

Abstract
Endothelial dysfunction plays a key role in the pathogenesis of diabetic vascular disease. The endothelium controls the tone of the underlying vascular smooth muscle through the production of vasodilator mediators. The endothelium-derived relaxing factors (EDRF) comprise nitric oxide (NO), prostacyclin, and a still elusive endothelium-derived hyperpolarizing factor (EDHF). Impaired endothelium-dependent vasodilation has been demonstrated in various vascular beds of different animal models of diabetes and in humans with type 1 and 2 diabetes. Several mechanisms of endothelial dysfunction have been reported, including impaired signal transduction or substrate availibility, impaired release of EDRF, increased destruction of EDRF, enhanced release of endothelium-derived constricting factors and decreased sensitivity of the vascular smooth muscle to EDRF. The principal mediators of hyperglycaemia-induced endothelial dysfunction may be activation of protein kinase C, increased activity of the polyol pathway, non-enzymatic glycation and oxidative stress. Correction of these pathways, as well as administration of ACE inhibitors and folate, has been shown to improve endothelium-dependent vasodilation in diabetes. Since the mechanisms of endothelial dysfunction appear to differ according to the diabetic model and the vascular bed under study, it is important to select clinically relevant models for future research of endothelial dysfunction.
Keywords: Advanced glycation end products, aldose reductase, diabetes, endothelial dysfunction, endothelium-dependent vasodilation, endothelium-derived hyperpolarizing factor, nitric oxide, oxidative stress, protein kinase C
Introduction
Macro- and microvascular disease are currently the principal causes of morbidity and mortality in patients with type I and type II diabetes mellitus. Loss of the modulatory role of the endothelium may be a critical and initiating factor in the development of diabetic vascular disease.
Endothelial cells actively regulate basal vascular tone and vascular reactivity in physiological and pathological conditions, by responding to mechanical forces and neurohumoral mediators with the release of a variety of relaxing and contracting factors (Furchgott & Vanhoutte, 1989). The endothelium-derived relaxing factors (EDRFs) include nitric oxide (NO), prostacyclin and an, as yet elusive, endothelium-derived hyperpolarizing factor (EDHF) (Feletou & Vanhoutte, 1999). The activity of the endothelium extends, however, far beyond the control of vascular tone and reactivity, and the release of vasodilating mediators clearly reflects only one aspect of the homeostatic and protective role of the endothelium. Nevertheless, endothelium-dependent vasodilatation is generally used as a reproducible and accessible parameter to probe endothelial function in different pathophysiological conditions.
The present communication reviews the extant experimental and clinical research on disordered endothelium-dependent vasodilatation in diabetes, with a focus on those studies ancillary to a better understanding of its mechanisms and aetiology.
Although strict glycaemic control delays the onset and slows down the progression of diabetic vascular complications (The DCCT Research Group, 2000), this strategy is not successful in all patients. The knowledge obtained from the studies on endothelial dysfunction has given impetus to the search for novel approaches in the prevention and treatment of diabetic vascular disease. These alternative strategies will be particularly suitable for those diabetic patients that are unable to achieve a strict metabolic control and will be addressed by the current review where appropriate.
Experimental and clinical evidence for the presence of impaired endothelium-dependent vasodilatation in type I and II diabetes
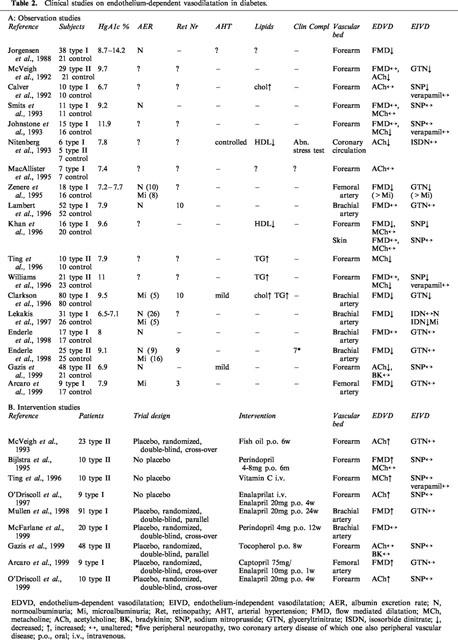
Whereas some of the earlier reports showed normal (Wakabayashi et al., 1987; Head et al., 1987; Mulhern & Docherty, 1989) or even enhanced (White & Carrier, 1986; Bhardwaj & Moore, 1988; Gebremedhin et al., 1988) endothelium-dependent vasodilatation, impaired responses to different endothelium-dependent agonists have been repeatedly and consistently demonstrated in different vascular beds of both chemically-induced and genetic models of type I diabetes (Table 1A–C). Similarly, impaired endothelium-dependent vasodilatation has been demonstrated in patients with type I diabetes in the absence of clinical complications, although several studies failed to confirm these findings (Table 2A). The discrepancies are most likely due to differences in the clinical characteristics of the study population. Not surprisingly, negative results were generally obtained in patients with normoalbuminuria and relatively good metabolic control (Smits et al., 1993; Lambert et al., 1996; Enderle et al., 1998). When microalbuminuric patients were included in the study population, impaired endothelium-dependent vasodilatation was consistently reported (Zenere et al., 1995; Clarkson et al., 1996; Lekakis et al., 1997; Arcaro et al., 1999).
Table 1.
Experimental studies of impaired endothelium-dependent vasodilatation with attempts to restore the defect.
