

Abstract
Sodium ions inhibit spontaneous Gi/Go-coupled receptor activity and promote agonist-induced responses in vitro. The effects of sodium on the relative efficacy of opioid agonists for G-protein activation was measured by guanosine-5′-O-(γ-35S)-triphosphate ([35S]-GTPγS) binding in membranes from two μ-opioid receptor-containing systems: CHO cells stably transfected with mouse μ receptors (mMOR-CHO cells) and rat thalamus.
NaCl inhibited basal [35S]-GTPγS binding in both systems, and this effect was partially mimicked by KCl. In mMOR-CHO membranes, net [35S]-GTPγS binding stimulated by partial but not full agonists was inhibited by NaCl with a potency that was inversely proportional to agonist efficacy. Monovalent cations were required for agonist-stimulated [35S]-GTPγS binding in this system, and increasing NaCl concentrations magnified relative efficacy differences among agonists.
In thalamic membranes, which contain a lower receptor:G-protein ratio than mMOR-CHO cells, similar monovalent cation effects were observed, with two exceptions: (1) [35S]-GTPγS binding stimulated by both full and partial agonists was inhibited by NaCl; and (2) monovalent cations were not required to observe agonist-stimulated [35S]-GTPγS binding.
Basal [35S]-GTPγS binding stimulated by the absence of monovalent cations resembled that of agonist-stimulated binding and was blocked by pretreatment of mMOR-CHO cells with pertussis toxin.
These results indicate that sodium inhibits spontaneous and agonist-occupied μ receptor-mediated G-protein activation in a manner inversely proportional to the efficacy of the agonist, and that spontaneous μ receptor activity and the relative efficacy of partial agonists acting at these receptors are both increased by increases in the stoichiometric ratio of receptors:G-proteins.
Keywords: G-protein, opioid receptor, sodium, agonist efficacy, [35S]-GTPγS binding, pertussis toxin
Introduction
Gi/o-coupled receptors can display spontaneous activity in the absence of agonist, as demonstrated by the effects of inverse agonists (negative antagonists) on basal G-protein activity (Costa et al., 1990; Hilf & Jakobs, 1992; Tian et al., 1994). This inverse agonism is most readily observable in the absence of sodium (Costa et al., 1990; Hilf & Jakobs, 1992; Tian et al., 1994). Although the structural basis for the inhibitory effect of sodium on receptor-mediated G-protein activation is not known, it may be due to interaction of the cation with a conserved aspartate residue in the second putative transmembrane domain (TM2) of G-protein-coupled receptors (Horstman et al., 1990). Mutation of this residue has been shown to result in a loss of sodium sensitivity of high affinity agonist binding, but the effect of these mutations on the ability of receptors to activate G-proteins is dependent on both the receptor type and endpoint (e.g. effector and/or G-protein activity) under investigation (Ceresa & Limbird, 1994; Kong et al., 1993a,1993b; Tao & Abood, 1998).
μ-type opioid receptors, which have a high affinity for opiate-derived analgesics (Corbett et al., 1993), are members of the superfamily of G-protein-coupled receptors (Chen et al., 1993; Thompson et al., 1993) and are primarily coupled to G-proteins of the pertussis toxin (PTX)-sensitive Gi/o-type (Childers, 1991). Different μ-opioid agonists display varying intrinsic efficacies for a variety of biological responses both in vivo (Adams et al., 1990; Mjanger & Yaksh, 1991) and in vitro (Carter & Medzihradsky, 1993; Yu & Sadee, 1988). Several laboratories have demonstrated that differences in μ-opioid agonist efficacy can be measured at the level of G-protein activation, as determined by their ability to maximally stimulate binding of the hydrolysis-resistant GTP analogue, guanosine-5′-O-(γ-35S)-triphosphate ([35S]-GTPγS), to membranes from cultured cell lines (Emmerson et al., 1996; Selley et al., 1997; 1998; Traynor & Nahorski, 1995) and brain (Selley et al., 1997; 1998; Sim et al., 1998). Moreover, relative efficacy differences among μ-opioid agonists depend on factors that affect the balance between G-protein activation and inactivation, such as the GDP concentration and the stoichiometric ratio of receptors:G-proteins (R:G ratio) (Selley et al., 1997; 1998), in a similar manner to other Gi/o-coupled receptors (Lorenzen et al., 1996; Newman-Tancredi et al., 1997; Pauwels et al., 1997). Studies from our laboratory have also shown that agonists of high and low intrinsic efficacies differ in their abilities to stimulate a maximal apparent affinity of [35S]-GTPγS binding and to activate a maximal number of G-proteins (Selley et al., 1997; 1998). These differences are probably related to differences in the ability of receptors to modulate G-protein affinity for GDP relative to that of [35S]-GTPγS when occupied by agonists of different intrinsic efficacies (Breivogel et al., 1998; Lorenzen et al., 1996; Selley et al., 1997).
Mathematical modelling of the inhibitory effect of sodium on receptor-coupled G-protein activity has predicted that relative agonist efficacy should be inversely proportional to the sodium concentration (Costa et al., 1992). In the present study, this hypothesis was tested by examining agonist-stimulated [35S]-GTPγS binding using opioid agonists of different intrinsic efficacies in membranes from two μ-opioid receptor-containing systems: CHO cells stably transfected with mouse μ receptor cDNA (mMOR-CHO cells) and which express high receptor levels (4–7 pmol mg−1) (Abood et al., 1995; Kaufman et al., 1995; Selley et al., 1998), and rat thalamus, which contains a moderate number of μ receptors (∼0.75 pmol mg−1) with few other opioid receptor types present (Herkenham & Pert, 1982; Selley et al., 1998). Furthermore, studies were performed in mMOR-CHO cell membranes to determine whether sodium inhibition of basal [35S]-GTPγS binding was due to inhibition of spontaneous μ receptor activity. These studies examine the mechanisms by which G-protein activity is regulated by receptors and their agonists, as well as by the cellular environment.
Methods
Materials
Male Sprague-Dawley rats (200 g) were purchased from Zivic-Miller (Zelienople, PA, U.S.A.). [35S]-GTPγS (1250 Ci mmol−1) was purchased from New England Nuclear Corp. (Boston, MA, U.S.A.). mMOR-CHO cells were generously provided by Drs Lawrence Toll and Christopher J. Evans. ScintiSafe Econo-1 scintillation fluid was obtained from Fisher Scientific (Pittsburgh, PA, U.S.A.). [D-Ala2,(N-Me)Phe4,Gly5-ol]-enkephalin (DAMGO), naloxone, penicillin-streptomycin and pertussis toxin (PTX) were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). All other non-peptide opioid agonists were obtained from the N.I.D.A. drug supply program (Research Triangle Institute, Research Triangle Park, NC, U.S.A.). Foetal bovine serum (FBS) and Geneticin (G-418) were purchased from Gibco/BRL (Grand Island, NY, U.S.A.). Guanosine-5′-O-(γ-thio)-triphosphate and guanosine-5′-diphosphate were purchased from Boehringer Mannheim (New York, NY, U.S.A.). All other chemicals (reagent grade) were obtained from Sigma Chemical Co. or from Fisher Scientific.
