

Abstract
The mechanisms underlying vasodilator effect of nicotine on mesenteric resistance blood vessels and the role of calcitonin gene-related peptide (CGRP)-containing (CGRPergic) vasodilator nerves were studied in the rat.
Mesenteric vascular beds isolated from Wistar rats were perfused with Krebs solution, and perfusion pressure was measured with a pressure transducer.
In preparations with intact endothelium and contracted by perfusion with Krebs solution containing methoxamine, perfusion of nicotine (1–100 μM) for 1 min caused a concentration-dependent vasodilator response without vasoconstriction.
The nicotine-induced vasodilation was markedly inhibited by hexamethonium (nicotinic cholinoceptor antagonist, 10 μM) and blocked by guanethidine (adrenergic neuron blocker, 5 μM).
Either denervation by cold storage (4°C for 72 h) or adrenergic denervation by 6-hydroxydopamine (toxin for adrenergic neurons, 2 mM for 20 min incubation, twice) blocked the nicotine-induced vasodilation.
Neither endothelium removal with perfusion of sodium deoxycholate (1.80 mg ml−1, for 30 s) nor treatment with Nω-nitro-L-arginine (nitric oxide synthase inhibitor, 100 μM), atropine (muscarinic cholinoceptor antagonist, 10 nM) or propranolol (β-adrenoceptor antagonist, 100 nM) affected the nicotine-induced vasodilation.
In preparations without endothelium, treatment with capsaicin (depleting CGRP-containing sensory nerves, 1 μM) or human CGRP[8–37] (CGRP receptor antagonist, 0.5 μM) markedly inhibited the nicotine-induced vasodilation.
These results suggest that, in the mesenteric resistance artery of the rat, nicotine induces vasodilation, which is independent of the function of the endothelium and is involved in activation of CGRPergic nerves. It is also suggested that nicotine stimulates presynaptic nicotinic cholinoceptors on adrenergic nerves to release adrenergic neurotransmitters, which then act on CGRPergic nerves to release endogenous CGRP from the nerve.
Keywords: Nicotine, adrenergic-dependent vasodilation, calcitonin gene-related peptide, nicotinic cholinoceptor, rat mesenteric resistance artery
Introduction
Although the tone of peripheral blood vessels is mainly regulated by vascular adrenergic nerves, recent studies have demonstrated that many blood vessels have nonadrenergic noncholinergic (NANC) innervation and these nerves contribute to vascular tone control (Beven & Brayden, 1987). Previously, we reported that periarterial nerve stimulation (PNS) of rat mesenteric resistance blood vessels induces neurogenic vasodilation, which is sensitive to the neurotoxic effect of tetrodotoxin (TTX) and capsaicin (depleting capsaicin-sensitive primary nerves) and that the mesenteric artery is innervated by NANC vasodilator nerves in which calcitonin gene-related peptide (CGRP), a potent vasodilator neuropeptide, acts as a neurotransmitter (Kawasaki et al., 1988). Thus, we proposed that the tone of the peripheral resistance vascular bed is controlled not only by sympathetic adrenergic nerves but also by CGRP-containing (CGRPergic) vasodilator nerves (Kawasaki et al., 1990a; Takenaga & Kawasaki, 1999).
CGRPergic nerves have been shown to be activated by various endogenous vasoactive substances such as prostaglandins (Geppetti et al., 1991), bradykinin (Geppetti et al., 1991) and histamine (Lou et al., 1991). We demonstrated that acetylcholine (cholinoceptor agonist, ACh) is capable of producing endothelium-independent vasodilation in the mesenteric resistance arteries of the rat, by activating the putative muscarinic receptors located on perivascular CGRPergic nerves to elicit release of CGRP (Takenaga et al., 1995). We also suggested that nicotinic cholinoceptors are not involved in the ACh-induced endothelium-independent vasodilation (Takenaga et al., 1995). However, in that study, we used guanethidine, an adrenergic neuron blocker, to block adrenergic neurotransmission. Recently, Zhang et al. (1998) demonstrated striking evidence that nicotine-induced vasodilation in the porcine basilar artery is dependent on intact adrenergic nerves, because the response is blocked by guanethidine and by chemical adrenergic denervation with 6-hydroxydopamine (6-OHDA). They proposed that nicotine stimulates presynaptic nicotinic cholinoceptors on perivascular adrenergic nerve terminals to release noradrenaline (NA) or related substances, which then stimulate release of nitric oxide (NO) from neighbouring NANC nitric oxidergic nerves to relax the blood vessels (Zhang et al., 1998).
Therefore, the present study was undertaken to investigate the mechanisms underlying nicotine-induced vascular response in mesenteric resistance blood vessels of the rat. We here indicate evidence that nicotine induces adrenergic-dependent and endothelium-independent vasodilation of the mesenteric resistance artery, which is mediated by endogenous CGRP released from CGRPergic nerves.
Methods
Animals
Male Wistar rats, weighing 250–350 g, were used in the present study. Animals were given food and water ad libitum. They were housed in the Animal Research Center of Okayama University at a controlled ambient temperature of 22°C with 50±10% relative humidity and with a 12-h light/12-h dark cycle (light on at 08.00 h).