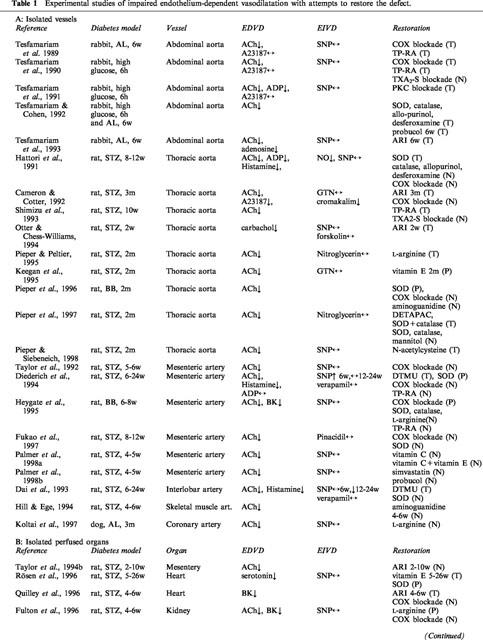

Table 2.
Clinical studies on endothelium-dependent vasodilatation in diabetes.
Studies investigating endothelial function in animal models of type II diabetes are scarce and have yielded conflicting results. Both impaired (Sakamoto et al., 1998) and preserved (Bohlen & Lash, 1995) endothelium-dependent responses have been reported. Clinical studies in patients with type II diabetes are almost inevitably confounded by the high prevalence of other cardiovascular risk factors that are known to affect endothelial function. Several authors demonstrated impaired endothelium-dependent vasodilatation in patients with type II diabetes (Table 2A), but even after rigorous patient selection, mild dyslipidaemia or hypertension were often present. Although probably irrelevant for practical purposes, it thus remains unclear whether diabetes type II per se affects endothelial function.
Mechanisms of impaired endothelium-dependent vasodilatation in diabetes
Impaired endothelium-dependent vasodilatation may arise from several mechanisms: decreased production of one of the EDRFs, enhanced inactivation of EDRF, impaired diffusion of EDRF to the underlying smooth muscle cells, decreased responsiveness of the smooth muscle to EDRF and enhanced generation of endothelium-derived constricting factors (EDCF) (Figure 1). For each of these mechanisms, both supporting and negative evidence have been presented. Whereas differences in diabetes model and in duration or severity of diabetes undoubtedly play a role in some of the discrepancies, the type of circulation, the size of the vessel and the conditions of study may be a much more important source of disparity. This has been typically illustrated by the presence of impaired endothelium-dependent vasodilatation in vivo in the mesenteric circulation or in the isolated perfused mesentery of diabetic rats, and by its absence in the isolated aorta of the same animals (Fortes et al., 1983; Taylor et al., 1994b). Similarly, bradykinin-mediated vasodilatation was depressed in the hindquarters vasculature, but was normal in the kidney and mesenterium of the same diabetic rats (Kiff et al., 1991).
Figure 1.
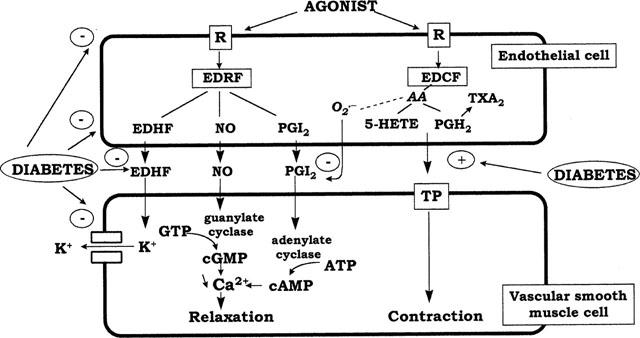
Mechanisms of endothelial dysfunction in diabetes. R, receptor; EDRF, endothelium-derived relaxing factor; EDHF, endothelium-derived hyperpolarizing factor; PGI2, prostacyclin; EDCF, endothelium-derived constricting factors; TXA2, thromboxane A2; PGH2, prostaglandin H2; 5-HETE, 5-hydroxyeicosatetraenoic acid; TP, prostanoid TP receptor; O2.−, superoxide anion.
Endothelial cells from different vascular beds exhibit metabolic and structural differences and may be affected differentially by hyperglycaemia (Sobrevia & Mann, 1997). Furthermore, the mechanisms of endothelium-dependent vasodilatation may be distinct, depending on the vascular preparation of study. Although NO has been generally considered as the principal mediator of endothelium-dependent relaxations, it has become clear that EDHF may also be an important regulator of vascular tone and reactivity, especially in small resistance vessels (Félétou & Vanhoutte, 1999). Several studies demonstrated a gradient in the release of EDRFs, with a progressively increasing contribution of EDHF in the more distal vessels. The identity of EDHF has been the subject of persistent controversy. It is likely that more than one ‘EDHF' exists, with substantial species and regional heterogeneity (Mombouli & Vanhoutte, 1997). Therefore, the relative contribution and the nature of the NO-independent vasodilator mechanisms may engender some of the observed discrepancies between the studies.
Most of the earlier research has concentrated on the study of isolated large conduit arteries. More recent research has shifted somewhat towards the study of isolated resistance vessels, which are of more direct relevance to the control of local blood flow. Studies of isolated perfused organs, although scarce, yield additional information, since vascular resistance and reactivity is determined by the whole circulation, including the smallest arterioles. Finally, although in vivo studies have limitations regarding the toxicity of certain pharmacological interventions, they allow for the study of endothelial function under physiologic flow conditions and in the presence of the diabetic extracellular fluid composition. It is therefore imperative to consider evidence from all types of experimental conditions before solid conclusions can be drawn.
