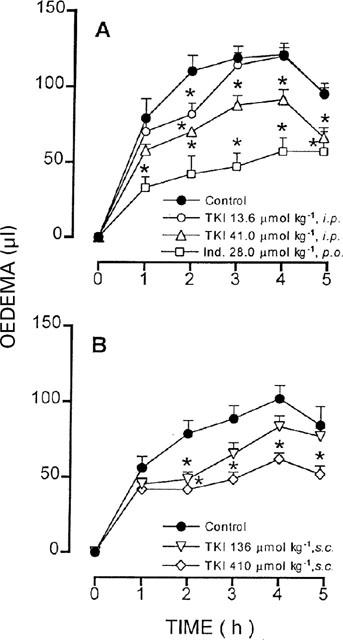
Effect of previous treatment with TKI injected intraperitoneally (13.6 and 41 μmol kg−1, i.p.) in (A) or subcutaneously (136 and 410 μmol kg−1, s.c.), in (B) on the paw oedema induced by carrageenin (0.5 mg paw−1) in mice. Control animals were injected i.p. or s.c. with the vehicle. The positive control group was treated orally with indomethacin (28 μmol kg−1, p.o.). Symbols and vertical bars are means±s.e.mean (eight animals in each group) of the volume difference between the paw injected with carrageenin and the contralateral paw injected with saline. *Different from control (P<0.05).
Bradykinin, histamine and serotonin-induced paw oedema
Injection of either bradykinin, histamine or serotonin (50 μg each) in control mice produced oedema of the hindpaw with maximal increase in volume within 30 min (respectively, 63.8±9.6, 72.9±7.5 and 65.0±9.9 μl, n=8). At a dose effective in reducing nociception and carrageenin-induced paw oedema, TKI (41 μmol kg−1, i.p.) did not affect the paw oedema induced by either phlogistic agent. A high dose of TKI (136 μmol kg−1, i.p.), however, reduced the bradykinin-induced paw oedema by 51% of control. In positive control animals pretreated with the antagonists of bradykinin B2 receptors, HOE-140 (0.5 μmol kg−1, i.p.), histamine H1 receptors, diphenhydramine (206 μmol kg−1, i.p.) or serotonin receptors, cyproheptadine (1.5 μmol kg−1, i.p.), the paw oedema induced by bradykinin, histamine or serotonin was decreased by 44, 50 and 51%, respectively.
Carrageenin-induced peritonitis
Intraperitoneal injection of carrageenin in control vehicle-treated mice induced migration of 9.6±1.1×106 leukocytes ml−1 (n=8). Previous treatment of mice with TKI (13.6, 41 and 136 μmol kg−1, i.p.) decreased the leukocytes infiltration by 55% (4.3±0.3×106 ml−1, n=7) only at the highest dose (Figure 6). In parallel experiments, treatment with the steroid antiinflammatory agent dexamethasone (1.3 μmol kg−1, i.p.) reduced the leukocytes migration by 40% (5.9±0.8×106 ml−1, n=7)
Ear oedema induced by croton oil and capsaicin
Topical application of croton oil increased the ear disk weight of control mice from 10.6±0.4 mg (ear injected with vehicle) to a maximum of 22.6±0.5 mg (n=8) after 30 min. Previous injection of TKI (41 μmol kg−1, i.p.) did not prevent the exudative process induced by croton oil (Control: 12.1±0.3 and TKI: 10.3±1.1 mg, n=8). Nevertheless, the same treatment with TKI was effective on the neurogenic inflammation induced by capsaicin in the mouse ear reducing its disk weight from 7.9±1.3 mg to 3.6±1.0 mg (n=6 each).
Oral treatment (2.5 μmol kg−1) or topical application (0.25 μmol ear−1) of dexamethasone in positive control groups reduced the ear oedema induced by croton oil from 12.0±1.0 to 5.5±1.5 mg (n=5) and from 12.1±0.3 to 3.7±0.7 mg, respectively.