Cell culture
mMOR-CHO cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air in 50% DMEM and 50% F-12 Nutrient Mixture (Ham) containing 100 units ml−1 penicillin, 100 μg ml−1 streptomycin, and 5% FBS (culture media). Cells were harvested by replacing the culture media with cold phosphate-buffered saline containing 0.04% EDTA, incubated for 5 min followed by agitation, and collected by centrifugation at 345×g for 10 min. For cell culture treatments, cells were incubated in culture media without serum in the presence and absence of 100 ng ml−1 PTX or 5 μM DAMGO for 18–24 h.
Membrane preparation
Rats were sacrificed by decapitation and the thalamus was dissected on ice. mMOR-CHO cells or rat thalami were homogenized in 20 volumes of ice-cold 50 mM Tris-HCl, 3 mM MgCl2, 1 mM EGTA, pH 7.4 (membrane buffer) with a Tissuemizer (Tekmar, Cincinnati, OH, U.S.A.). The homogenate was centrifuged at 48,000×g at 4°C for 10 min, resuspended in membrane buffer, centrifuged again at 48,000×g at 4°C for 10 min and finally resuspended in 50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4 (assay buffer).
[35S]-GTPγS binding assays
Agonist-stimulated [35S]-GTPγS binding was assayed as previously described with slight modifications (Selley et al., 1997; 1998). mMOR-CHO cell (25 μg protein) or rat thalamic (10 μg protein) membranes were incubated for 1 h at 30°C in a final volume of 1 ml. The incubation mixture included 0.05-0.1 nM [35S]-GTPγS and 10 μM (mMOR-CHO) or 30 μM (rat thalamus) GDP with and without maximally effective concentrations of various agonists, in assay buffer. Unless otherwise indicated, agonist concentrations were: 100 nM levallorphan, 50 nM buprenorphine, 10 μM morphine, and 5 μM (mMOR-CHO) or 10 μM (rat thalamus) DAMGO. In some experiments, the NaCl or GDP concentrations were varied or omitted from the reaction mixture. In other experiments, the total monovalent cation concentration was maintained at 120 mM by replacing NaCl with equimolar concentrations of KCl. In all experiments, basal binding was assessed in the absence of drug, and nonspecific binding was measured in the presence of 10 μM unlabelled GTPγS. For GTPγS saturation analyses, mMOR-CHO cell membranes were incubated in assay buffer (1 ml final volume) with 0.08–0.1 nM [35S]-GTPγS and 0.02–2000 nM unlabelled GTPγS in the presence of 10 μM GDP, with and without various drugs, for 1 h at 30°C. All incubations were terminated by rapid filtration under vacuum through Whatman GF/B glass fibre filters, followed by three washes with 3 ml ice-cold 50 mM Tris-HCl, pH 7.4. Bound radioactivity was determined by liquid scintillation spectrophotometry at 95% efficiency for 35S, after overnight extraction of the filters in 5 ml ScintiSafe Econo-1 scintillation fluid.
Receptor binding assays
mMOR-CHO cell membranes (50 μg protein) were incubated for 1 h at 30°C in assay buffer with 0.8 nM [3H]-naloxone and 0–50 nM unlabelled naloxone. Nonspecific binding was determined with 10 μM unlabelled levallorphan. Reactions were terminated by rapid filtration through glass fibre filters, and the filters were rinsed three times with ice-cold Tris buffer. Bound radioactivity was determined by liquid scintillation spectrophotometry at 45% efficiency for 3H, after overnight extraction of the filters in scintillation fluid.
Data analysis
Unless otherwise indicated, data are reported as mean values±s.e. of at least three separate experiments that were each performed in triplicate. Net stimulated [35S]-GTPγS binding is defined as stimulated binding minus basal binding. Per cent stimulation is defined as net stimulated binding divided by basal binding (×100%). Non-linear regression analysis of concentration-effect curves was performed with JMP (SAS Institute, Cary, NY, U.S.A.) using an iterative model. Statistical significance of the data was determined by analysis of variance followed by the non-paired 2-tailed Student's t-test or Dunnett's test, also performed with JMP. IC50 values of GDP were determined by log-logit (Hill) analysis. Saturation analyses of absolute and net-stimulated [35S]-GTPγS binding were conducted using EBDA and LIGAND, as previously described (Selley et al., 1997; 1998). Because the presence of GDP precludes determination of absolute KD and Bmax values from [35S]-GTPγS saturation analyses, these values are termed apparent KD and Bmax values.
Results
Effects of sodium on relative agonist efficacy
To examine the effects of sodium on the relative efficacy of μ-opioid agonists, saturating concentrations of agonists were assayed for stimulation of [35S]-GTPγS binding in membranes prepared from mMOR-CHO cells (Kaufman et al., 1995). Agonists were chosen on the basis of different relative and intrinsic efficacies for G-protein activation: DAMGO, sufentanil, morphine, buprenorphine and levallorphan (Emmerson et al., 1996; Selley et al., 1997; 1998 Traynor & Nahorski, 1995). Agonist-stimulated [35S]-GTPγS binding was measured with excess (10 μM) GDP and varying concentrations of NaCl. Sodium inhibited basal [35S]-GTPγS binding with an IC50 value of 30±5.7 mM (Figure 1A). No significant stimulation of [35S]-GTPγS binding was observed with any agonist in the absence of NaCl. However, the potency of NaCl in inhibiting [35S]-GTPγS binding was decreased in the presence of agonists. Levallorphan increased the concentration of NaCl required for half-maximal inhibition by approximately 2.5 fold, whereas buprenorphine increased it by approximately 4 fold. These potency values could only be estimated because maximal effects of sodium could not be observed in the presence of these partial agonists. The high efficacy agonists, DAMGO, sufentanil and morphine, eliminated NaCl inhibition of [35S]-GTPγS binding up to 125 mM NaCl. Figure 1B shows net agonist-stimulated [35S]-GTPγS binding as a function of NaCl concentration. For the partial agonists levallorphan and buprenorphine, agonist-stimulated [35S]-GTPγS binding increased as a function of NaCl concentration up to 25 and 50 mM, respectively. Net [35S]-GTPγS binding stimulated by levallorphan and buprenorphine was decreased by 100–125 mM NaCl (P<0.05 versus 25 and 50 mM NaCl, respectively). With the higher efficacy agonists, DAMGO, sufentanil and morphine, net agonist-stimulated [35S]-GTPγS binding increased with NaCl concentrations up to 75 mM, but was not significantly inhibited by NaCl concentrations up to 125 mM. When the data were calculated as per cent stimulation over basal [35S]-GTPγS binding (Figure 1C), stimulation by the high efficacy agonists increased with increasing NaCl concentrations up to approximately 100 mM, due in part to the decrease in basal [35S]-GTPγS binding. However, per cent stimulation by levallorphan and buprenorphine increased to a maximum at 50 and 75 mM NaCl, respectively, and decreased with 125 mM NaCl. (P<0.05 versus 50 and 75 mM NaCl, respectively). Thus, relative agonist efficacy for G-protein activation via μ-opioid receptors was dependent on the sodium concentration, with increasing sodium concentrations magnifying relative efficacy differences.