Signal transduction pathway
Reduced expression and structural modifications of G-proteins, with reversal upon insulin treatment, have been described in diabetic rat retina (Sobrevia & Mann, 1997). Impaired ACh-induced relaxation with normal responses to bradykinin has been reported in isolated resistance vessels from patients with type I diabetes (McNally et al., 1994), in the forearm circulation of type II diabetes patients (Gazis et al., 1999) and in mesenteric and hindlimb arteries of streptozotocin (STZ)-rats (Lash & Bohlen, 1991; Taylor et al., 1995), suggesting an abnormality at the level of the G-proteins. However, several other studies found equally suppressed responses to different endothelium-dependent agonists (Heygate et al., 1995; Fulton et al., 1996; Costa e Forti & Fonteles, 1998; Mayhan & Patel, 1995; 1998; Mayhan, 1997) or impaired relaxation to the calcium-ionophore A23187 (Oyama et al., 1986; Durante et al., 1988; Cameron & Cotter, 1992; Fukao et al., 1997), making a disturbance of receptors or receptor-coupled mechanisms unlikely as a common mechanism of endothelial dysfunction.
Substrate availability
Although the supply of L-arginine is not a rate-limiting factor for NO synthesis in normal circumstances, reduced availability or impaired transport or metabolism of L-arginine could be a mechanism of endothelial dysfunction in diabetic vessels. Markedly reduced serum arginine levels have been observed in diabetic rats (Pieper & Peltier, 1995; Rösen et al., 1996; Angulo et al., 1998) and have been attributed to enhanced consumption of L-arginine due to an increased NO synthesis. In accordance, an increased NO synthase activity, measured by conversion of 3H-L-arginine to 3H-L-citrulline, was demonstrated in diabetic rat heart endothelium (Rösen et al., 1996). Exogenous L-arginine (partially) restored endothelium-dependent vasodilatation in certain (Pieper & Peltier, 1995; Fulton et al., 1996; Matsunaga et al., 1996; Angulo et al., 1998), but not all studies (Heygate et al., 1995; Koltai et al., 1997; Mayhan et al., 1997). These variable results may relate to an aspecific effect of L-arginine: this amino acid is known to release insulin, which by itself may stimulate endothelium-dependent vasodilatation (MacAllister et al., 1995).
Increased destruction of EDRF
Much of the attention has focused on the extent of the endothelium-dependent vasodilatation. However, a more transient relaxation has also been reported in the aorta of diabetic rats, even though the degree of relaxation was normal (Hattori et al., 1991). Superoxide dismutase (SOD) restored the duration of the aortic relaxation, suggesting inactivation of EDRF by oxygen-derived free radicals. In a EDRF bioassay experiment, the perfusion of diabetic aorta produced less relaxation of a bioassay ring, as compared to control aorta. Infusion of SOD at a site proximal to the donor segment normalized the relaxations, suggesting that similar levels of EDRF are released by diabetic aorta, but that their action is attenuated by reactive oxygen species (Pieper et al., 1992). The role of free radicals will be more extensively discussed under the subheading ‘Oxidative stress'.
EDCF
Several observations implicate an overproduction of endothelium-derived vasoconstrictors, most likely prostanoids, in the pathophysiology of endothelial dysfunction, e.g. in pial arterioles of diabetic rats in vivo (Mayhan et al., 1991) and in diabetic isolated aorta (Shimizu et al., 1993; Tesfamariam et al., 1989). Similar mechanisms may play a role after in vitro exposure of rabbit aorta to high glucose concentrations (Tesfamariam et al., 1990). These EDCFs are thought to be released together with the EDRFs and oppose their effects on the smooth muscle cells. The impaired relaxations are restored by non-specific cyclo-oxygenase blockade and prostanoid TP receptor antagonists, but not by thromboxane A2 synthase blockers, suggesting that the culprit is a prostaglandin endoperoxide (Tesfamariam et al., 1989; 1990; Mayhan et al., 1991; Shimizu et al., 1993). On the other hand, cyclo-oxygenase inhibition did not restore impaired endothelium-dependent relaxations in isolated mesenteric arteries (Taylor et al., 1992; Diederich et al., 1994; Fukao et al., 1997), in the isolated perfused heart and kidney (Quilley et al., 1996; Fulton et al., 1996) and in the renal microcirculation in vivo (De Vriese et al., 1999), indicating that vasoactive prostanoids do not play an important contributory role to the endothelial dysfunction in these vascular beds.
EDHF
Few studies have focused on the contribution of EDHF to endothelial dysfunction in diabetes. In the absence of specific inhibitors of EDHF, most of the current evidence is inevitably indirect. Decreased ACh-induced hyperpolarization and NO synthase- and cyclo-oxygenase-resistant relaxation were observed in isolated mesenteric arteries (Fukao et al., 1997). In the Langendorff perfused heart (Quilley et al., 1996), in the isolated perfused kidney (Fulton et al., 1996) as well as in the renal microcirculation in vivo (De Vriese et al., 1999), the NO synthase- and cyclo-oxygenase-resistant vasodilatation to bradykinin or ACh was profoundly impaired. Other authors observed a more pronounced deficit of the ACh-induced relaxations in the presence of NO synthase- and cyclo-oxygenase-blockade in mesenteric arteries (Taylor et al., 1992) or a decreased NO synthase-resistant ACh-induced relaxation in isolated renal arteries of diabetic rats (Dai et al., 1993), but they did not link their findings to an impaired EDHF-mediated influence. In the isolated rat aorta, no evidence was found for a decreased contribution of EDHF to the endothelium-dependent relaxations (Endo et al., 1995). Since the contribution of EDHF is most pronounced in smaller vessels, it is not surprising that evidence for a role for EDHF in diabetic endothelial dysfunction is restricted to resistance artery and whole organ studies.
Decreased responsiveness of the vascular smooth muscle to EDRF
The large majority of the studies demonstrate an impaired vasodilatation to endothelium-dependent agonists in the presence of preserved responses to endothelium-independent vasodilatators. This suggests that the diabetic state does not cause a generalized reduction in the sensitivity of the smooth muscle to EDRF–at least not initially. In one of the few experimental studies where a decreased response to nitrovasodilators was observed, it was preceded by a disturbed response to ACh (Dai et al., 1993). An impaired response to nitrovasodilators was more frequently found in humans (McVeigh et al., 1992; Calver et al., 1992; Zenere et al., 1995; Williams et al., 1996; Clarkson et al., 1996; Lekakis et al., 1997), perhaps due to the more advanced state of the diabetes. In support of this contention, the dilatation to isosorbide dinitrate was decreased in microalbuminuric, but not in normoalbuminuric patients (Lekakis et al., 1997). These findings suggest that distinct mechanisms may mediate the impaired response to endothelium-independent agonists.