Discussion
The pharmacological activity of phenylacetyl-FSR-EDDnp, a novel tissue kallikrein inhibitor (TKI) synthetized according to the specificities of the enzyme extended binding site (Juliano et al., 1995; Portaro et al., 1997), was assessed in nociceptive and inflammatory processes. Mice were used because in this species the active tissue kallikrein liberates Lys-bradykinin (kallidin) rather than bradykinin (Bhoola et al., 1992a), as occurs in humans. The tissue kallikrein is an acidic glycoprotein with catalytic reaction steps similar to those described for other serine proteases, but sharing antigenic and structural identities distinct from those of plasma kallikrein (Bhoola et al., 1992a). Detection of this kininogenase in synovial fluids, neutrophils and bronchoalveolar lavage fluid of asthmatic subjects has suggested an important role for the tissue kallikrein system in inflammatory processes (Christiansen et al., 1987; Selwyn et al., 1989; Bhoola et al., 1992b; William et al., 1997).
TKI was active upon intraperitoneal or subcutaneous administration in mice without inducing signs of toxicity or changes in the spontaneous motor activity, as compared with control animal. Short lasting abdominal constrictions were observed in a few animals after i.p. injection of the highest dose tested. These occasional and transitory effects were attributed to peritoneal irritation at the injection site, and possibly reflecting the non specific effects at high doses of TKI.
The antinociceptive activity of TKI was evaluated on the acetic acid or kaolin-induced writhes, on the formalin-induced paw licking, on the capsaicin-induced neurogenic pain and on the tail flick test. Pretreatment of mice with TKI reduced the acetic acid-induced writhing after either i.p. or s.c. administration, with ID50 value five times higher after upon the former treatment than that obtained after the latter. The effect was related to the dose, reached maximal value after 30 min i.p. injection, and lasted for about 2 h.
The acetic acid-induced writhing is a standard test for pain, sensitive to opiates as well as non-opiates analgesics (Siegmund et al., 1957; Steranka et al., 1987). The associated nociceptive response is believed to involve the release of endogenous substances such as bradykinin (BK) and prostanoids among others, that stimulate nociceptive endings (Whittle, 1964; Berkenkopf & Weichman, 1988). The antinociceptive effect of TKI determined on this assay was unaffected by previous administration of naloxone or bilateral adrenalectomy, excluding opioid mediation or corticosteroids release from the adrenal cortex by either direct or indirect actions of the drug. The effect, however, could be explained by inhibition of kinin formation by tissue kallikrein, as previously shown in in vitro and in vivo studies (Juliano et al., 1995; Bizeto et al., 1996).
Both PKSI-527 and SBTI, a specific and non-specific plasma kallikrein inhibitors, respectively, also reduced the mouse writhing response indicating involvement of the kallikrein–kinin system (KKS), in agreement with other studies (Steranka et al., 1987; Chau et al., 1991; Heapy et al., 1993). In addition, the BK B2 receptor antagonist HOE-140 reduced the acetic acid-induced writhes, confirming other reports (Steranka et al., 1987; Heapy et al., 1993). These data indicated activation of both tissue and plasma kinin releasing pathways in the mouse writhing response. The ID50 value of TKI determined in this assay was 10 times lower than that determined for PKSI-527, indicating a higher potency of the tissue kallikrein inhibitor in the writhing response. TKI was shown to inhibit human tissue kallikrein in vitro with Ki=0.7 μM, but it did not inhibit human plasma kallikrein at concentrations 1000 fold higher than its Ki value (Portaro et al., 1997). PKSI-527 in its turn, inhibited plasma kallikrein in vitro with Ki value of 0.81 μM, while it inhibited tissue kallikrein with Ki values higher than 500 μM (Okada et al., 1999). Assuming that both TKI and PKSI-527 exhibit the same specifity in vitro and in vivo, our results suggest a predominant role of tissue kallikrein in the mouse writhing response.
At a dose inhibiting the acetic acid-induced writhing, TKI also reduced writhing induced by kaolin in mice, supporting a role of the tissue kinin release in nociception. Kaolin is an activator of Factor XII reported to induce nociception mainly through activation of the KKS and release of BK (Fujiyoshi et al., 1989). Treatment with the kininase inhibitor enalapril prevents degradation of BK, leading to potentiation of the writhing reaction and increase of the assay specificity (Fujiyoshi et al., 1989). Potentiation of the kaolin-induced writhing in enalapril-treated mice was reduced by injection of the serine protease inhibitor SBTI, favouring a role of the KKS in this model. This possibility is reinforced by the reported inhibition of the kaolin-induced writhing by BK B2 receptor antagonists (Corrêa et al., 1996). Although the plasma kinin releasing pathway is important in this assay, a possible endogenous activation of tissue kallikrein by plasma kallikrein (Takada et al., 1985; Proud & Kaplan, 1988) can not be excluded, which may explain the TKI antinociceptive effect in enalapril-treated mice.