Perfusion of mesenteric vascular beds
The animals were anaesthetized with pentobarbital-Na (50 mg kg−1, intraperitoneally) and mesenteric vascular beds were isolated and prepared for perfusion as described previously (Kawasaki et al., 1988; 1990a; Kawasaki & Takasaki, 1984). The superior mesenteric artery was cannulated and flushed gently with Krebs-Ringer bicarbonate solution (Krebs solution) in order to eliminate blood in the vascular bed. After removal of the entire intestine and associated vascular bed, the mesenteric vascular bed was separated from intestine by cutting close to the intestinal wall. Only four main arterial branches from the superior mesenteric trunk running to the terminal ileum were perfused. All other branches of the superior mesenteric artery were tied off. The isolated mesenteric vascular bed was then placed in a water jacketed organ bath maintained at 37°C and perfused with a modified (see below) Krebs solution at a constant flow rate of 5 ml min−1 with a peristaltic pump (model AC-2120, ATTO Co., Tokyo, Japan). The preparation was also superfused with the same solution at a rate of 0.5 ml min−1 to prevent drying. The Krebs solution was bubbled with a mixture of 95% O2–5% CO2 before passage through a warming coil maintained at 37°C. The modified Krebs solution was of the following composition (mM): NaCl 119.0, KCl 4.7, CaCl2 2.4, MgSO4 1.2, NaHCO3 25.0, KH2PO4 1.2, disodium EDTA 0.03 and dextrose 11.1 (pH 7.4). Changes in the perfusion pressure were measured with a pressure transducer (model TP-400T, Nihon Kohden) and recorded on a pen recorder (model U-228, Nippon Denshi Kagaku, Tokyo, Japan).
Perfusion of nicotine and PNS
After the basal perfusion pressure had been allowed to stabilize, preparations were perfused with Krebs solution containing methoxamine (α1-adrenoceptor agonist) at concentrations of 2–7 μM to induce submaximal vasoconstriction. After stabilization of the elevated perfusion pressure, preparations were subjected to perfusion of nicotine and PNS.
The final concentration of nicotine at concentrations of 1–100 μM, which was made by dilution of Krebs solution containing methoxamine, was perfused for 1 min. To avoid tachyphylaxis, the perfusion of nicotine was carried out at 20-min intervals. At the end of each experiment, preparations were perfused with 100 μM papaverine to cause complete relaxation. Vasodilator activity is expressed as a percentage of the perfusion pressure at maximum relaxation induced by papaverine.
PNS was applied for 30 s by using bipolar platinum ring electrodes placed around the superior mesenteric artery. Rectangular pulses of 1 ms and supramaximal voltage (50 V) were applied at 2, 8 and 12 Hz by using an electronic stimulator (model SEN 3301, Nihon Kohden).
Chemical removal of vascular endothelium
To remove the vascular endothelium, preparations with resting tone were perfused with a 1.80 mg ml−1 solution of sodium deoxycholate (SD) in saline for 30 s as described by Takenaga et al. (1995). This caused a transient increase (20–30 mmHg) in perfusion pressure. The preparations were then rinsed with SD-free Krebs solution for 40 min. After the preparations were contracted by perfusion with Krebs solution containing methoxamine, chemical removal of the endothelium was assessed by the lack of a relaxant effect after a bolus injection of 1 nmol ACh, which was injected directly into the perfusate proximal to the arterial cannula with an infusion pump (model 975, Harvard Apparatus, Holliston, MA., U.S.A.). Volumes were 100 μl for 10 s.
Chemical denervation with 6-OHDA or treatment with capsaicin
In vitro adrenergic denervation was carried out by incubation with 6-OHDA as described by Zhang et al. (1998). The isolated mesenteric vascular bed was incubated in Krebs solution containing 6-OHDA (2 mM) for 20 min twice with a 30 min-interval in 6-OHDA-free Krebs solution. Successful denervation of adrenergic nerves was confirmed by the lack of the PNS-induced vasoconstriction (8 and 12 Hz) and NA release in the perfusate (12 Hz) in the resting tension.
In vitro depletion of CGRPergic nerve was performed according to the method described by Kawasaki et al. (1998; 1990a). The isolated mesenteric vascular bed was perfused with Krebs solution containing capsaicin (1 μM) for 20 min and then rinsed with capsaicin-free Krebs solution. After being rinsed for 60 min, the preparation was contracted by perfusion with Krebs solution containing methoxamine (2 μM) and subjected to perfusion of nicotine. After the elevated perfusion pressure stabilized, PNS at 2 Hz was performed to check for the presence of CGRPergic nerves. Successful loss of CGRPergic nerve function was confirmed by the lack of a relaxant effect to PNS (2 Hz).
Cold-storage denervation
Isolated mesenteric vascular beds were stored in cold Krebs solution at 4°C for 72 h to achieve cold-storage denervation (Kawasaki et al., 1991). To determine the intact responsiveness of the smooth muscle, a bolus injection of ACh or CGRP was carried out to cause vasodilation. Successful denervation of periarterial nerves was confirmed by the lack of the PNS-induced vasoconstriction (8 and 12 Hz) and vasodilation (2 Hz).
Catecholamine assay
The perfusate was collected for 3 min before and during PNS (12 Hz) in perfused mesenteric vascular beds treated by guanethidine or denervated by in vitro 6-OHDA incubation. Activated alumina (30 mg) and 100 μl internal standard (I.S.; 3,4-dihydroxybenzylamine, 100 ng ml−1) were added to each sample to absorb catecholamines and I.S. The alumina collected was washed with pure water and both catecholamines and I.S. were eluted with 0.2 M perchloric acid. NA and I.S. in the elute was assayed by high performance liquid chromatography (HPLC) with an electrochemical detector (model ECD-300, Eicom Co., Kyoto, Japan).