Interestingly, a selectively decreased responsiveness to ATP-operated potassium channel openers has been reported by several authors (Cameron & Cotter, 1992; Mayhan & Faraci, 1993; Crijns et al., 1998), although a preserved response was noted in other studies (Fukao et al., 1997; De Vriese et al., 1999).
Conclusion
As demonstrated in this section, a broad spectrum of altered properties is potentially responsible for disordered endothelium-dependent vasodilatation in diabetes. In the aorta, impaired endothelium-dependent vasodilatation can largely be attributed to production of vasoconstrictor prostanoids and/or oxygen-derived free radicals. An important action of the latter may be the rapid destruction of NO, initially leading to an increased NO synthase activity. NO production may ultimately become compromised, perhaps by limited availability of the substrate L-arginine. In the smaller vessels, the situation is less clear-cut, and several types of interventions have failed to restore the defect. One reason may be that most of the research has focused on impaired NO-mediated vasodilatation with less concern for NO-independent vasodilatory mechanisms. Often, decreased endothelium-dependent vasodilatation was automatically reported as decreased NO-mediated vasodilatation, without knowledge of the relative contribution of the different endothelium-derived vasodilator mechanisms. As outlined above, EDHF is gaining importance as an alternative regulator of vascular tone and reactivity. This new level of understanding will hopefully give impetus to more research into NO-independent mechanisms of endothelial dysfunction, which may be especially important in small arteries.
Aetiology of endothelial dysfunction in diabetes
Although the nature of the pathogenic link between high ambient glucose concentrations and diabetic complications remains a matter of debate, hyperglycaemia is clearly recognized as the primary culprit in the pathogenesis of diabetic complications. Hyperglycaemia induces repeated acute changes in intracellular metabolism (activation of polyol pathway, activation of diacylglycerol-protein kinase C, increased oxidative stress), as well as cumulative long-term changes in the structure and function of macromolecules through formation of advanced glycation end products (AGEs). The present part of the review examines the evidence for the involvement of these pathways in the pathogenesis of endothelial dysfunction (Figure 2). The different pathways intersect at several points and potential interactions will be discussed when relevant.
Figure 2.
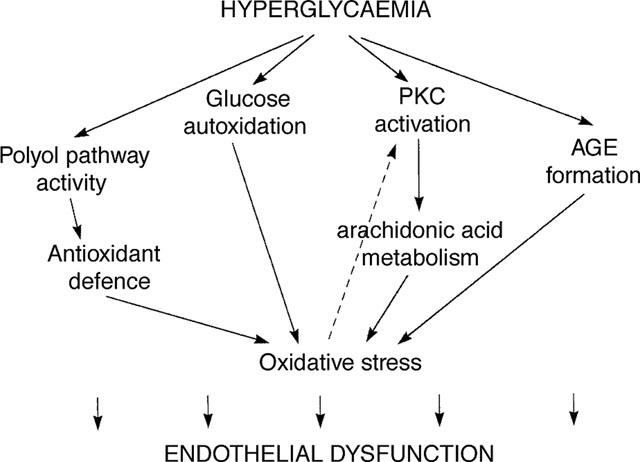
Outline and interactions of hyperglycaemia-induced metabolic pathways potentially involved in the pathophysiology of endothelial dysfunction.
Hyperglycaemia
Impaired ACh-induced relaxation was reversed by chronic insulin treatment (Wang et al., 1993; Taylor et al., 1994a), but not by acute insulin administration, even though glycaemia was normalized (Wang et al., 1993; Bucala et al., 1991). Defective endothelium-dependent relaxation was restored 4 weeks after pancreatic transplantation, performed in rats after 12 weeks of diabetes (Pieper et al., 1998a). A close relationship between endothelial dysfunction and metabolic control was found in streptozotocin-diabetic rats in which the degree of hyperglycaemia was manipulated with subcutaneous insulin implants (Angulo et al., 1998). Conversely, acute exposure to high glucose concentrations induces endothelial dysfunction similar to that in diabetic animals (Tesfamariam et al., 1990; 1991; Tesfamariam & Cohen, 1992; Bohlen & Lash, 1993; Mayhan & Patel, 1995). Since little research on endothelial dysfunction has been conducted in animal models of type II diabetes, it is unknown whether hyperglycaemia, in the presence of hyperinsulinaemia and insulin resistance, has the same deleterious effects on endothelial cell metabolism as in type I diabetes. In Otsuka Long-Evans Tokushima Fatty rats, endothelial dysfunction was improved by exercise training but not by food restriction, although both measures similarly improved hyperglycaemia and serum lipid levels, lessened abdominal fat content, and increased sensitivity to insulin, suggesting that the beneficial effect of exercise was unrelated to these factors (Sakamoto et al., 1998).
In human diabetes, the evidence is less straightforward. Glycaemic control is a predictor of micro- and macrovascular complications, although the relationship is relatively weak, especially in type II diabetes. More pronounced endothelial dysfunction was reported in type I patients with poor glycaemic control as compared with patients with better haemoglobin A1c (Jorgensen et al., 1988). However, other studies found no correlation between haemoglobin A1c values and degree of endothelium-dependent vasodilatation (Johnstone et al., 1993; Clarkson et al., 1996; Lambert et al., 1996; Mullen et al., 1998). These observations may indicate that, in human diabetes, hyperglycaemia-induced cellular alterations are substantially modulated downstream. Alternatively, the coexistence of other risk factors may be required for the full expression of the damaging effects of hyperglycaemia.