Administered either i.p. or s.c., TKI reduced both the neurogenic (first phase) and inflammatory (second phase) pain in the formalin test. The first phase is attributed to direct activation of nociceptors and primary afferents fibres by formalin, causing the release of BK and tachykinins (Shibata et al., 1989; Corrêa & Calixto, 1993). This phase is inhibited by opioid antagonists and BK B2 receptor antagonists (Corrêa et al., 1996). The second phase is due to an inflammatory reaction caused by tissue injury, involving the release of histamine, serotonin, prostaglandins, bradykinin, and excitatory aminoacids (Coderre & Melzack, 1992; Damas & Liegeois, 1999). This late phase is inhibited by non-steroidal anti-inflammatory drugs (NSAID), opioid analgesics (Hunskaar & Hole, 1987), and BK B2 receptor antagonists (Corrêa & Calixto, 1993). Although TKI reduced both neurogenic and inflammatory responses, the effect was more prominent on the latter phase. The ID50 value of TKI determined in the second phase (20.4 μmol kg−1) was four times lower than that obtained in the first phase, and similar to that determined on the acetic acid assay (17.7 μmol kg−1), indicating that the antinociceptive effect of TKI is related to inhibition of the release of kinins.
A number of studies have shown that BK receptor antagonists strongly inhibit the second phase of the formalin-induced pain, while they reduce the first phase only at high doses (Corrêa & Calixto, 1993, Campos et al., 1996). BK was also reported to produce pain and hyperalgesia by activation of B2 receptors in sensory neurons (Steranka et al., 1987; Dray, 1995), implicating the KKS on the formalin-induced excitation of nociceptive afferents. Accordingly, inhibition of the release of endogenous proinflammatory mediators by tissue kallikrein may explain the inhibitory effect of TKI on both neurogenic and inflammatory pain in the formalin test.
Further evidence of the TKI effectiveness on neurogenic pain was obtained in the capsaicin-induced pain test. Intraplantar injection of capsaicin in the mouse hindpaw induces a single phase nociceptive response similar to that displayed in the former phase of the formalin test (Sakurada et al., 1992). The response to capsaicin is attributed to direct activation of specific receptors leading to stimulation of Aδ and C afferent nociceptive fibres, and to the release of glutamate and neuropeptides (Dray, 1995). This response was inhibited by BK B2 receptor antagonists (Corrêa et al., 1996), and related to activation of the KKS (Shibata et al., 1989). Treatment of mice with TKI, i.p., reduced the capsaicin-induced nociceptive response with ID50 value (23 μmol kg−1) similar to those values obtained in the acetic acid assay (17.7 μmol kg−1) and the second phase of the formalin test (20.4 μmol kg−1), indicating common underlying mechanisms in all three tests, probably related to inhibition of tissue kallikrein.
At a dose producing pronounced antinociceptive effect, TKI (136 μmol kg−1, i.p.) did not affect the nociceptive responses in the mouse tail flick test. This model involves spinal nociceptive reflexes (Laneuville et al., 1988; Bauer et al., 1992) and it is sensitive to opioid modulation (Suh et al., 1989). The lack of effect of TKI in this test was expected since no evidence of BK participation on the transmission of spinal nociceptive reflexes was reported (Corrêa et al., 1996), providing further support for involvement of the KKS in the TKI-induced antinociceptive effect.
To evaluate the effect of TKI on acute inflammatory responses, standard models were used: the paw oedema induced by different phlogistic agents, carrageenin-induced peritonitis and the ear oedema induced by croton oil or capsaicin. Carrageenin is a polysaccharide known to activate the Hageman factor and to liberate kallikrein from its inactive precursor prekallikrein (Hargreaves et al., 1988). At doses effective in nociception, TKI reduced the paw swelling induced by carrageenin with the effect lasting for 5 h at the highest doses (41 μmol kg−1, i.p., and 410 μmol kg−1, s.c.). The effect could also be related to inhibition of kinins release, which have been implicated as major proinflammatory mediators in addition to histamine and prostaglandins (Di Rosa et al., 1971; Hargreaves et al., 1988). In the same model, tissues kinins were shown to be maximal after 1 h injection of carrageenin and to induce the release of other mediators that interact in synergism producing the oedema. The effect was reported to be mediated by B2 receptors and to last for 8 h (Décarie et al., 1996). Specific BK B2 receptor antagonists inhibited the carrageenin-induced paw oedema in rats (Burch & DeHaas, 1990; Décarie et al., 1996) and mice (Corrêa et al., 1996), confirming involvement of kinins in this model of acute inflammation.