Experimental protocols
To assess the underlying mechanisms of the vascular effect of nicotine, the effects of various agents were examined. An active tone of the preparation was produced by methoxamine (7 μM), and after the elevated perfusion pressure stabilized, Krebs solution containing the final concentration of nicotine and 5 μM guanethidine (adrenergic neuron blocker) or 10 μM hexamethonium (nicotinic cholinoceptor antagonist) was perfused. In preparations without endothelium, the active tone of the preparation was produced by perfusion with Krebs solution containing methoxamine (2 μM). The effects of 10 nM atropine (muscarinic cholinoceptor antagonist), 100 nM propranolol (β-adrenoceptor antagonist) and 100 μM Nω-nitro-L-arginine (nitric oxide synthase inhibitor, L-NA) on the vasodilator response to nicotine were examined in preparations without endothelium.
In another series of experiments, the vascular effect of nicotine was examined in preparations with intact endothelium and denervated with 6-OHDA or cold-storage. The nicotine effect in preparations treated with capsaicin was measured without endothelium.
In another series of experiments, the effect of human CGRP[8–37] on nicotine-induced responses was examined in preparations without endothelium. The Krebs solution containing methoxamine (2 μM), nicotine (10, 30 and 100 μM) and CGRP[8–37] (0.5 μM) was perfused into the preparation and upon vasopressure stabilization, a higher concentration of nicotine was perfused.
Statistical analysis
Experimental results are presented as mean±s.e.mean. Statistical analysis was performed by use of Student's unpaired or paired t-test and one-way analysis of variance followed by Tukey's test. A P value less than 0.05 was considered statistically significant.
Drugs
The following drugs were used: ACh chloride (Daiichi Pharmaceutical Co., Tokyo, Japan), atropine sulphate (Ishizu Seiyaku Ltd, Tokyo, Japan), capsaicin (Sigma Chemical Co., St. Louis, MO, U.S.A), guanethidine sulphate (Sigma), hexamethonium bromide (Sigma), 6-OHDA hydrobromide (Sigma), human CGRP[8–37] (Peptide Institute, Osaka, Japan), isoproterenol hydrochloride (Nikken Kagaku Co., Tokyo, Japan), methoxamine hydrochloride (Nihon Shinyaku Co., Kyoto, Japan), nicotine tartrate salt (Sigma), L-NA (Sigma), papaverine hydrochloride (Dainippon Pharmaceutical Co., Osaka, Japan), propranolol hydrochloride (Sigma), rat CGRP (Peptide Institute), SD (Ishizu Seiyaku). All drugs, except capsaicin and SD, were dissolved in distilled water and diluted with Krebs solution containing 2–7 μM methoxamine. Capsaicin was dissolved in 50% ethanol and diluted with Krebs solution (final alcohol concentration, 0.4 mg ml−1). SD was dissolved in 0.9% saline.
Results
Vascular responses to nicotine perfusion
When the active tone was produced by perfusion of Krebs solution containing 7 μM methoxamine, PNS at 2 Hz caused a transient pressor response followed by long-lasting depressor response as shown in Figure 1A and Table 1. The long-lasting vasodilation has been shown to be mediated by CGRPergic nerves (Kawasaki et al., 1998; 1990a). A bolus injection of ACh caused a sharp drop in perfusion pressure, due to vasodilation (Figure 1A).
Figure 1.
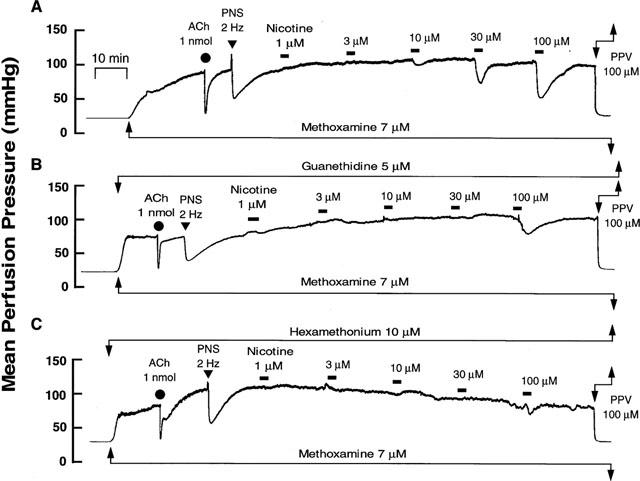
Typical records showing vascular responses to nicotine perfusion (A) and effects of guanethidine (B, 5 μM) and hexamethonium (C, 10 μM) in rat perfused mesenteric vascular beds with active tone produced by methoxamine (7 μM). Nicotine (1–100 μM) was perfused for 1 min at horizontal bars. ACh, bolus injection of acetylcholine (1 nmol). PNS, periarterial nerve stimulation (2 Hz). PPV, perfusion of papaverine (100 μM).
Table 1.
Effects of various drugs and treatments on vasoconstriction and vasodilation induced by PNS (2 Hz) in rat perfused mesenteric vascular beds with active tone

In preparations with active tone, perfusion of nicotine at concentrations of 1–100 μM for 1 min decreased the perfusion pressure, in a concentration-dependent manner, due to vasodilation (Figures 1A and 2). The nicotine-induced vasodilation was long-lasting and returned to preperfusion level within 5–10 min as shown in Figure 1A. A very slight vasoconstriction by nicotine at lower concentrations of 1–10 μM preceded the vasodilation.
Figure 2.