Aldose reductase
In tissues that do not require insulin for cellular glucose uptake, such as the kidney, retina, nerves and blood vessels, hyperglycaemia activates the polyol pathway, resulting in the formation of sorbitol (Gabbay, 1973). Aldose reductase is the first and rate-limiting enzyme in the polyol pathway and reduces the aldehyde form of glucose to sorbitol. Several experimental and clinical studies have evidenced a link between the increased polyol pathway activity and the occurrence of chronic diabetic complications. Interestingly, only the classic target organs of diabetic complications were found to be sensitive to damage associated with elevated levels of human aldose reductase gene expression in transgenic mice carrying human aldose reductase cDNA (Giugliano et al., 1996). Aldose reductase inhibitors were effective in the prevention of experimental diabetic neuropathy, albuminuria and cataracts (Zenon et al., 1990). Consequently, aldose reductase inhibitors were tested for their ability to improve endothelial dysfunction in experimental models of diabetes. Chronic oral treatment with structurally dissimilar aldose reductase inhibitors restored abnormal endothelium-dependent vasodilatation in all (Cameron & Cotter, 1992; Tesfamariam et al., 1993; Otter & Chess-Williams, 1994; Quilley et al., 1996) but one (Taylor et al., 1994b) study. The mechanisms responsible for the beneficial effects of aldose reductase inhibitors have not been elucidated, but several hypotheses have been formulated. Aldose reductase utilizes NADPH for the conversion of glucose to sorbitol and may thus deplete the cellular stores of NADPH (Gabbay, 1973). Reduced NADPH is required for the functioning of many endothelial enzymes, including NO synthase and cytochrome P450, as well as for the antioxidant activity of glutathione reductase. Alternatively, a high polyol pathway flux consumes large quantities of ATP and may thus compromise the energy supply required for EDRF production (Cameron & Cotter, 1992). Aldose reductase inhibitors prevent the consumption of NADPH and energy in the polyol pathway and by virtue of this, may restore impaired EDRF production and endogenous antioxidant protection. So far, no studies have evaluated the potential beneficial effect of aldose reductase inhibitors on endothelial function in human diabetes. Although initial studies with aldose reductase inhibitors in experimental diabetic neuropathy were promising, it should be noted that these drugs have consistently failed to demonstrate a clinically meaningful improvement of diabetic neuropathy in humans (Pfeifer et al., 1997).
Protein kinase C
Another glucose-induced alteration in cellular metabolism that may account for endothelial dysfunction is activation of protein kinase C. Hyperglycaemia causes de novo synthesis of diacylglycerol, leading to activation of protein kinase C -preferentially the β-isoform- , a pathway now demonstrated in all vascular tissues involved in diabetic complications (Craven et al., 1995; Koya & King, 1998). The consequences of protein kinase C activation are multiple, since it is involved in a variety of cellular functions (Koya & King, 1998). Of relevance to impaired responses to endothelium-dependent agonists are the activation of phospholipase A2 with increased production of arachidonic acid metabolites, and the inhibition of Na+-K+-ATPase. The adverse effects of elevated glucose levels on ACh-induced relaxation of rabbit aorta and rat pial arterioles were restored by the addition of protein kinase C-inhibitors (Tesfamariam et al., 1991; Mayhan & Patel, 1995). In addition, the glucose-induced release of vasoconstrictor prostanoids was prevented by protein kinase C-inhibition (Tesfamariam et al., 1991). In experimental diabetes, protein kinase C-inhibitors improved endothelial dysfunction in pial arterioles in vivo (Pelligrino et al., 1994), but not in isolated mesenteric arteries (Diederich et al., 1994).
Vitamin E was reported to prevent diacylglycerol-protein kinase C-mediated vascular dysfunction in diabetes (Kunisaki et al., 1995), indicating a link between oxidative stress and the protein kinase C pathway.
AGEs
Glucose is known to bind non-enzymatically to free amino groups on proteins or to lipids. Through a series of oxidative and non-oxidative reactions, AGEs are formed irreversibly and accumulate in tissues over time. Recently, the concept of non-enzymatic protein modification by glucose has been broadened to include a variety of reactive carbonyl compounds that are capable of AGE formation, and the term ‘carbonyl stress' has been put forward (Miyata et al., 1999). Although AGE formation occurs during the normal ageing process, it is markedly accelerated during diabetes, as a consequence of an increase in substrate, e.g. glucose, and in the prevailing oxidant stress in this disease (Baynes & Thorpe, 1999). The pathogenicity of AGEs is related to their ability to accumulate in tissues with the formation of cross-links, and to generate oxygen-derived free radicals. In addition, the interaction of AGEs with their cellular receptors (RAGEs) may trigger sustained cellular activation and a further increase of the oxidative stress (Schmidt et al., 1999). Treatment with aminoguanidine, an inhibitor of AGE formation, has proven beneficial on the progression of a broad range of diabetic complications in animal models and is currently under study in human diabetes (Friedman, 1999). Interpretation of the effects of aminoguanidine may be complicated by other actions of the component, including inhibition of NO synthase (Tilton et al., 1993).
AGEs are known to quench NO (Bucala et al., 1991), but the relevance of this in vitro phenomenon to the in vivo situation has not been demonstrated. Aminoguanidine partially prevented the time-dependent progression of impaired vasodilatation to acetylcholine and nitroglycerin in STZ-diabetes (Bucala et al., 1991). Since no other vasodilator responses were tested, it is unclear whether this protective effect was related to decreased NO quenching or to an aspecific improvement of vascular distensibility. Several authors found no beneficial effect of aminoguanidine on disordered endothelium-dependent vasodilatation in experimental diabetes (Hill & Ege, 1994; Pieper et al., 1996; Crijns et al., 1998). In contrast, aminoguanidine prevented diabetes-induced changes in arteriolar mechanical behaviour, as defined by decreased passive compliance and impaired myogenic reactivity of the arteriolar wall (Huijberts, et al., 1993; Hill & Ege, 1994). Taken together, the deleterious effects of AGE accumulation in vascular tissues are more likely related to alterations in the connective tissue composition of the microvascular wall resulting in increased tissue rigidity, rather than to functional interference with vascular smooth muscle reactivity.