At doses effective on the carrageenin-induced paw oedema. TKI did not affect the paw oedema produced by either BK, histamine or serotonin suggesting its specific action in those models involving kinins release. The BK-induced paw oedema, however, was reduced by a high dose of TKI (136 μmol kg−1, i.p.) indicating additional mechanisms, possibly nonspecific. In fact, being a protease inhibitor one can not exclude such action at high doses of TKI. This action could also explain the decrease in cell migration of the carrageenin-induced peritonitis produced by a high dose of TKI, although the tissue kallikrein-dependent pathway is implicated in this model (Figueroa et al., 1989, William et al., 1997).
TKI did not influence the cutaneous inflammation induced by croton oil, a model strongly inhibited by steroid anti-inflammatory agents (Tubaro et al., 1985), excluding once more a possible drug influence on the endogenous corticosteroids system. In contrast, TKI reduced the ear oedema induced by capsaicin in mice, a model of neurogenic inflammation reported to involve primarily the release of neuropeptides through activation of sensory nerves (Inoue et al., 1993), suggesting implication of tissue kallikrein also in this model. Taken together, the results showed that the effects of TKI were more prominent on the nociceptive than on the acute inflammatory models, indicating the relevance of the tissue-kallikrein pathway in nociception in mice.
In conclusion, upon parenteral administration, the tissue kallikrein inhibitor phenylacetyl-FSR-EDDnp exerted antinociceptive and anti-inflammatory actions only in those models where injuries are related to kinin formation by tissue kallikrein, and in those sensitive to nonsteroidal anti-inflammatory agents. The TKI-induced antinociceptive effect was unrelated to opioid mediation or corticosteroids release from the adrenal cortex. These results indicated that the tissue kallikrein-dependent pathway of kinin generation is involved in nociception and inflammatory reactions in mice, and probably in humans too because of the enzyme similarity. Our data finally show that specific tissue kallikrein inhibitors could be useful analgesic and anti-inflammatory agents different from BK B2 receptors antagonists since they might inhibit the release of kinins as well as of other proinflammatory mediators in pathological processes.
Acknowledgments
This work was supported by grants from Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). J.A.S. Emim and M.S.A. Castro received a fellowship from Coordenação de Aperfeiçoamento do Pessoal de Nivel Superior (CAPES).
Abbreviations
- BK
bradykinin
- HMWK
high molecular weight kininogen
- HOE-140
D-Arg-(Hyp3-Thi5-D-Tic7-Oic8)-bradykinin
- KKS
kallikrein-kinin system
- LMWK
low molecular weight kininogen
- Lys-BK
kallidin
- PBS
phosphate buffered saline
- PKSI-527
N-(trans-4-aminomethylcyclohexanecarbonyl)-phenylalanine-4-carboxy methylanilide)
- SBTI
soybean trypsin inhibitor
- TKI
phenylacetyl-Phe-Ser-Arg-N-(2,4-dinitrophenyl)-ethylenediamine
References
- AHLUWALIA A., PERRETTI M. B1 receptors as a new inflammatory target. Could this B the 1. Trends Pharmacol. Sci. 1999;20:100–104. doi: 10.1016/s0165-6147(99)01321-8. [DOI] [PubMed] [Google Scholar]
- BAUER M.B., MELLER S.T., GEBHART G.F. Bradykinin modulation of a spinal nociceptive reflex in the rat. Brain Res. 1992;578:186–196. doi: 10.1016/0006-8993(92)90247-7. [DOI] [PubMed] [Google Scholar]