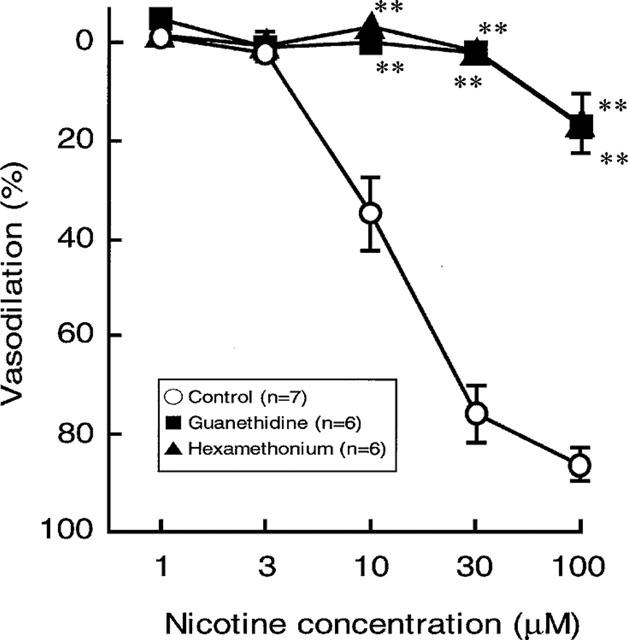
Effects of guanethidine (5 μM) and hexamethonium (10 μM) on the nicotine-induced vasodilation in rat perfused mesenteric vascular beds with active tone. Each point represents mean±s.e.mean from 5–7 experiments. **P<0.01, compared with control.
Effects of hexamethonium and guanethidine on vasodilator response to nicotine perfusion
In preparations with intact endothelium, as shown in Figures 1B and 2, the nicotine-induced vasodilation was markedly inhibited by guanethidine (5 μM). Furthermore, hexamethonium (10 μM) blocked the nicotine-induced vasodilation (Figures 1C and 2). In the presence of guanethidine, both the PNS-induced release of NA (12 Hz; control release, 157.1±17.0 pg ml−1, n=4; guanethidine treatment, 23.0±22.9 pg ml−1, n=3; P<0.01) and vasoconstrictor res-ponses (2, 8 and 12 Hz), but not vasodilator response (2 Hz), were abolished (Table 1). However, hexamethonium did not affect the PNS (2 Hz)-induced vascular responses (Figures 1B, C and Table 1).
Effects of chemical and cold-storage denervation on vasodilator response to nicotine perfusion
In perfused mesenteric vascular beds with intact endothelium and with resting tone, PNS at 8 and 12 Hz caused an increase in perfusion pressure, due to vasoconstriction (Figure 3A). This vasoconstrictor response has been shown to be due to stimulation of vascular adrenergic nerves (Kawasaki & Takasaki, 1984; Takenaga & Kawasaki, 1999). Cold-storage denervation abolished both the PNS (2, 8 and 12 Hz)-induced vasoconstriction and vasodilation (2 Hz) (Figure 3B and Table 1). In this preparation, the nicotine-induced vasodilation was markedly attenuated and the highest concentration of nicotine (100 μM) induced a slight vasodilation (16.9±1.1%) (Figures 3B and 4).
Figure 3.
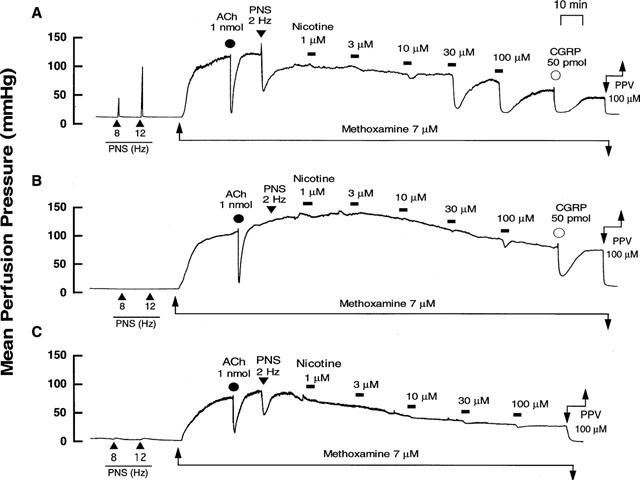
Typical records showing effects of cold-storage denervation and in vitro 6-hydroxydopamine (6-OHDA) incubation on vascular responses to nicotine perfusion in rat perfused mesenteric vascular beds with active tone produced by methoxamine (7 μM). (A) Control responses. (B) Responses after cold-storage denervation (4°C for 72 h). (C) Responses after 6-OHDA incubation (2 mM for 20 min, twice). Nicotine (1–100 μM) was perfused for 1 min at horizontal bars. ACh and CGRP, bolus injection of acetylcholine (1 nmol) and calcitonin gene-related peptide (50 pmol), respectively. PNS, periarterial nerve stimulation at 2, 8 and 12 Hz. PPV, perfusion of papaverine (100 μM).
Figure 4.

Effects of denervation by cold-storage and in vitro 6-hydroxydopamine (6-OHDA) incubation on vasodilator responses to nicotine perfusion in rat perfused mesenteric vascular beds with active tone produced by methoxamine (7 μM). Each point represents mean±s.e.mean from 5–7 experiments. *P<0.05, **P<0.01 compared with control.