Figure 1.

Effect of NaCl on relative agonist efficacy for stimulation of [35S]-GTPγS binding to mMOR-CHO cell membranes. Membranes were incubated with 0.1 nM [35S]-GTPγS and varying concentrations of NaCl in the presence of 10 μM GDP with and without maximally effective concentrations of opioid agonists. Data are mean values±s.e. of: (A) per cent of control [35S]-GTPγS binding (measured in the absence of NaCl or agonist); (B) net agonist-stimulated [35S]-GTPγS binding; and (C) per cent stimulation of [35S]-GTPγS binding by agonist (measured at each NaCl concentration). Control [35S]-GTPγS binding was 206±16.9 fmol mg−1.
In rat thalamic membranes, previous studies (Selley et al., 1997; 1998) showed that morphine and sufentanil acted as partial agonists relative to DAMGO, buprenorphine was a lower efficacy partial agonist, and levallorphan was an antagonist. In this system, NaCl inhibited basal and agonist-stimulated [35S]-GTPγS binding (Figure 2A). The IC50 value of NaCl in inhibiting basal [35S]-GTPγS binding was 40±4.4 mM, similar to mMOR-CHO cell membranes. However, in contrast to mMOR-CHO membranes, all agonists stimulated [35S]-GTPγS binding in thalamic membranes by approximately 40% in the absence of sodium, and increasing NaCl concentrations inhibited [35S]-GTPγS binding stimulated by all agonists. Similarly, net agonist-stimulated [35S]-GTPγS binding was inhibited by increasing concentrations of NaCl (Figure 2B). Net levallorphan-stimulated binding was inhibited by all NaCl concentrations tested (P<0.01 versus no NaCl), reaching a minimum at 100 mM NaCl, where the compound produced no stimulation. Net buprenorphine-stimulated binding was also inhibited by all NaCl concentrations tested (P<0.01 versus no NaCl), while net sufentanil- and morphine-stimulated [35S]-GTPγS binding was decreased by 50–125 mM NaCl (P<0.05 versus no NaCl), and net DAMGO-stimulated binding was only significantly inhibited by 100–125 mM NaCl (P<0.05 versus no NaCl). When the data were calculated as per cent stimulation (Figure 2C), stimulation by DAMGO increased with increasing NaCl concentrations, reaching a maximum at 75 mM NaCl. A similar effect was observed with sufentanil and morphine, but the maximal per cent stimulation by these agonists was achieved at 25 mM NaCl, and did not increase further with increasing NaCl concentrations. The per cent stimulation by levallorphan was decreased by all NaCl concentrations (P<0.01 versus no NaCl), and was abolished by 100–125 mM NaCl. Similarly, NaCl concentrations of 25–125 mM inhibited the per cent stimulation by buprenorphine (P<0.05 versus no NaCl), reaching a minimum at 100 mM NaCl. Thus, increasing sodium concentrations also increased the relative efficacy differences among agonists in rat thalamus.
Figure 2.
Effect of NaCl on relative agonist efficacy for stimulation of [35S]-GTPγS binding to rat thalamic membranes. Membranes were incubated with 0.1 nM [35S]-GTPγS and varying concentrations of NaCl in the presence of 30 μM GDP with and without maximally effective concentrations of the various opioid agonists. Data are mean values±s.e. of: (A) per cent of control [35S]-GTPγS binding (measured in the absence of NaCl or agonist); (B) net agonist-stimulated [35S]-GTPγS binding; and (C) per cent stimulation of [35S]-GTPγS binding by agonist (measured at each NaCl concentration). Control [35S]-GTPγS binding was 588±74.9 fmol mg−1.
The inhibitory effect of NaCl on stimulation of [35S]-GTPγS binding by partial agonists was due to decreases in the relative efficacy of these compounds, rather than decreases in their potency (Table 1). In mMOR-CHO membranes, increasing the NaCl concentration from 20 to 120 mM increased the DAMGO and levallorphan EC50 values by 150 fold and 6.5 fold, respectively, but also increased the efficacy of DAMGO relative to levallorphan by 10 fold. Similar results were obtained comparing DAMGO and morphine in thalamic membranes, with a 14 fold increase in the EC50 value of DAMGO and a 4 fold increase in the EC50 value of morphine, while the efficacy of DAMGO increased by 2 fold compared to morphine.
Table 1.
Potency and efficacy values of selected partial agonists compared to the full agonist DAMGO in mMOR-CHO cell and rat thalamic membranes

The effect of NaCl was partially selective to sodium, as demonstrated by replacement of NaCl with equal concentrations of KCl. Basal [35S]-GTPγS binding in mMOR-CHO membranes was effectively suppressed by K+: basal [35S]-GTPγS binding increased only 83% when 120 mM Na+ was replaced with 120 mM K+ (Figure 3), in contrast to the 5 fold increase observed in the absence of monovalent cations (Figure 1A). DAMGO- and morphine-stimulated [35S]-GTPγS binding, however, remained unaffected when K+ was replaced by Na+ (Figure 3). In contrast, Na+ decreased the levels of both buprenorphine- and levallorphan-stimulated [35S]-GTPγS binding. These results show that, unlike DAMGO and morphine, the relative efficacies of buprenorphine and levallorphan were sharply decreased by increasing the Na+:K+ ratio. Similar results were obtained in rat thalamic membranes (not shown): replacement of Na+ with K+ increased basal [35S]-GTPγS binding by 36% (compared to the 5 fold increase in the absence of monovalent cations), and increasing the Na+:K+ ratio sharply decreased the relative efficacy of partial agonists, especially buprenorphine and levallorphan. Thus, K+ only partially substituted for Na+ with regard to its effects on basal and agonist-stimulated [35S]-GTPγS binding in both mMOR-CHO and thalamic membranes.
Figure 3.

Effect of Na+:K+ ratio on relative agonist efficacy for stimulation of [35S]-GTPγS binding to mMOR-CHO cell membranes. Membranes were incubated with 0.05 nM [35S]-GTPγS in the presence of 10 μM GDP with and without maximally effective concentrations of opioid agonists and 120 mM KCl only, 60 mM KCl+60 mM NaCl, or 120 mM NaCl only. Data are mean fmol [35S]-GTPγS bound per mg membrane protein±s.e.