- BERKENKOPF J.W., WEICHMAN B.M. Production of prostacyclin in mice following intraperitoneal injection of acetic acid, phenylbenzoquinone and zymosan: its role in the writhing response. Prostaglandins. 1988;36:693–709. doi: 10.1016/0090-6980(88)90014-7. [DOI] [PubMed] [Google Scholar]
- BHOOLA K.D., ELSON C.J., DIEPPE P.A. Kinins-key mediators in inflammatory arthritis. Br. J. Rheum. 1992a;31:509–518. doi: 10.1093/rheumatology/31.8.509. [DOI] [PubMed] [Google Scholar]
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens and kininases. Pharmacol. Rev. 1992b;44:1–80. [PubMed] [Google Scholar]
- BIZETO L., ANTUNES E., PORTARO F.C.V., JULIANO M.A., PRADO E.S., DE NUCCI G. Pharmacological characterization of novel tissue kallikrein inhibitors in vivo. Immunopharmacology. 1996;32:111–114. doi: 10.1016/0162-3109(95)00067-4. [DOI] [PubMed] [Google Scholar]
- BLACKBERG M., OHLSSON K. Interactions in vitro and in vivo between porcine tissue kallikrein and porcine plasma proteinase inhibitors. Scand. J. Clin. Lab. Invest. 1994;54:643–651. doi: 10.3109/00365519409087545. [DOI] [PubMed] [Google Scholar]
- BOND A.P., LEMON M., DIEPPE P.A., BHOOLA K.D. Generation of kinins in synovial fluid from patients with arthropathy. Immunopharmacology. 1997;36:209–216. doi: 10.1016/s0162-3109(97)00023-4. [DOI] [PubMed] [Google Scholar]
- BURCH R.M., DEHAAS C. A bradykinin antagonist inhibits carrageenan edema in rats. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:189–193. doi: 10.1007/BF00166963. [DOI] [PubMed] [Google Scholar]
- BURTON J., BENETOS A. The design of specific inhibitors of tissue kallikrein and their effect on the blood pressure of the rat. Adv. Exp. Med. Biol. 1989;247B:9–13. doi: 10.1007/978-1-4615-9546-5_2. [DOI] [PubMed] [Google Scholar]
- CAMPOS R.O., ALVES R.V., KYLE D.J., CHAKRAVARTY S., MAVUNKEL B.J., CALIXTO J.B. Antioedematogenic and antinociceptive actions of NPC 18521, a novel bradykinin B2 receptor antagonist. Eur. J. Pharmacol. 1996;316:277–286. doi: 10.1016/s0014-2999(96)00661-9. [DOI] [PubMed] [Google Scholar]
- CHAGAS J.R., JULIANO L., PRADO E.S. Intramolecularly quenched fluorogenic tetrapeptide substrates for tissue and plasma kallikreins. Anal. Biochem. 1991;192:419–425. doi: 10.1016/0003-2697(91)90558-b. [DOI] [PubMed] [Google Scholar]
- CHAU T.T., LEWIN AC., WALTER T.L., CARLSON R.P., WEICHMAN B.M. Evidence for a role of bradykinin in experimental pain models. Agents Actions. 1991;34:235–238. doi: 10.1007/BF01993290. [DOI] [PubMed] [Google Scholar]
- CHRISTIANSEN S.C., PROUD D, COCHRANE C.G. Detection of tissue kallikrein in the bronchoalveolar lavage fluid of asthmatic subjects. J. Clin. Invest. 1987;79:188–197. doi: 10.1172/JCI112782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CODERRE T.J., MELZACK R. The contribution of excitatory aminoacids to central sensibilization and persistent nociception after formalin-induced tissue injury. J. Neuroscience. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRÊA C.R., CALIXTO J.B. Evidence for participation of B1 and B2 kinin receptors in formalin-induced nociceptive response in the mouse. Br. J. Pharmacol. 1993;110:193–198. doi: 10.1111/j.1476-5381.1993.tb13791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRÊA C.R., KYLE D.J., CHAKRAVERTY S., CALIXTO J.B. Antinociceptive profile of the pseudopeptide B2 bradykinin receptor antagonist NPC 18688 in mice. Br. J. Pharmacol. 1996;117:552–558. doi: 10.1111/j.1476-5381.1996.tb15226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAMAS J., LIEGEOIS J.F. The inflammatory reaction induced by formalin in the rat paw. Naunyn Schmiedebergs Arch. Pharmacol. 1999;359:200–207. doi: 10.1007/pl00005345. [DOI] [PubMed] [Google Scholar]