In perfused mesenteric vascular bed treated with 6-OHDA, PNS (8 and 12 Hz) at resting tone caused a very slight vasoconstrictor response (Figure 3C), but PNS (2 Hz) induced a vasodilator response when the perfusion pressure was increased by methoxamine (Figure 3C and Table 1). In addition, release of NA in the perfusate by PNS (12 Hz) was markedly decreased after 6-OHDA treatment (control rel- ease, 157.1±17.0 pg ml−1, n=4; 6-OHDA treatment, 21.2±21.1 pg ml−1, n=4; P<0.01). In this preparation, the nicotine-induced vasodilation at concentrations of 1–30 μM was markedly inhibited, as shown in Figures 3C and 4. A moderate vasodilation (47.3±7.2%) was observed at the highest concentration of 100 μM nicotine perfusion, as observed in preparations denervated by cold-storage. The vasodilation remaining after 6-OHDA treatment was further inhibited by 5 μM guanethidine (15.1±2.5%).
Effect of chemical denudation of vascular endothelium on vasodilator response to nicotine perfusion
Since chemical denudation of vascular endothelium increased the vasoconstriction induced by methoxamine, the concentration of methoxamine required to raise the tone was reduced to 2 μM after denudation. In this preparation, the ACh-induced vasodilation was abolished (control response, 91.1±1.4%, n=7; response after chemical denudation, 11.6±1.7%, n=9; P<0.01), indicating that the endothelium was successfully removed. However, PNS (2 Hz) still induced vasodilation (Table 1).
As shown in Figure 5A, endothelium removal did not affect the vasodilation induced by nicotine.
Figure 5.
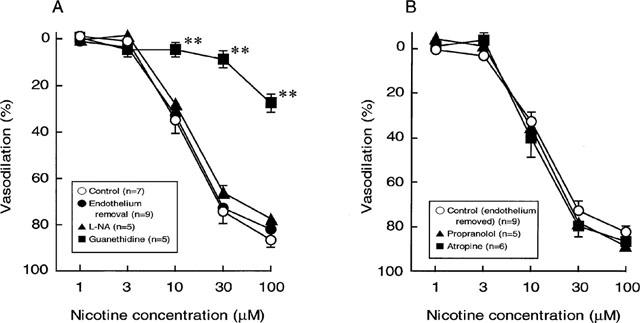
Effects of endothelium removal (A), endothelium removal plus L-nitro-arginine (100 μM) (A), propranolol (0.1 μM) (B) and atropine (10 nM) (B) on the nicotine-induced vasodilation in rat perfused mesenteric vascular beds with active tone. Each point represents mean±s.e.mean from 5–9 experiments. **P<0.01, compared with control.
Effects of various drugs on vasodilator or response to nicotine perfusion
To assess the possible mechanisms underlying the vasodilator response to perfusion of nicotine, the effects of L-NA, atropine and propranolol were examined. In preparations without endothelium, the nicotine-induced vasodilation was not altered by L-NA (100 μM) (Figure 5A). In addition, neither atropine (10 nM) nor propranolol (100 nM) had an effect on the nicotine-induced vasodilation in preparations without endothelium (Figure 5B). However, propranolol significantly inhibited vasodilator responses to a bolus injection of 1 nmol isoprenaline (β-adrenoceptor agonist) (control response, 49.1±8.8%; response in the presence of propranolol, 6.3±2.2%, n=5, P<0.01). Also, atropine at 10 nM concentration has been shown to abolish vasodilator response to a bolus injection of 1 nmol ACh (Takenaga et al., 1995).
Effects of capsaicin and CGRP[8–37] on vasodilator response to nicotine perfusion
Capsaicin treatment in the endothelium-removed preparation abolished the vasodilator responses to PNS (2 Hz) without affecting vasoconstrictor response to PNS (2 Hz) (Figure 6B and Table 1). Additionally, the nicotine-induced vasodilation was markedly attenuated after treatment with capsaicin, as shown in Figures 6B and 7.
Figure 6.
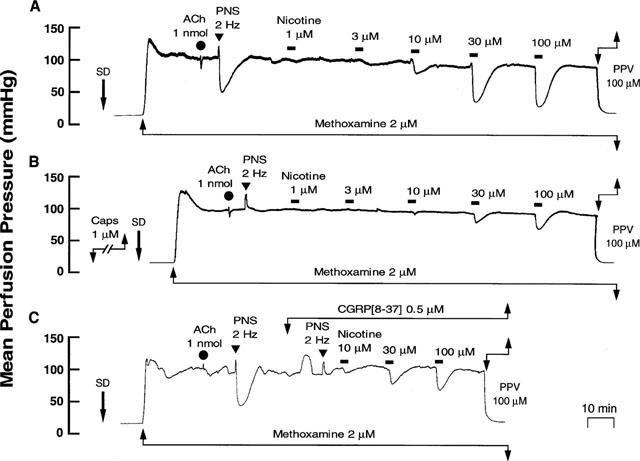
Typical records showing effects of capsaicin (1 μM) and CGRP[8–37] (0.5 μM) treatment on the nicotine-induced vasodilation in rat perfused mesenteric vascular beds without endothelium. (A) Control responses. (B) Responses after treatment with capsaicin. (C) Responses in the presence of CGRP[8–37]. Nicotine (1–100 μM) was perfused for 1 min at horizontal bars. SD, sodium deoxycholate perfusion for 30 s. ACh, bolus injection of acetylcholine (1 nmol). PNS, periarterial nerve stimulation (2 Hz). PPV, perfusion of papaverine (100 μM).
Figure 7.