Effects of pertussis toxin (PTX) on agonist- and sodium-modulated [35S]-GTPγS binding
To determine whether [35S]-GTPγS binding stimulated by either DAMGO or the absence of NaCl was blocked by uncoupling of Gi/o proteins from receptors, mMOR-CHO cells were treated with PTX and assayed for [35S]-GTPγS binding. This treatment completely blocked PTX-catalyzed ADP-ribosylation of G-proteins in mMOR-CHO membranes, as measured with [32P]-NAD+ (data not shown). In untreated cells, [35S]-GTPγS binding was stimulated approximately 5 fold by either the absence of monovalent cations or by the addition of DAMGO and 100 mM NaCl (Figure 4). Stimulation by DAMGO and by the absence of monovalent cations was not additive. PTX pretreatment eliminated the stimulation of [35S]-GTPγS binding by either the absence of monovalent cations or by DAMGO (with 100 mM NaCl). In untreated cells, naloxone blocked the stimulation of [35S]-GTPγS binding by DAMGO (with 100 mM NaCl), but did not significantly affect basal [35S]-GTPγS binding in the presence or absence of NaCl (data not shown).
Figure 4.
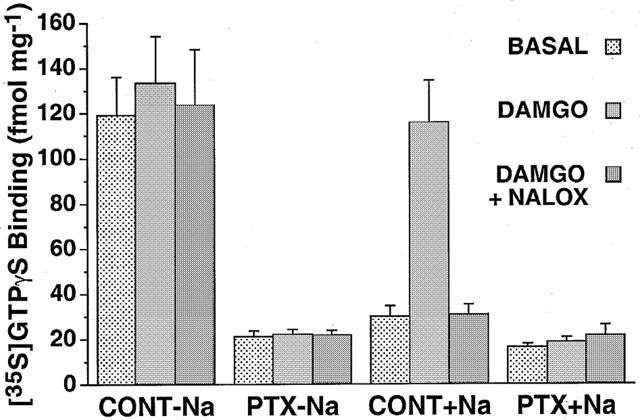
Effect of PTX pretreatment on modulation of [35S]-GTPγS binding by NaCl and agonist. Membranes from control and PTX-pretreated mMOR-CHO cells were incubated with 0.05 nM [35S]-GTPγS and 10 μM GDP in the presence and absence of 100 mM NaCl with and without 5 μM DAMGO and/or 1 μM naloxone present. Data are mean fmol [35S]-GTPγS bound per mg membrane protein±s.e.
Effects of DAMGO, sodium and PTX on competition by GDP for [35S]-GTPγS binding to mMOR-CHO membranes
Agonists stimulate [35S]-GTPγS binding by decreasing the affinity of Gα for GDP, correlating with agonist efficacy (Breivogel et al., 1998; Lorenzen et al., 1996; Selley et al., 1997). To examine whether [35S]-GTPγS binding stimulated by the absence of monovalent cations produced the same shift in GDP affinity as a full agonist, mMOR-CHO membranes were incubated with varying concentrations of GDP in the presence and absence of NaCl and DAMGO. Results showed that the potency of GDP in competing for [35S]-GTPγS binding was decreased by approximately 8 fold by either the presence of DAMGO or the absence of monovalent cations, compared to binding in the presence of 100 mM NaCl alone (Table 2). DAMGO did not further decrease the potency of GDP in the absence of monovalent cations. Furthermore, the effects of DAMGO and of the absence of monovalent cations on GDP potency were both blocked by prior treatment of the cells with PTX (Table 2).
Table 2.
IC50 values for GDP inhibition of [35S]-GTPγS binding in control and PTX-pretreated mMor-CHO cell membranes

Saturation analysis of [35S]-GTPγS binding to mMOR-CHO membranes
Previous studies showed that DAMGO stimulated both the apparent affinity and Bmax values of high affinity (but not low affinity) [35S]-GTPγS binding to mMOR-CHO cell membranes (Selley et al., 1997; 1998). To determine whether similar effects are produced by the absence of monovalent cations, saturation analysis of [35S]-GTPγS binding was conducted in mMOR-CHO membranes (Figure 5). These experiments showed that DAMGO decreased the apparent KD of high affinity [35S]-GTPγS binding by 4 fold, and increased the apparent Bmax by 2.5 fold, in the presence of NaCl (Table 3). The absence of monovalent cations produced a similar effect, decreasing the apparent KD and increasing the apparent Bmax of high affinity [35S]-GTPγS binding by 3 fold. Prior treatment of mMOR-CHO cells with PTX eliminated high affinity [35S]-GTPγS binding in either the absence (Figure 5, Table 3) or presence (data not shown) of NaCl or DAMGO. PTX pretreatment, DAMGO and NaCl all had no affect on low affinity [35S]-GTPγS binding.
Figure 5.

Effect of NaCl and agonist on saturation binding of [35S]-GTPγS presented as a Scatchard plot. Membranes from control and PTX-pretreated mMOR-CHO cells were incubated with 0.1 nM [35S]-GTPγS, 10 μM GDP and varying concentrations of unlabelled GTPγS in the presence and absence of 100 mM NaCl with and without 5 μM DAMGO. Data shown are from a typical experiment that was performed in triplicate and replicated at least three times with similar results.
Table 3.
Apparent KD and Bmax values from saturation analysis of [35S]-GTPγS binding in mMOR-CHO cell membranes

Although a slight increase in basal [35S]-GTPγS binding was observed in the absence of monovalent cations without GDP (data not shown), this effect was not statistically significant (P=0.16) and not reversed by PTX. Nonetheless, to rule out a direct effect of sodium on G-protein binding of [35S]-GTPγS, saturation analysis of [35S]-GTPγS binding was conducted in mMOR-CHO membranes in the absence of GDP (not shown). No significant differences in apparent KD or Bmax values of high affinity [35S]-GTPγS binding were found in the presence or absence of 100 mM NaCl (2.35±0.23 nM and 21.6±1.89 pmol mg−1 versus 1.96±0.18 nM and 22.2±1.86 pmol mg−1, respectively). Similar results were obtained in the presence of DAMGO, which itself had no significant effect on high affinity [35S]-GTPγS binding in the absence of GDP. Thus, similar to effects of agonists, the absence of monovalent cations increased both the apparent affinity and Bmax of high affinity [35S]-GTPγS binding only in the presence of GDP.