- D'AMOUR F.E., SMITH J. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- DÉCARIE A., ADAM A., COUTURE R. Effects of captopril and icatibant on bradykinin (BK) and des[Arg9]BK in carrageenan-induced edema. Peptides. 1996;17:1009–1015. doi: 10.1016/0196-9781(96)00145-3. [DOI] [PubMed] [Google Scholar]
- DESHANDRA M.R., BHOOLA K.D. Pathophysiology of the kallikrein-kinin system in mammalian nervous tissue. Pharmacol. Ther. 1998;79:105–127. doi: 10.1016/s0163-7258(98)00011-4. [DOI] [PubMed] [Google Scholar]
- DI ROSA M., GIROUD J.P., WILLOUGHBY D.A. Studies of the mediators of the acute inflammatory response induced in rats in different sites by carrageenin and turpetine. J. Path. 1971;104:15–29. doi: 10.1002/path.1711040103. [DOI] [PubMed] [Google Scholar]
- DRAY A. Inflammatory mediators of pain. Br. J. Anaesth. 1995;75:125–131. doi: 10.1093/bja/75.2.125. [DOI] [PubMed] [Google Scholar]
- EMIM J.A., OLIVEIRA A.B., LAPA A.J. Pharmacological evaluation of the anti-inflammatory activity of a citrus bioflavonoid, hesperidin, and the isoflavonoids, duartin and claussequinone, in rats and mice. J. Pharm. Pharmacol. 1994;46:118–122. doi: 10.1111/j.2042-7158.1994.tb03753.x. [DOI] [PubMed] [Google Scholar]
- FERRÁNDIZ M.L., ALCARAZ M.J. Anti-inflammatory activity and inhibition of arachidonic acid metabolism by flavonoids. Agents & Actions. 1991;32:283–288. doi: 10.1007/BF01980887. [DOI] [PubMed] [Google Scholar]
- FIGUEROA C.D., MACIVER A.G., DIEPPE P., MACKENZIE J.C., BHOOLA K.D. Presence of immunoreactive tissue kallikrein in human polymorphonuclear (PMN) leucocytes. Adv. Exp. Med. Biol. 1989;247B:207–210. doi: 10.1007/978-1-4615-9546-5_34. [DOI] [PubMed] [Google Scholar]
- FUJIYOSHI T., HAYASHI I., OH-ISHI S., KUWASHIMA M., IIDA H., DOZEN M., TANIGUCHI N., IKEDA K., OHNISHI H. Kaolin-induced writhing in mice, a new model of possible bradykinin-induced pain for assessment of analgesic agents. Agents & Actions. 1989;27:332–334. doi: 10.1007/BF01972814. [DOI] [PubMed] [Google Scholar]
- GUILLÉN M.E.N., EMIM J.A.S., SOUCCAR C., LAPA A.J. Analgesic and antiinflammatory activities of the aqueous extract of Plantago major L. Int. J. Pharmacognosy. 1997;35:99–104. [Google Scholar]
- HARGREAVES K.M., TROULLOS E.S., DIONNE R.A., SCHMIDT E.A., SCHAFER S.C., JORIS J.L. Bradykinin is increased during acute and chronic inflammation: therapeutic implications. Clin Pharmacol Ther. 1988;44:613–621. doi: 10.1038/clpt.1988.202. [DOI] [PubMed] [Google Scholar]
- HEAPY C.G., SHAW S., FARMER S.C. Differential sensitivity of antinociceptive assays to the bradykinin antagonist Hoe 140. Br. J. Pharmacol. 1993;108:209–213. doi: 10.1111/j.1476-5381.1993.tb13464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENRIQUES M.G.M.O., SILVA P.M.R., MARTINS M.A., FLORES C.A., CUNHA F.Q., ASSREUY-FILHO J., CORDEIRO R.S.B. Mouse paw edema. A new model for inflammation. Braz. J. Med. Biol. Res. 1987;20:243–249. [PubMed] [Google Scholar]
- HUNSKAAR S., FASMER O.B., HOLE K. Formalin test in mice, a useful technique for evaluating mild analgesics. J. Neurosci. Meth. 1985;14:69–76. doi: 10.1016/0165-0270(85)90116-5. [DOI] [PubMed] [Google Scholar]
- HUNSKAAR S.A., HOLE K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain. 1987;30:103–114. doi: 10.1016/0304-3959(87)90088-1. [DOI] [PubMed] [Google Scholar]