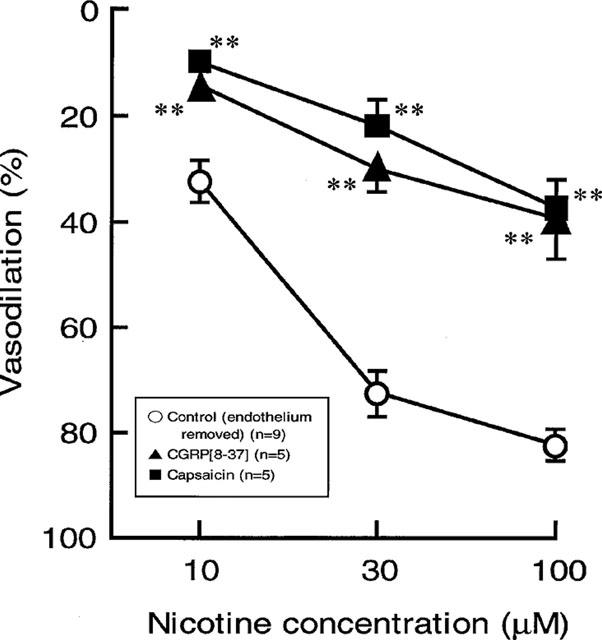
Effects of capsaicin and CGRP[8–37] treatments on the nicotine-induced vasodilation in rat perfused mesenteric vascular beds without endothelium. Each point represents mean±s.e.mean from 5–9 experiments. **P<0.01, compared with control.
In preparations without endothelium, perfusion of Krebs solution containing 0.5 μM CGRP[8–37] resulted in a transient increase in mean perfusion pressure and markedly attenuated vasodilator responses both to perfusion of nicotine and to PNS (2 Hz) (Figures 6C and 7, and Table 1).
Discussion
The present study demonstrated that, in the rat mesenteric artery with active tone produced by methoxamine, perfusion of nicotine induced a concentration-dependent vasodilator response. The nicotine-induced vasodilation was inhibited by hexamethonium, a nicotinic cholinoceptor antagonist, but was not affected by atropine, a muscarinic cholinoceptor antagonist, suggesting that the response is mediated by activation of nicotinic cholinoceptors. Furthermore, the major finding of this study is that the nicotine-induced vasodilation was markedly attenuated by guanethidine, which blocks adrenergic neurotransmission by inhibiting NA release from adrenergic nerve terminals. Also, the nicotine-induced vasodilation was markedly but not completely blocked by cold-storage denervation or selective chemical destruction of adrenergic neurons by 6-OHDA. Although the reason for the residual vasorelaxation after the denervation is not clear, it may be direct effect of nicotine on the vascular smooth muscle cells. Taken together, the present findings indicate that nicotine-induced vasodilator response requires the intact adrenergic nerve function. The present findings are supported by a recent report that the nicotine-induced vasodilator was blocked by guanethidine or 6-OHDA-induced chemical destruction of sympathetic terminals in the porcine basilar artery and was dependent on intact adrenergic innervation (Zhang et al., 1998). Also, similar findings were reported by Hua et al. (1994), who showed that the effect of nicotine was blocked by destruction of adrenergic terminals in the rat trachea.
Nicotine, as well as transmural nerve stimulation (TNS), has been shown to cause vasodilation in cerebral arteries such as the basilar artery, middle cerebral artery and the circle of Willis in several species (pig, rabbit, cat, dog) (Toda & Okamura, 1991; Toda, 1975; Lee & Sarwinski, 1991; Lee et al., 1996). The mechanism underlying the action of nicotine is different from that of TNS in that its action is nicotinic cholinoceptor-mediated and TTX-insensitive (Toda & Okamura, 1991; Toda, 1975; Lee & Sarwinski, 1991; Lee et al., 1996). However, the vasodilation induced by either nicotine or TNS in cerebral arteries is abolished by L-NA, a NO synthase inhibitor, indicating that nicotine-induced vasorelaxation was mediated by NO-containing nerves (nitric oxidergic nerves) (Toda & Okamura, 1991; Lee & Sarwinski, 1991; Lee et al., 1996). In the present study, the nicotine-induced vasodilation is not affected by endothelium-removal or by endothelium-removal in the presence of L-NA, suggesting that neither endothelium nor nitric oxidergic nerves is involved in the nicotine-induced vasodilation in rat mesenteric arteries. These findings indicate that nicotine induces endothelium-independent vasodilation in rat mesenteric vascular beds, and it is unlikely that NO was involved in the mechanisms underlying the nicotine-induced vasodilation.
Evidence has been accumulated that many blood vessels are innervated by NANC nerves (Bevan & Brayden, 1987; Kawasaki et al., 1988; Toda & Okamura, 1991; Toda, 1975; Lee & Sarwinski, 1991; Lee et al., 1996). We have reported previously that the mesenteric resistance arteries of the rat are innervated by NANC vasodilator nerves, in which CGRP, a potent vasodilator neuropeptide, acts as a vasodilator neurotransmitter (Kawasaki et al., 1988; 1990b). Since CGRPergic vasodilator nerves are widely distributed throughout the rat mesenteric vascular bed, PNS induces neurogenic vasorelaxation, which is sensitive to the neurotoxic effect of TTX and is abolished by capsaicin (Kawasaki et al., 1988; 1990a). Capsaicin has been shown to deplete neuropeptides such as CGRP, substance P and vasoactive intestinal polypeptide (VIP) from primary sensory neurons which leads to depleting capsaicin-sensitive primary nerves (Fujimori et al., 1989; Holzer, 1991). Furthermore, the PNS-induced neurogenic vasorelaxation in the rat mesenteric artery is also inhibited by human CGRP[8–37], a C-terminal fragment of CGRP and a CGRP receptor antagonist, indicating that the vasorelaxation is mediated by endogenous CGRP released from CGRPergic nerves (Kawasaki et al., 1991; Takenaga et al., 1995). In the present study, the vasodilator response to nicotine perfusion was sensitive to the effect of capsaicin. Thus, it is very likely that the capsaicin-sensitive peptidergic nerves are responsible for the vasorelaxation induced by nicotine. Furthermore, the present study showed that the nicotine-induced vasodilation was inhibited by the CGRP receptor antagonist, CGRP[8–37], which also blocked the PNS-induced CGRPergic nerve-mediated vasodilation. These findings strongly suggest that CGRPergic vasodilator nerves in the mesenteric artery are involved in the nicotine-induced vasodilation. Since guanethidine and 6-OHDA blocked the vasorelaxation induced by nicotine and did not affect the neurotransmission of CGRPergic nerves, it is unlikely that nicotine directry acts on CGRPergic nerves to cause vasodilation. The nicotine-induced vasodilation is likely to be elicited indirectly by neighbouring neurons, probably adrenergic neurons, via neurotransmitters released from adrenergic nerve terminals.