Effects of chronic agonist treatment of mMOR-CHO cells on modulation of [35S]-GTPγS binding by DAMGO and sodium
Previous research (Breivogel et al., 1997) has established that apparent Bmax values of net agonist-stimulated [35S]-GTPγS binding are decreased by agonist-induced receptor desensitization following chronic agonist treatment of cells. If stimulation of [35S]-GTPγS binding by the absence of monovalent cations was due to spontaneous μ-opioid receptor activity, it might be possible to decrease the apparent Bmax of these net-stimulated [35S]-GTPγS binding sites by pretreatment of the cells with DAMGO to induce μ receptor desensitization. To examine this possibility, net-stimulated [35S]-GTPγS binding was examined in GTPγS saturation experiments in membranes from control and DAMGO-pretreated mMOR-CHO cells. The results showed that both DAMGO (with 100 mM NaCl) and the absence of monovalent cations, yielded similar populations of net-stimulated [35S]-GTPγS binding sites when basal binding in the presence of 100 mM NaCl alone was subtracted (Table 4). Pretreatment of mMOR-CHO cells for 18 h with 5 μM DAMGO desensitized the DAMGO-stimulated [35S]-GTPγS binding in membranes, resulting in a 64% decrease in the apparent Bmax of net DAMGO-stimulated [35S]-GTPγS binding, with no significant change in the apparent KD (Table 4). This attenuation of DAMGO-stimulated [35S]-GTPγS binding was not due to a decrease in receptor Bmax (downregulation), since the Bmax values of [3H]-naloxone binding (∼4 pmol mg−1) were not significantly different between control and DAMGO-pretreated mMOR-CHO cells (not shown). The apparent Bmax of net [35S]-GTPγS binding stimulated by the absence of monovalent cations showed a 30% decrease in DAMGO-pretreated cells, with no significant change in apparent KD (Table 4). These decreases were probably not due to changes in basal (receptor-independent) G-protein activity, since the binding of 0.1 nM [35S]-GTPγS in the presence of 100 mM NaCl was not affected by DAMGO pretreatment (52.6±2.5 fmol mg−1 versus 54.3±2.2 fmol mg−1 in control and DAMGO-pretreated mMOR-CHO cell membranes, respectively). Thus, DAMGO pretreatment decreased the number of receptor-activated G-proteins measured in either the pesence of DAMGO or the absence of monovalent cations, but the magnitude of this desensitization was about twice as great for DAMGO-stimulated [35S]-GTPγS binding than for that stimulated by the absence of monovalent cations.
Table 4.
Apparent KD and Bmax values from saturation analysis of net-stimulated [35S]-GTPγS binding in control and DAMGO-pretreated mMOR-CHO cells

One explanation for this difference is the presence of other Gi/o-coupled receptors native to CHO cells, whose spontaneous activity was stimulated by the absence of monovalent cations. To test this possibility, saturation analysis of net [35S]-GTPγS binding was conducted in membranes from untransfected CHO cells. Although DAMGO did not affect [35S]-GTPγS binding in untransfected CHO cell membranes (data not shown), basal [35S]-GTPγS binding was stimulated by the absence of monovalent cations. The apparent Bmax values from untransfected CHO cells (Table 4) were not significantly different from the apparent Bmax value obtained in DAMGO-pretreated mMOR-CHO cells in the absence of monovalent cations, but were significantly lower than the apparent Bmax values obtained in control mMOR-CHO cells in the presence of DAMGO and NaCl or in the absence of monovalent cations. As expected, DAMGO pretreatment had no effect on [35S]-GTPγS binding in untransfected CHO cells. Interestingly, the apparent KD values of net [35S]-GTPγS binding stimulated by the absence of monovalent cations in untransfected CHO cells were approximately 3 fold lower than those stimulated by DAMGO in control or DAMGO-pretreated mMOR-CHO cells, and than that stimulated by the absence of monovalent cations in control mMOR-CHO cells, but were only 2 fold lower than those from DAMGO-pretreated mMOR-CHO cells in the absence of monovalent cations.
Discussion
The present results support previous studies of sodium effects on spontaneous Gi/o-coupled receptor activity and relative agonist efficacy (Costa et al., 1990; 1992). In both mMOR-CHO cell and rat thalamic membranes, the potency of sodium in inhibiting [35S]-GTPγS binding was inversely related to the agonist efficacy. The ability of sodium to promote agonist stimulation of [35S]-GTPγS binding also varied inversely with agonist efficacy, such that relative efficacy differences between agonists were magnified by increasing sodium concentrations. The effect of sodium on relative efficacy was due to a decrease in maximal stimulation by partial agonists compared to the full agonist DAMGO, rather than to a decrease in the potency of partial agonists. This finding agrees with previous studies of δ opioid and somatostatin sst5 receptor-stimulated [35S]-GTPγS binding (Szekeres & Traynor, 1997; Williams et al., 1997), and agrees with the well-established finding that sodium has a greater effect on binding of full versus partial agonists to opioid receptors in radioligand binding studies (Pert & Snyder, 1974). The present study expands upon previous findings by providing a quantitative examination of the effects of a range of sodium concentrations on the relative efficacy of several full and partial opioid agonists for G-protein activation. Moreover, this is the only report to examine the effects of sodium on agonist efficacy for stimulation of [35S]-GTPγS binding with the μ-opioid receptor, which is primarily responsible for mediating the analgesic, reinforcing and dependence effects of clinically relevant opiates such as morphine (Matthes et al., 1996).
The effects of sodium on basal and agonist-stimulated G-protein activation were partially mimicked by potassium. Potassium was nearly as effective as sodium in promoting receptor-mediated G-protein activation compared to the absence of monovalent cations, but was much less effective in reducing the relative efficacy of partial agonists. These results are similar to those previously reported for modulation of low Km GTPase activity by the δ-opioid receptor in NG108-15 cell membranes (Costa et al., 1990). In contrast to these previous results, however, potassium was nearly as effective as sodium in reducing basal G-protein activity in the present study. This difference may be due to different assay conditions, such as the presence of excess GDP in the [35S]-GTPγS binding assay. Indeed, Szekeres & Traynor (1997) reported that in NG108-15 membranes, replacement of sodium with potassium at 100 mM increased basal [35S]-GTPγS binding by 48%, similar to results obtained in this study.
Two important differences between mMOR-CHO cells and rat thalamus may be related to the fact that transfected CHO cells have a higher ratio of μ receptors to activated G-proteins (R:G) than rat thalamus (Selley et al., 1997; 1998). First, the ability of sodium to inhibit net agonist-stimulated [35S]-GTPγS binding was greater in the system with the lower R:G ratio; for example, sodium did not inhibit net [35S]-GTPγS binding stimulated by the full agonist DAMGO in mMOR-CHO membranes, compared to nearly a 50% decrease in thalamus. Second, the sodium requirement for agonist-stimulated [35S]-GTPγS binding was greater in the system with the higher R:G ratio. This finding supports the prediction of theoretical models showing that increasing the R:G ratio should increase spontaneous receptor activity (Costa et al., 1992), and agrees with experimental evidence that increasing receptor expression increases the spontaneous activity of other G-protein-coupled receptors, including α2-adrenergic (Tian et al., 1994), 5-HT1A (Newman-Tancredi et al., 1997) and β2-adrenergic (Adie & Milligan, 1994).