- INOUE H., NAGATA N., KOSHIHARA Y. Profile of capsaicin-induced mouse ear oedema as neurogenic inflammatory model: comparison with arachidonic acid-induced ear oedema. Br. J. Pharmacol. 1993;110:1614–1620. doi: 10.1111/j.1476-5381.1993.tb14009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JULIANO M.A., JULIANO L. Synthesis and kinetic parameters of hydrolysis by trypsin of some acyl-arginyl-p-nitroanilides and peptides containing arginyl-p-nitroanilide. Braz. J. Med. Biol. Res. 1985;18:435–445. [PubMed] [Google Scholar]
- JULIANO L., PORTARO F.C.V., CHAGAS J.R., HIRATA I.Y., JULIANO M.A., BIZETO L., ANTUNES E., DE NUCCI G., PRADO E.S. Design of inhibitors for tissue kallikreins based on their extended binding site specificities. Peptides. 1995;1994:885–886. [Google Scholar]
- KATORI M., MAJIMA M., HARADA Y., UENO A. A significant role of plasma kallikrein-kinin system in plasma exudation of rat carrageenin-induced pleurisy. Adv. Exp. Med. Biol. 1989;247A:137–144. doi: 10.1007/978-1-4615-9543-4_19. [DOI] [PubMed] [Google Scholar]
- KOSTER R., ANDERSON M., DE BEER E.J. Acetic acid for analgesic screening. Fed. Proc. 1959;18:412. [Google Scholar]
- LANEUVILLE O, , DORAIS J., COUTURE R. Characterization of the effects produced by neurokinins and three agonists selective for neurokinin receptor subtypes in a spinal nociceptive reflex of the rat. Life Sci. 1988;42:1295–1305. doi: 10.1016/0024-3205(88)90223-8. [DOI] [PubMed] [Google Scholar]
- OKADA Y., TSUDA Y., TADA M., WANAKA K., HIJIKATA-OKUNOMIYA A., OKAMOTO U., OKAMOTO S. Development of plasma kallikrein selective inhibitors. Biopolymers. 1999;51:41–50. doi: 10.1002/(SICI)1097-0282(1999)51:1<41::AID-BIP5>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- OKUNISHI H., SPRAGG J., BURTON J., TODA N. In vivo inhibition of tissue kallikreins by kininogen sequence analogue peptides. Adv. Exp. Med. Biol. 1989;247B:23–28. doi: 10.1007/978-1-4615-9546-5_4. [DOI] [PubMed] [Google Scholar]
- PERKINS M.N., KELLY D., DAVIS A.J. Bradykinin B1 and B2 receptor mechanisms and cytokine-induced hyperalgesia in the rat. Can J Physiol Pharmacol. 1995;73:832–836. doi: 10.1139/y95-113. [DOI] [PubMed] [Google Scholar]
- POLOSA R. Role of the kinin-kallikrein pathway in allergic diseases. Allergy. 1993;48:217–225. doi: 10.1111/j.1398-9995.1993.tb00719.x. [DOI] [PubMed] [Google Scholar]
- POOLE S., LORENZETTI B.B., CUNHA F.Q., FERREIRA S.H. Bradykinin B1 and B2 receptors, tumor necrosis factor alpha and inflammatory hyperalgesia. Br. J. Pharmacol. 1999;126:649–656. doi: 10.1038/sj.bjp.0702347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PORTARO F.C., CEZARI M.H., JULIANO M.A., JULIANO L., WALMSLEY A.R., PRADO E.S. Design of kallidin-releasing tissue kallikrein inhibitors based on the specificities of the enzyme's binding subsites. Biochem. J. 1997;323:167–171. doi: 10.1042/bj3230167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROUD D., KAPLAN A.P. Kinin formation: mechanisms and role in inflammatory disorders. Annu. Rev. Immunol. 1988;6:49–83. doi: 10.1146/annurev.iy.06.040188.000405. [DOI] [PubMed] [Google Scholar]
- SAKURADA T.L., KATSUMATA K., TAN-NO K., SAKURADA S., KISARA K. The capsaicin test in mice for evaluating tachykinin antagonists in the spinal cord. Neuropharmacology. 1992;31:1279–1285. doi: 10.1016/0028-3908(92)90057-v. [DOI] [PubMed] [Google Scholar]
- SCHIANTERELLI P., CADEL S., ACERBI D., PAVESI L. Antiinflammatory activity and bioavailability of percutaneous piroxicam. Arzneim.-Forsch., Drug. Res. 1982;32:230–235. [PubMed] [Google Scholar]