Nicotine stimulates release of neurotransmitters such as NA (Su & Bevan, 1970; Richardt et al., 1988), neuropeptide Y (NPY) (Richardt et al., 1988) and adenosine triphosphate (ATP) (von Kugelgen & Starke, 1991) from adrenergic nerve terminals. Therefore, although it is possible that these neurotransmitters released from adrenergic terminals by nicotine are involved in nicotine-induced vasodilation, it is unlikely, because they are well known to be vasoconstrictors. An alternative explanation is that the nicotine-induced vasorelaxation is mediated by NA released from adrenergic nerves, which acts directly on postsynaptic β-adrenoceptors (Diana et al., 1990). However, this explanation is also unlikely, because the β-adrenoceptor antagonist, propranolol, does not affect the nicotine-induced vasodilation. Therefore, it is possible that nicotine acts directly on nicotinic cholinoceptors located on vascular adrenergic nerves to release certain adrenergic neurotransmitters which then act on the neighbouring CGRPergic vasodilator nerves to cause release of CGRP and vasodilation. Recently, Zhang et al. (1998) reported the possibility that the nicotine-induced relaxation of the porcine basilar artery may be mediated by NA, which is released from adrenergic nerve terminals by nicotine. However, we have demonstrated that both NA and NPY inhibit the CGRP release from the CGRPergic nerve terminals of the mesenteric artery by presynaptic mechanisms via activation of α2-adrenoceptors and NPY receptors (Kawasaki et al., 1990b; 1991). Additionally, the present study showed that perfusion of nicotine caused little vasoconstriction in the mesenteric artery, suggesting that only a small amount of NA or related vasoconstrictor transmitter may be released by nicotine. Thus, the involvement of neurally released NA and NPY is ruled out of the mechanisms underlying the nicotine-induced vasodilation in the rat mesenteric artery. However, further studies are needed to clarify the identity of the neurotransmitter(s) involved.
In conclusion, the present study suggests that nicotine induces endothelium-independent vasodilation through nicotinic cholinoceptors located on adrenergic nerve terminals in mesenteric resistance blood vessels. It is also suggested that the nicotine-induced vasodilation is dependent on intact adrenergic nerves and is mediated by endogenous CGRP released from CGRPergic nerves. It appears that nicotine acts on presynaptic nicotinic cholinoceptors to release adrenergic neurotransmitters or related substances, which then activate CGRPergic nerves to cause CGRP release and vasodilation.
Acknowledgments
This study was supported by grants from the Smoking Research Foundation and in part by a Grant-in-Aid (Nos. 086772611, 09672326 and 10557244) for Scientific Research from the Ministry of Education, Science and Culture of Japan.
Abbreviations
- ACh
acetylcholine
- ATP
adenosine triphosphate
- CGRP
calcitonin gene-related peptide
- HPLC
high performance liquid chromatography
- I.S.
internal standard
- L-NA
Nω-nitro-L-arginine
- NA
noradrenaline
- NANC
nonadrenergic noncholinergic
- NO
nitric oxide
- NPY
neuropeptide Y
- 6-OHDA
6-hydroxydopamine
- PNS
periarterial nerve stimulation
- PPV
papaverine
- SD
sodium deoxycholate
- TNS
transmural nerve stimulation
- TTX
tetrodotoxin
- VIP
vasoactive intestinal polypeptide
References
- BEVAN J.A., BRAYDEN J.E. Nonadrenergic neural vasodilator mechanisms. Circ. Res. 1987;60:309–326. doi: 10.1161/01.res.60.3.309. [DOI] [PubMed] [Google Scholar]
- DIANA J.N., QIAN S.F., HEESCH C.M., BARRON K.W., CHIEN C.Y. Nicotine-induced skeletal muscle vasodilation is mediated by release of epinephrine from nerve terminals. Am. J. Physiol. 1990;259:H1718–H1729. doi: 10.1152/ajpheart.1990.259.6.H1718. [DOI] [PubMed] [Google Scholar]
- FUJIMORI A., SAITO A., KIMURA S., WATANABE T., UCHIYAMA Y., KAWASAKI H., GOTO K. Neurogenic vasodilator and release of calcitonin gene-related peptide (CGRP) from perivascular nerve from the rat mesenteric artery. Biochem. Biophs. Res. Commun. 1989;165:1391–1398. doi: 10.1016/0006-291x(89)92758-7. [DOI] [PubMed] [Google Scholar]