Oxidative stress
A considerable body of evidence implicates oxidative stress as an important pathogenic element in diabetic endothelial dysfunction. Oxidative stress is defined as an increase in the steady-state levels of reactive oxygen species and may occur as a result of increased free radical generation and/or decreased anti-oxidant defence mechanisms. Although there is controversy about the antioxidant status in diabetes, several studies have reported decreased plasma or tissue concentrations of superoxide dismutase, catalase, glutathione and ascorbic acid in both clinical and experimental diabetes (Giugliano et al., 1996). Diabetic aorta was found to be more sensitive to free radical exposure than normal aorta (Pieper & Gross, 1988). In addition, diabetes has been associated with an increased generation of oxygen-derived free radicals (Giugliano et al., 1996). Sources of reactive oxygen species in diabetes may include autoxidation of glucose (Wolff & Dean, 1987), AGE-formation and the binding of AGEs to their receptors (Yan, et al., 1994; Ceriello, 1999), increased substrate flux through the polyol pathway (Giugliano et al., 1996) and stimulation of eicosanoid metabolism (Kontos, 1987; Tesfamariam & Cohen, 1992). Oxygen-derived free radicals may impair endothelium-dependent vasodilatation through inactivation of NO or by serving as an EDCF (Rubanyi & Vanhoutte, 1986; Katusic & Vanhoutte, 1989). Acute administration of scavengers of superoxide anion, including superoxide dismutase (Hattori et al., 1991; Tesfamariam & Cohen, 1992; Pieper et al., 1996; Bohlen & Lash, 1993) and the combination of superoxide dismutase with catalase (Pieper et al., 1997) improved or normalized the abnormal endothelium-dependent responses in different models of diabetes and during high glucose exposure. Similarly, chronic treatment with probucol (Tesfamariam & Cohen, 1992), N-acetylcysteine (Pieper & Siebeneich, 1998b), vitamin E (Keegan et al., 1995; Rösen et al., 1996) and vitamin C (Ting et al., 1996) prevented the development of endothelial dysfunction in clinical and experimental diabetes. In an in vivo study of high glucose exposure of the mesenteric circulation, superoxide dismutase and catalase were equally or more effective than cyclo-oxygenase inhibition in restoring the impaired ACh-induced vasodilatation, suggesting that the oxygen-derived radicals produced during prostanoid synthesis, rather than the prostanoids themselves, were responsible for the endothelial dysfunction (Bohlen & Lash, 1993). In some studies, hydroxyl radical scavengers restored endothelium-dependent vasodilatation, while superoxide dismutase had less or no effect (Dai et al., 1993; Diederich et al., 1994; Pieper et al., 1997; Mayhan & Patel, 1998), suggesting a more important role of the hydroxyl radical in eliciting endothelial dysfunction.
In contrast, several studies failed to demonstrate a beneficial effect of antioxidant administration in resistance arteries (Heygate et al., 1995; Matsunaga et al., 1996; Palmer et al., 1998a,1998b). This may be related to the more limited contribution of NO to endothelium-dependent vasodilatation in these vessels. In the forearm circulation of patients with type II diabetes, vitamin E supplementation during 8 weeks did not improve endothelium-dependent vasodilatation (Gazis et al., 1999). Surprisingly, high dose vitamin E supplementation caused a further attenuation of endothelium-dependent vasodilatation in mesenteric arteries of the rat, despite a decrease of the 8-epi-prostaglandin F2α level, an indicator of oxidative stress (Palmer et al., 1998a), suggesting that exaggerated antioxidant supplementation may even be deleterious.
Further studies examining the long-term effects of antioxidant supplementation will be required, before antioxidant vitamins can be recommended for the prevention of vascular complications in diabetes.
Conclusion
There exist several intersections and areas of overlap between the principal mediators of glucose-induced damage to the vascular endothelium. It is therefore not surprising that correction of any of them may result in amelioration of endothelial dysfunction and the efficiency of one approach does not necessarily preclude that other mechanisms are at play as well.
Endothelial dysfunction as a therapeutic target
Recent interest has focused on strategies to reverse or retard endothelial dysfunction in order to modify the natural history of diabetic vascular disease. As outlined above, various interventions have proven effective in restoring impaired endothelium-dependent vasodilatation in certain vascular beds in animal or human diabetes. Most benefit may, however, be derived from those therapies that have an aspecific or broad beneficial action on endothelial cell metabolism.
ACE inhibitors were shown to ameliorate endothelial dysfunction in patients with diverse cardiovascular risk factors (Anderson, 1999). Similarly, ACE inhibition improved endothelium-dependent vasodilatation in type I and type II diabetes patients without affecting the response to nitrovasodilators (Bijlstra et al., 1995; O'Driscoll et al., 1997; 1999; Arcaro et al., 1999), but other authors did not confirm these findings (Mullen et al., 1998; McFarlane et al., 1999) (Table 2B). Although the mechanisms responsible for the beneficial effects of ACE inhibitors have not been settled, a number of potential explanations have been put forward. ACE inhibition may decrease angiotensin II-induced NADH oxidase activity and by virtue of this, decrease vascular production of superoxide anions. In addition, ACE inhibitors may stimulate basal NO production by suppression of bradykinin breakdown or perhaps by potentiation of the vascular effects of insulin (Vanhoutte et al., 1995).