- SELWYN B., FIGUEROA C.D., FINK E., SWAN A., DIEPPE P.A., BHOOLA K.D. A tissue kallikrein in the synovial fluid of patients with rheumatoid arthritis. Ann. Rheum. Dis. 1989;48:128–133. doi: 10.1136/ard.48.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARMA J.N. Therapeutic prospects of bradykinin receptor antagonists. Gen Pharmacol. 1993;24:267–274. doi: 10.1016/0306-3623(93)90302-e. [DOI] [PubMed] [Google Scholar]
- SHIBATA M., OHKUBO T., TAKAHASHI H., INOKI R. Modified formalin test: characteristic biphasic pain response. Pain. 1989;38:347–352. doi: 10.1016/0304-3959(89)90222-4. [DOI] [PubMed] [Google Scholar]
- SIEGMUND E., CADMUS R., LU G. A method for evaluating both non-narcotic and narcotic analgesics. Proc. Soc. Exp. Biol. Med. 1957;95:729–731. doi: 10.3181/00379727-95-23345. [DOI] [PubMed] [Google Scholar]
- SOKAL R.R., ROHLF F.J. Biometry - The Principles and Practice of Statistics 1981New York: W.H. Freeman; 8592nd edn [Google Scholar]
- STERANKA L.R., DEHAAS C.J., VAVREK R.J., STEWART J.M., ENNA S.J., SNYDER S.H. Antinociceptive effects of bradykinin antagonist. Eur. J. Pharmacol. 1987;136:261–262. doi: 10.1016/0014-2999(87)90723-0. [DOI] [PubMed] [Google Scholar]
- SUH H.H., FUJIMOTO J.M., TSENG L.L. Differential mechanisms mediating beta-endorphin- and morphine-induced analgesia in mice. Eur. J. Pharmacol. 1989;168:61–70. doi: 10.1016/0014-2999(89)90633-x. [DOI] [PubMed] [Google Scholar]
- SZELKE M., EVANS D.M., JONES D.M., FAWCETT L., ASHWORTH D., OLSSON H., FEATHERSTONE R.L., CHURCH M.K. Synthetic inhibitors of tissue kallikrein effects in vivo in a model of allergic inflammation. Braz. J. Med. Biol. Res. 1994;27:1943–1947. [PubMed] [Google Scholar]
- TAKADA Y., SKIDGEL R.A., ERDOS E.G. Purification of human urinary prokallikrein. Identification of the site of activation by the metalloproteinase thermolysin. Biochem. J. 1985;232:851–858. doi: 10.1042/bj2320851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TENO N., WANAKA K., OKADA Y., TAGUCHI H., OKAMOTO U., HIJIKATA-OKUNOMIYA A., OKAMOTO S. Development of active center-directed plasmin and plasma kallikrein inhibitors and studies on the structure-inhibitory activity relationship. Chem. Pharm. Bull. 1993;41:1079–1090. doi: 10.1248/cpb.41.1079. [DOI] [PubMed] [Google Scholar]
- TUBARO A., DRI P., DELBELLO G., ZILLI C., LOGGIA R.D. The croton oil ear test revisited. Agents and Actions. 1985;17:347–349. doi: 10.1007/BF01982641. [DOI] [PubMed] [Google Scholar]
- WALKER K., PERKINS M., DRAY A. Kinins and kinin receptors in the nervous system. Neurochem. Int. 1995;26:1–16. doi: 10.1016/0197-0186(94)00114-a. [DOI] [PubMed] [Google Scholar]
- WHITTLE B.A. Release of a kinin by intraperitoneal injection of chemical agents in mice. Int. J. Neuropharmacol. 1964;3:369–378. doi: 10.1016/0028-3908(64)90066-8. [DOI] [PubMed] [Google Scholar]
- WILLIAMS R.J., HENDERSON L.M., NAIDOO Y., CASSIM B., ELSON C.J., BHOOLA K.D. Immunocytochemical analysis of tissue kallikrein and the kinin moiety in rheumathoid synovial fluid neutrophils. Br. J. Rheumatol. 1997;36 4:420–425. doi: 10.1093/rheumatology/36.4.420. [DOI] [PubMed] [Google Scholar]
- WIRTH K., HOCK F.J., ALBUS U., LINZ W., ALPERMANN H.G., ANAGNOSTOPOULOS H., HENKE S.T., BREIPOHL G., KÖNING W., KNOLL W.J., SCHÖLKENS B.A. Hoe 140 a new potent and long acting bradykinin-antagonist: in vivo studies. Br. J. Pharmacol. 1991;102:774–777. doi: 10.1111/j.1476-5381.1991.tb12249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIMMERMANN M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]