Interpretation of the present study was affected by the lack of an inverse μ-opioid agonist. Thus, several alternate approaches were utilized to demonstrate that effects of sodium on μ receptor-stimulated [35S]-GTPγS binding were due to an inhibitory effect of the cation on the ability of μ receptors to assume an active (G-protein-activating) conformation. First, pretreatment of mMOR-CHO cells with PTX blocked the stimulation of [35S]-GTPγS binding both by DAMGO (with sodium present) and by the absence of monovalent cations. This finding is important because the mechanism by which PTX-catalyzed ADP ribosylation of Gi/o-proteins disrupts signal transduction is reportedly due to uncoupling from receptor-stimulated GDP/GTP exchange, and not to inhibition of the basal G-protein activation rate itself (Sunyer et al., 1989). Second, the potency of GDP in inhibiting [35S]-GTPγS binding to mMOR-CHO membranes was decreased to the same extent by DAMGO (with sodium) and by the absence of monovalent cations (with or without DAMGO), and the rightward shifts in the GDP curves produced by either agonist or the absence of monovalent cations were both eliminated by prior treatment of the cells with PTX. Moreover, similar to the effect of agonist, the absence of monovalent cations did not significantly stimulate [35S]-GTPγS binding in the absence of GDP, indicating that sodium did not directly effect G-protein affinity for [35S]-GTPγS. Third, biphasic saturation analysis of [35S]-GTPγS binding revealed that both DAMGO (with sodium) and the absence of monovalent cations increased the apparent affinity and Bmax values of high affinity [35S]-GTPγS binding without affecting low affinity [35S]-GTPγS binding sites. Moreover, PTX eliminated high affinity [35S]-GTPγS binding both in the absence of monovalent cations and presence of agonist. As with agonist-stimulated [35S]-GTPγS binding, the effect of the absence of monovalent cations on apparent KD and Bmax values of high affinity [35S]-GTPγS binding was observed only in the presence of GDP. Fourth, DAMGO (with sodium) or the absence of monovalent cations produced identical apparent KD and Bmax values for [35S]-GTPγS binding, while chronic DAMGO-pretreatment of cells desensitized stimulation produced by DAMGO (with sodium) and by the absence of monovalent cations.
The attenuation of μ receptor-mediated G-protein activation observed after DAMGO pretreatment of mMOR-CHO cells was apparently due to desensitization rather than downregulation of the μ receptor, as determined by the lack of a significant decrease in the Bmax of [3H]-naloxone. This observation differed from previous reports of DAMGO-induced downregulation of μ receptors expressed in CHO cells (Kato et al., 1998; Pak et al., 1996). Variation between the results of the present and previous studies may have been due to differences in cell culture and agonist pretreatment conditions, species of cloned μ receptor or method of membrane preparation. The desensitization observed in the this study was likely due to a homologous uncoupling of μ receptors from G-proteins, rather than a decrease in basal G-protein activity or levels, because no difference in basal [35S]-GTPγS binding was observed in the presence of 100 mM NaCl. This conclusion is supported by a previous report that chronic pretreatment of μ receptor-containing neuroblastoma cells with DAMGO did not affect the levels or basal activity of Gi/o-proteins (Ammer & Schulz, 1993).
The smaller magnitude of desensitization observed in the absence of monovalent cations was probably due to other Gi/o-coupled receptors in CHO cells, whose spontaneous activities were also stimulated by the absence of monovalent cations. Indeed, sodium inhibition of spontaneous receptor activity has been reported for other Gi/o-coupled receptors (Costa et al., 1990; Hilf & Jakobs, 1992; Mullaney et al., 1996; Szekeres & Traynor, 1997; Tian et al., 1994; Wenzel-Seifert et al., 1998). This possibility was supported by data in untransfected CHO cells showing that apparent Bmax values of net [35S]-GTPγS binding in the absence of monovalent cations were similar to the apparent Bmax value in the absence of monovalent cations in DAMGO-pretreated mMOR-CHO cells, but were lower than in control mMOR-CHO cells. Furthermore, in the present study, there was no increase in the apparent Bmax of activated G-proteins in the absence of monovalent cations compared to the value obtained and DAMGO with sodium, suggesting that there were enough μ receptors present to fully activate the Gi/o-protein pool without additional receptor activity. These results are consistent with the hypothesis that high relative efficacies of partial agonists in mMOR-CHO membranes are due to the high R:G ratio; i.e., there is a ‘ceiling effect' on the maximal number of G-proteins that can be activated by μ receptors in this system (Selley et al., 1997; 1998), since the number of μ receptors in mMOR-CHO membranes exceeds the number of receptor-activated G-proteins (Selley et al., 1998). In support of this interpretation, several opioids that were full agonists in mMOR-CHO membranes demonstrated receptor reserve for G-protein activation in this system, but not in rat thalamus (Selley et al., 1998). One finding inconsistent with this interpretation is that the Bmax value of high affinity [35S]-GTPγS binding measured in the absence of GDP was 3 fold higher than that measured with GDP in the absence of monovalent cations or presence of DAMGO and NaCl. However, not all high affinity [35S]-GTPγS binding sites are sensitive to the modulatory effects of Gi/o-coupled receptors on GDP affinity. High affinity [35S]-GTPγS binding sites such as Gαs, Gαq/11, Gα12/13 or other GTP-binding proteins in CHO cell membranes may remain occupied by GDP, while GDP dissociates from receptor-activated Gαi/o.
The finding that the apparent KD values of net [35S]-GTPγS binding stimulated by the absence of monovalent cations in untransfected CHO cells were lower than those in mMOR-CHO cells may indicate small differences in the ability of different receptor types to decrease the affinity of receptor-coupled G-proteins for GDP. The apparent KD value obtained in DAMGO-pretreated mMOR-CHO cells in the absence of monovalent cations was intermediate between the value obtained in untreated mMOR-CHO and untransfected CHO cells, further suggesting that [35S]-GTPγS binding stimulated by the absence of monovalent cations in DAMGO-pretreated mMOR-CHO cells was due to spontaneous activity of non-opioid Gi/o-coupled receptors along with the remaining (undesensitized) μ receptors.
What is the physiological role of sodium in modulating the activation state of Gi/o-coupled receptors? Previous studies with the δ-opioid inverse agonist ICI-174,864 suggest that spontaneous receptor activity is minimal in intact NG108-15 cells (Costa et al., 1992), but that δ-opioid receptors display significant spontaneous activity in transfected cells with high levels of δ receptors (Chiu et al., 1996; Merkouris et al., 1997). Even if spontaneous receptor activity is minimal in most physiological systems due to the lower naturally-occurring R:G ratios than transfected cells, the present study indicates that sodium can significantly modulate relative agonist efficacy by its ability to produce non-equivalent suppression of the responses stimulated by agonists of varying intrinsic efficacies. This effect of sodium was observed even when high monovalent cation concentrations were maintained with potassium, indicating that the sodium:potassium ratio was a significant factor in the determination of relative efficacy. However, the question remains whether intracellular or extracellular sodium concentrations regulate receptor activity. If intracellular sodium is more important, then changes in the sodium:potassium ratio at the internal membrane surface (such as those occurring transiently during a neuronal action potential) may regulate agonist efficacy in vivo. Alternatively, if the primary site of sodium action on the receptor is extracellular, then the minimal fluctuations in the extracellular sodium:potassium ratio that would be expected to occur in mammals may mean that sodium would not significantly modulate agonist efficacy in vivo. Unfortunately, the literature has been conflicting with regard to this question (Puttfarcken et al., 1986; Yabaluri & Medzihradsky, 1997). Considering previous findings that sodium sensitivity of receptor-coupled G-protein activity varies among different brain regions (Pacheco et al., 1994), it will be important to further examine the effects of both intracellular and extracellular sodium on receptor-coupled G-protein activity and agonist efficacy.