Treatment with folate normalizes endothelial function in patients with familial hypercholesterolaemia (Verhaar et al., 1999). Furthermore, folate restored endothelial dysfunction during a methionine load test in healthy volunteers without affecting the rise in plasma homocysteine levels (Usui et al., 1999). In STZ-diabetes in the rat, folate acutely improved endothelial dysfunction in the renal microcirculation (De Vriese et al., 1999). If these initial observations could be confirmed in humans, folate may act as a universal tool for the prevention of vascular complications associated with different cardiovascular risk factors. The mechanism of action of folate is at present unknown.
In addition to the development of therapies that may restore the function of the endothelium, we may have to alter our thinking on the use of treatment modalities that have the potential to destroy the endothelium, such as the commonly used balloon dilatation for vascular stenoses. Since regenerating endothelium is known to be dysfunctional (Shibano & Vanhoutte, 1994), the ultimate benefit of these therapies may be questionable.
Conclusions and future perspectives
The present communication reviews the reported studies on the pathophysiology of endothelium-dependent vasodilatation in experimental and clinical diabetes. The high number of available studies and the disparity of the findings highlight the complex pathophysiology of disordered endothelium-dependent vasodilatation in diabetes. Several metabolic pathways overlap and intersect in their adverse effects on endothelial cell homeostasis. In addition, the susceptibility of tissues to the damaging effects of hyperglycaemia may vary. Finally, the mechanisms of endothelium-dependent vasodilatation may be quite different according to the size of the vessel and its anatomical location. In the light of these considerations, it may be hazardous to extrapolate conclusions drawn in one vessel type or diabetes model to another. Such observations emphasize the importance to select clinically relevant models for future studies on endothelial dysfunction.
First, the prevalence of type II diabetes has been rising dramatically over the past few decades. Currently, diabetes type II accounts for more than 90% of the diabetic population. The natural history of vascular disease in type II diabetes may differ substantially from that in type I diabetes. Experimental research should therefore shift from STZ-diabetes to animal models of type II diabetes, as are now commonly available and increasingly better characterized (Perico & Remuzzi, 1999).
Second, experimental research has mainly focused on large conduit arteries such as the aorta and resistance vessels from the mesenteric circulation, whereas clinical research was largely conducted in the forearm circulation. It may be more relevant to study endothelial dysfunction in the typical target organs responsible for the clinical complications of diabetes, such as the circulations of the kidney, heart, retina and brain.
Third, although altered vascular reactivity and compliance per se may influence target organ functioning, disordered endothelium-dependent vasodilatation is primarily a marker of endothelial dysfunction. A particular intervention that improves endothelium-dependent vasodilatation is likely to confer commensurate benefit in other aspects of endothelial function. Nevertheless, it is imperative to link the effect on endothelium-dependent vasodilatation to a therapeutic impact on long-term target organ functioning and ultimately on survival.
The progress that has been made in the understanding of the complex pathophysiology of disordered endothelium-dependent vasodilatation in diabetes has set the stage for further investigation of therapeutic interventions to restore endothelial function. Hopefully, these ‘endothelial cell replacement therapies' will have the potential to improve the dismal prognosis of diabetic vascular disease.
Abbreviations
- ACh
acetylcholine
- AGE
advanced glycation end product
- EDCF
endothelium-derived constricting factors
- EDHF
endothelium-derived hyperpolarizing factor
- EDRF
endothelium-derived relaxing factor
- NO
nitric oxide
- SOD
superoxide dismutase
- STZ
streptozotocin
References
- ANDERSON T.J. Assessment and treatment of endothelial dysfunction in humans. J. Am. Coll. Cardiol. 1999;34:631–638. doi: 10.1016/s0735-1097(99)00259-4. [DOI] [PubMed] [Google Scholar]
- ANGULO J., RODRIGUEZ-MANAS L., PEIRO C., NEIRA M., MARIN J., SANCHEZ-FERRER C.F. Impairment of nitric oxide-mediated relaxations in anaesthetized autoperfused streptozotocin-induced diabetic rats. Naunyn Schmiedeberg's Arch. Pharmacol. 1998;358:529–537. doi: 10.1007/pl00005289. [DOI] [PubMed] [Google Scholar]
- ARCARO G., ZENERE B.M., SAGGIANI F., ZENTI M.G., MONAUNI T., LECHI A., MUGGEO M., BONADONNA R.C. ACE inhibitors improve endothelial function in type 1 diabetic patients with normal arterial pressure and microalbuminuria. Diabetes Care. 1999;22:1536–1542. doi: 10.2337/diacare.22.9.1536. [DOI] [PubMed] [Google Scholar]
- BAYNES J.W., THORPE S.R. Role of oxidative stress in diabetic complications. A new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- BHARDWAJ R., MOORE P.K. Increased vasodilator response to acetylcholine of renal blood vessels from diabetic rats. J. Pharm. Pharmacol. 1988;40:739–742. doi: 10.1111/j.2042-7158.1988.tb07009.x. [DOI] [PubMed] [Google Scholar]