- GEPPETTI P., BIANCO E.D., TRAMONTANA M., VIGANO T., FOLCO G.C., MAGGI C.A., MANZINI S., FANCIULLACI M. Arachidonic acid and bradykinin share a common pathway to release neuropepide from capsaicin-sensitive sensory nerve fibers of the guinea-pig heart. J. Pharmacol. Exp. Ther. 1991;259:759–765. [PubMed] [Google Scholar]
- HOLZER P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol. Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- HUA X.-Y., JINNO S., BACK M., TAM E.K., YAKSH T.L. Multiple mechanisms for the effects of capsaicin, bradykinin and nicotine on CGRP release from tracheal afferent nerves: Role of prostaglandins, sympathetic nerves and mast cells. Neuropharmacology. 1994;33:1147–1154. doi: 10.1016/s0028-3908(05)80004-8. [DOI] [PubMed] [Google Scholar]
- KAWASAKI H., NUKI C., SAITO A., TAKASAKI K. Role of calcitonin gene-related peptide-containing nerves in the vascular adrenergic neurotransmission. J. Pharmacol. Exp. Ther. 1990a;252:403–409. [PubMed] [Google Scholar]
- KAWASAKI H., NUKI C., SAITO A., TAKASAKI K. Adrenergic modulation of calcitonin gene-related peptide (CGRP)-containing nerve-mediated vasodilation in the rat mesenteric resistance vessels. Brain Res. 1990b;506:287–290. doi: 10.1016/0006-8993(90)91263-g. [DOI] [PubMed] [Google Scholar]
- KAWASAKI H., NUKI C., SAITO A., TAKASAKI K. NPY modulates neurotransmission of CGRP-containing vasodilator nerves in rat mesenteric arteries. Am. J. Physiol. 1991;261:H683–H690. doi: 10.1152/ajpheart.1991.261.3.H683. [DOI] [PubMed] [Google Scholar]
- KAWASAKI H., TAKASAKI K. Vasoconstrictor response induced by 5-hydroxytryptamine released from vascular adrenergic nerves by periarterial nerve stimulation. J. Pharmacol. Exp. Ther. 1984;229:816–822. [PubMed] [Google Scholar]
- KAWASAKI H., TAKASAKI K., SAITO S., GOTO K. Calcitonin gene-related peptide acts as a novel vasodilator neurotransmitter in mesenteric resistance vessels of the rat. Nature. 1988;335:54–56. doi: 10.1038/335164a0. [DOI] [PubMed] [Google Scholar]
- LEE T.J.-F., SARWINSKI S. Nitric oxidergic neurogenic vasodilation in the porcine basilar artery. Blood Vessels. 1991;28:407–412. doi: 10.1159/000158887. [DOI] [PubMed] [Google Scholar]
- LEE T.J.-F., SARWINSKI S., ISHINE T., LAI C.C., CHEN F.Y. Inhibition of cerebral neurogenic vasodilation by L-glutamine and nitric oxide synthase inhibitors and its reversal by L-citrulline. J. Pharmacol. Exp. Ther. 1996;276:1–6. [PubMed] [Google Scholar]
- LOU Y.P., KARLSSON J.A., FRANCO-CERECEDA A., LUNDBERG J.M. Selectivity of ruthenium red in inhibiting bronchoconstriction and CGRP release induced by afferent C-fiber activation in the guinea-pig lung. Acts Physiol. Scand. 1991;142:191–199. doi: 10.1111/j.1748-1716.1991.tb09147.x. [DOI] [PubMed] [Google Scholar]
- RICHARDT G., HAASS M., NEEB S., HOCK M., LANG R.E., SCHOMIG A. Nicotine-induced release of noradrenaline and neuropeptide Y in guinea pig heart. Klin. Wochenschr. 1988;66:21–27. [PubMed] [Google Scholar]
- SU C., BEVAN J.A. Blockade of the nicotine-induced norepinephrine release by cocaine phenoxybenzamine and desipramine. J. Pharmacol. Exp. Ther. 1970;175:533–540. [PubMed] [Google Scholar]
- TAKENAGA M., KAWASAKI H. Endogenous calcitonin gene-related peptide suppresses vasoconstriction mediated by adrenergic nerves in rat mesenteric resistance blood vessels. Eur. J. Pharmacol. 1999;367:239–245. doi: 10.1016/s0014-2999(98)00949-2. [DOI] [PubMed] [Google Scholar]
- TAKENAGA M., KAWASAKI H., WADA A., ETO T. Calcitonin gene-related peptide mediates acetylcholine-induced endothelium-independent vasodilation in mesenteric resistance blood vessels of the rat. Circ. Res. 1995;76:935–941. doi: 10.1161/01.res.76.6.935. [DOI] [PubMed] [Google Scholar]
- TODA N. Nicotine-induced relaxation in isolated canine cerebral arteries. J. Pharmacol. Exp. Ther. 1975;193:376–384. [PubMed] [Google Scholar]
- TODA N., OKAMURA T. Role of nitric oxide in neurally induced cerebroarterial relaxation. J. Pharmacol. Exp. Ther. 1991;258:1027–1032. [PubMed] [Google Scholar]
- VON KUGELGEN I., STARKE K. Release of noradrenaline and ATP by electrical stimulation and nicotine in guinea-pig vas deferens. Naunyn-Schmiedeberg's Arch. Pharmacol. 1991;344:419–429. doi: 10.1007/BF00172581. [DOI] [PubMed] [Google Scholar]
- ZHANG W., EDVINSSON L., LEE T.J.-F. Mechanism of nicotine-induced relaxation in the porcine basilar artery. J. Pharmacol. Exp. Ther. 1998;284:790–797. [PubMed] [Google Scholar]