Acknowledgments
The authors thank Jennifer A. Schwegel and Ruoyu Xiao for excellent technical assistance, and Dr Christopher J. Evans and Duane Keith for development of the mMOR-CHO cell line. This work was partially supported by USPHS grants DA10770 (D.E. Selley), and DA02904 and DA-06634 (S.R. Childers) from the National Institute on Drug Abuse.
Abbreviations
- DAMGO
[D-Ala2,(N-Me)Phe4,Gly5-ol]-enkephalin
- DMEM
Dulbecco's modified eagle's medium
- FBS
foetal bovine serum
- GTPγS
guanosine-5′-O-(γ-thio)-triphosphate
- PTX
pertussis toxin
References
- ABOOD M., NOEL M., CARTER R., HARRIS L. Evaluation of a series of N-alkyl benzomorphans in cell lines expressing transfected δ- and μ-opioid receptors. Biochem. Pharmacol. 1995;50:851–859. doi: 10.1016/0006-2952(95)02007-y. [DOI] [PubMed] [Google Scholar]
- ADAMS J., PARONIS C., HOLTZMAN S. Assessment of relative intrinsic activity of mu-opioid analgesics in vivo by using β-funaltrexamine. J. Pharmacol. Exp. Ther. 1990;255:1027–1032. [PubMed] [Google Scholar]
- ADIE E.J., MILLIGAN G. Regulation of basal adenylate cyclase activity in neuroblastoma x glioma hybrid, NG108-15, cells transfected to express the human β2 adrenoceptor: evidence for empty receptor stimulation of the adenylate cyclase cascade. Biochem. J. 1994;303:803–808. doi: 10.1042/bj3030803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMMER H., SCHULZ R. Alterations in the expression of G-proteins and regulation of adenylate cyclase in human neuroblastoma SH-SY5Y cells chronically exposed to low-efficacy μ-opioids. Biochem. J. 1993;295:263–271. doi: 10.1042/bj2950263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SELLEY D.E., CHILDERS S.R. Acute and chronic effects of opioids on delta and mu receptor activation of G-proteins in NG108-15 and SK-N-SH cell membranes. J. Neurochem. 1997;68:1462–1472. doi: 10.1046/j.1471-4159.1997.68041462.x. [DOI] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SELLEY D.E., CHILDERS S.R. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPγS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 1998;273:16865–16873. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- CARTER B.D., MEDZIHRADSKY F. Receptor mechanisms of opioid tolerance in SH-SY5Y human neural cells. Mol. Pharmacol. 1993;43:465–473. [PubMed] [Google Scholar]
- CERESA B.P., LIMBIRD L.E. Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of α2-adrenergic receptor-G-protein interactions: A negative charge at amino acid residue 79 forecasts α2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. Journal of Biol. Chem. 1994;269:29557–29564. [PubMed] [Google Scholar]
- CHEN Y., MESTEK A., LIU J., HURLEY J.A., YU L. Molecular cloning and functional expression of a μ-opioid receptor from rat brain. Mol. Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- CHILDERS S.R. Opioid receptor-coupled second messengers. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- CHIU T., YUNG L., WONG Y. Inverse agonistic effect of ICI-174,864 on the cloned δ-opioid receptor: role of G-protein and adenylyl cyclase activation. Mol. Pharmacol. 1996;50:1651–1657. [PubMed] [Google Scholar]
- CORBETT A.D., PATERSON S.J., KOSTERLITZ H.W. Selectivity of ligands for opioid receptors {it}Handbook of Experimental Pharmacology: Opioids I 1993Berlin: Springer-Verlag; 645–679.ed. Herz, A. pp [Google Scholar]
- COSTA T., LANG J., GLESS C., HERZ A. Spontaneous association between opioid receptors and GTP-binding proteins in native membranes: specific regulation by antagonists and sodium ions. Mol. Pharmacol. 1990;37:383–394. [PubMed] [Google Scholar]
- COSTA T., OGINO Y., MUNSON P., ONARAN H., RODBARD D. Drug efficacy at guanine nucleotide-binding regulatory protein linked receptors: Thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol. Pharmacol. 1992;41:549–560. [PubMed] [Google Scholar]
- EMMERSON P.J., CLARK M.J., MANSOUR A., AKIL H., WOODS J.H., MEDZIHRADSKY F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the μ opioid receptor. J. Pharmacol. Exp. Ther. 1996;278:1121–1127. [PubMed] [Google Scholar]
- HERKENHAM M., PERT C.B. Light microscopic localization of brain opiate receptors: A general autoradiographic method which preserves tissue quality. J. Neurosc. 1982;2:1129–1149. doi: 10.1523/JNEUROSCI.02-08-01129.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILF G., JAKOBS K.H. Agonist-independent inhibition of G protein activation by muscarinic acetylcholine receptor antagonists in cardiac membranes. European J. Pharmacol. 1992;225:245–252. doi: 10.1016/0922-4106(92)90026-r. [DOI] [PubMed] [Google Scholar]
- HORSTMAN D.A., BRANDON S., WILSON A.L., GUYER C.A., CRAGOE C.A., LIMBIRD L.E. An aspartate conserved among G-protein receptors confers allosteric regulation of alpha(2)-adrenergic receptors by sodium. J. Biol. Chem. 1990;265:21590–21595. [PubMed] [Google Scholar]
- KATO S., FUKUDA K., MORIKAWA H., SHODA T., MIMA H., MORI K. Adaptations to chronic agonist-exposure of mu-opioid receptor-expressing Chinese hamster ovary cells. Europ. J. Pharmacol. 1998;345:221–228. doi: 10.1016/s0014-2999(98)00023-5. [DOI] [PubMed] [Google Scholar]
- KAUFMAN D., KEITH D., ANTON B., TIAN J., MAGENDZO K., NEWMAN D., TRAN T., LEE D., WEN C., XIA Y.-R., LUSIS A., EVANS C. Characterization of the murine μ opioid receptor gene. J. Biol. Chem. 1995;270:15877–15883. doi: 10.1074/jbc.270.26.15877. [DOI] [PubMed] [Google Scholar]
- KONG H., RAYNOR K., YASUDA K., BELL G., REISINE T. Mutation of an aspartate at residue 89 somatostatin receptor subtype 2 prevents Na+ regulation of agonist binding but does not alter receptor-G protein association. Mol. Pharmacol. 1993a;44:380–384. [PubMed] [Google Scholar]
- KONG H., RAYNOR K., YASUDA K., MOE S., PORTOGHESE P., BELL G., REISINE T. A single residue, aspartic acid 95, in the δ opioid receptor specifies selective high affinity agonist binding. J. Biol. Chem. 1993b;286:23055–23058. [PubMed] [Google Scholar]
- LORENZEN A., GUERRA L., VOGT H., SCHWABE U. Interaction of full and partial agonists of the A1 adenosine receptor with receptor/G protein complexes in rat brain membranes. Mol. Pharmacol. 1996;49:915–926. [PubMed] [Google Scholar]
- MATTHES H.W.D., MALDONADO R., SIMONIN F., VALVERDE O., SLOWE S., KITCHEN I., BEFORT K., DIERICH A., LEMEUR M., DOLLE P., TZAVARA E., HANOUNE J., ROQUES B.P., KIEFFER B.L. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- MERKOURIS M., MULLANEY I., GEORGOUSSI Z., MILLIGAN G. Regulation of spontaneous activity of the δ-opioid receptor: Studies of inverse agonism in intact cells. J. Neurochem. 1997;69:2115–2122. doi: 10.1046/j.1471-4159.1997.69052115.x. [DOI] [PubMed] [Google Scholar]
- MJANGER E., YAKSH T.L. Characteristics of dose-dependent antagonism by beta-funaltrexamine of the antinociceptive effects intrathecal mu agonists. J. Pharmacol. Exp. Ther. 1991;258:544–550. [PubMed] [Google Scholar]
- MULLANEY I., CARR I., MILLIGAN G. Analysis of inverse agonism at the δ opioid receptor after expression in Rat 1 fibroblasts. Biochem. J. 1996;315:227–234. doi: 10.1042/bj3150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWMAN-TANCREDI A., CONTE C., CHAPUT C., VERRIELE L., MILLAN M. Agonist and inverse agonist efficacy at human recombinant serotonin 5-HT1A receptors as a function of receptor:G-protein stoichiometry. Neuropharmacol. 1997;36:451–459. doi: 10.1016/s0028-3908(97)00022-1. [DOI] [PubMed] [Google Scholar]
- PACHECO M.A., WARD S.J., CHILDERS S.R. Differential requirements of sodium for coupling of cannabinoid receptors to adenylyl cyclase in rat brain membranes. J. Neurochem. 1994;62:1773–1782. doi: 10.1046/j.1471-4159.1994.62051773.x. [DOI] [PubMed] [Google Scholar]
- PAK Y., KOUVELAS A., SCHEIDLER M., RASMUSSEN J., O'DOWD B., GEORGE S. Agonist-induced functional desensitization of the μ-opioid receptor is mediated by loss of membrane receptors rather than uncoupling from G protein. Mol. Pharmacol. 1996;50:1214–1222. [PubMed] [Google Scholar]
- PAUWELS P., TARDIF S., WURCH T., COLPAERT F. Stimulated [35S]GTPgS binding by 5-HT1A receptor agonists in recombinant cell lines: Modulation of apparent efficacy by G-protein activation state. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:551–561. doi: 10.1007/pl00005090. [DOI] [PubMed] [Google Scholar]
- PERT C.B., SNYDER S.H. Opiate receptor binding of agonists and antagonists affected differentially by sodium. Mol. Pharmacol. 1974;10:868–879. [Google Scholar]
- PUTTFARCKEN P., WERLING L., BROWN S., COTE T., COX B. Sodium regulation of agonist binding at opioid receptors. I. Effects of sodium replacement on binding at μ- and δ-type receptors in 7315c and NG108-15 cells and cell membranes. Mol. Pharmacol. 1986;30:81–89. [PubMed] [Google Scholar]
- SELLEY D., LIU Q., CHILDERS S. Signal transduction correlates of mu opioid agonist intrinsic efficacy: Receptor-stimulated [35S]GTPγS binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- SELLEY D.E., SIM L.J., XIAO R., LIU Q., CHILDERS S.R. Mu opioid receptor-stimulated [35S]GTPγS binding in rat thalamus and cultured cell lines: Signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- SIM L.J., LIU Q.X., CHILDERS S.R., SELLEY D.E. Endomorphin-stimulated guanosine-5′-O-(3-[35S]thio)triphosphate binding in rat brain: Evidence for partial agonist activity at mu opioid receptors. J. Neurochem. 1998;70:1567–1576. doi: 10.1046/j.1471-4159.1998.70041567.x. [DOI] [PubMed] [Google Scholar]
- SUNYER T., MONASTIRSKY B., CODINA J., BIRNBAUMER L. Studies on nucleotide and receptor regulation of Gi proteins: Effects of pertussis toxin. Mol. Endocrinol. 1989;3:1115–1124. doi: 10.1210/mend-3-7-1115. [DOI] [PubMed] [Google Scholar]
- SZEKERES P., TRAYNOR J. Delta opioid modulation of the binding of guanosine-5′-O-(3-[35S]thio)triphosphate to NG108-15 cell membranes: Characterization of agonist and inverse agonist effects. J. Pharmacol. Exp. Ther. 1997;283:1276–1284. [PubMed] [Google Scholar]
- TAO Q., ABOOD M. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J. Pharmacol. Exp. Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- THOMPSON R.C., MANSOUR A., AKIL H., WATSON S.J. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- TIAN W.-N., DUZIC E., LANIER S.M., DETH R.C. Determinants of α2-adrenergic receptor activation of G proteins: evidence for a precoupled receptor/G protein state. Mol. Pharmacol. 1994;45:524–531. [PubMed] [Google Scholar]
- TRAYNOR J.R., NAHORSKI S.R. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995;47:848–854. doi: 10.1016/S0026-895X(25)08634-1. [DOI] [PubMed] [Google Scholar]
- WENZEL-SEIFERT K., HURT C., SEIFERT R. High constitutive activity of the human formyl peptide receptor. J. Biol. Chem. 1998;273:24181–24189. doi: 10.1074/jbc.273.37.24181. [DOI] [PubMed] [Google Scholar]
- WILLIAMS A., MICHEL A., FENIUK W., HUMPHREY P. Somatostatin5 receptor-mediated [35S]guanosine-5′-O-(3-thio)triphosphate binding: Agonist potencies and the influence of sodium chloride on intrinsic activity. Mol. Pharmacol. 1997;51:1060–1069. doi: 10.1124/mol.51.6.1060. [DOI] [PubMed] [Google Scholar]
- YABALURI N., MEDZIHRADSKY F. Regulation of mu-opioid receptor in neural cells by extracellular sodium. J. Neurochem. 1997;68:1053–1061. doi: 10.1046/j.1471-4159.1997.68031053.x. [DOI] [PubMed] [Google Scholar]
- YU V.C., SADEE W. Efficacy and tolerance of narcotic analgesics at the mu opioid receptor in differentiated human neuroblastoma cells. J. Pharmacol. Exp. Ther. 1988;245:350–355. [PubMed] [Google Scholar]