- BIJLSTRA P.J., SMITS P., LUTTERMAN J.A., THIEN T. Effect of long-term angiotensin-converting enzyme inhibition on endothelial function in patients with the insulin-resistance syndrome. J. Cardiovasc. Pharmacol. 1995;25:658–664. doi: 10.1097/00005344-199504000-00021. [DOI] [PubMed] [Google Scholar]
- BOHLEN H.G., LASH J.M. Topical hyperglycemia rapidly suppresses EDRF-mediated vasodilation of normal rat arterioles. Am. J. Physiol. 1993;265:H219–H225. doi: 10.1152/ajpheart.1993.265.1.H219. [DOI] [PubMed] [Google Scholar]
- BOHLEN H.G., LASH J.M. Endothelium-dependent vasodilation is preserved in non-insulin-dependent Zucker fatty diabetic rats. Am. J. Physiol. 1995;268:H2366–H2374. doi: 10.1152/ajpheart.1995.268.6.H2366. [DOI] [PubMed] [Google Scholar]
- BUCALA R., TRACEY K.J., CERAMI A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilation in experimental diabetes. J. Clin. Invest. 1991;87:432–438. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALVER A., COLLIER J., VALLANCE P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin-dependent diabetes. J. Clin. Invest. 1992;90:2548–2554. doi: 10.1172/JCI116149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMERON N.E., COTTER M.A. Impaired contraction and relaxation in aorta from streptozotocin-diabetic rats: role of polyol pathway. Diabetologia. 1992;35:1011–1019. doi: 10.1007/BF02221675. [DOI] [PubMed] [Google Scholar]
- CERIELLO A. Hyperglycemia: the bridge between non-enzymatic glycation and oxidative stress in the pathogenesis of diabetic complications. Diabetes Nutr. Metab. 1999;12:42–46. [PubMed] [Google Scholar]
- CLARKSON P., CELERMAJER D.S., DONALD A.E., SAMPSON M., SORENSEN K.E., ADAMS M., YUE D.K., BETTERIDGE D.J., DEANFIELD J.E. Impaired vascular reactivity in insulin-dependent diabetes mellitus is related to disease duration and low density lipoprotein cholesterol levels. J. Am. Coll. Cardiol. 1996;28:573–579. doi: 10.1016/0735-1097(96)82380-1. [DOI] [PubMed] [Google Scholar]
- COSTA E FORTI A., FONTELES M.C. Decreased endothelium dependent relaxation (nitric oxide) in diabetic kidneys. Horm. Metab. Res. 1998;30:55–57. doi: 10.1055/s-2007-978831. [DOI] [PubMed] [Google Scholar]
- CRAVEN P.A., STUDER R.K., NEGRETE H., DERUBERTIS F.R. Protein kinase C in diabetic nephropathy. J. Diabetes Complic. 1995;9:241–245. doi: 10.1016/1056-8727(95)80012-4. [DOI] [PubMed] [Google Scholar]
- CRIJNS F.R., STRUIJKER BOUDIER H.A., WOLFFENBUTTEL B.H. Arteriolar reactivity in conscious diabetic rats. Influence of aminoguanidine treatment. Diabetes. 1998;47:918–923. doi: 10.2337/diabetes.47.6.918. [DOI] [PubMed] [Google Scholar]
- DAI F., DIEDERICH A., SKOPEC J., DIEDERICH D. Diabetes-induced endothelial dysfunction in streptozotocin-treated rats: role of prostaglandin endoperoxides and free radicals. J. Am. Soc. Nephrol. 1993;4:1327–1336. doi: 10.1681/ASN.V461327. [DOI] [PubMed] [Google Scholar]
- DE VRIESE A., VAN DE VOORDE J., VANHOLDER R., LAMEIRE N. Impaired endothelium-derived hyperpolarizing factormediated renal vasodilatory response in diabetes: restoration with folate. J. Am. Soc. Nephrol. 1999;10:394A. [Google Scholar]
- DIEDERICH D., SKOPEC J., DIEDERICH A., DAI F. Endothelial dysfunction in mesenteric resistance arteries of diabetic rats: role of free radicals. Am. J. Physiol. 1994;266:H1153–H1161. doi: 10.1152/ajpheart.1994.266.3.H1153. [DOI] [PubMed] [Google Scholar]
- DURANTE W., SEN A.K., SUNAHARA F.A. Impairment of endothelium-dependent relaxation in aortae from spontaneously diabetic rats. Br. J. Pharmacol. 1988;94:463–468. doi: 10.1111/j.1476-5381.1988.tb11548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENDERLE M.D., BENDA N., SCHMUELLING R.M., HAERING H.U., PFOHL M. Preserved endothelial function in IDDM patients, but not in NIDDM patients, compared with healthy subjects. Diabetes Care. 1998;21:271–277. doi: 10.2337/diacare.21.2.271. [DOI] [PubMed] [Google Scholar]
- ENDO K., ABIRU T., MACHIDA H., KASUYA Y., KAMATA K. Endothelium-derived hyperpolarizing factor does not contribute to the decrease in endothelium-dependent relaxation in the aorta of streptozotocin-induced diabetic rats. Gen. Pharmacol. 1995;26:149–153. doi: 10.1016/0306-3623(94)00159-k. [DOI] [PubMed] [Google Scholar]
- FÉLÉTOU M., VANHOUTTE P.M. The alternative: EDHF. J. Moll. Cell. Cardiol. 1999;31:15–22. doi: 10.1006/jmcc.1998.0840. [DOI] [PubMed] [Google Scholar]
- FORTES Z.B., LEME J.G., SCIVOLETTO R. Vascular reactivity in diabetes mellitus: role of the endothelial cell. Br. J. Pharmacol. 1983;79:771–781. doi: 10.1111/j.1476-5381.1983.tb10016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIEDMAN E.A. Advanced glycosylated end products and hyperglycemia in the pathogenesis of diabetic complications. Diabetes Care. 1999;22 Suppl 2:B65–B71. [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M, , SAKUMA I., KITABATAKE A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1997;121:1383–1391. doi: 10.1038/sj.bjp.0701258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FULTON D., MCGIFF J.C., QUILLEY J. Cytochrome P450 arachidonate metabolites: deficit in diabetes mellitus. FASEB J. 1996;9:A113. [Google Scholar]
- FURCHGOTT R.F., VANHOUTTE P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- GABBAY K.H. The sorbitol pathway and the complications of diabetes. N. Engl. J. Med. 1973;288:831–836. doi: 10.1056/NEJM197304192881609. [DOI] [PubMed] [Google Scholar]
- GAZIS A., WHITE D.J., PAGE S.R., COCKCROFT J.R. Effect of oral vitamin E (α-tocopherol) supplementation on vascular endothelial function in type 2 diabetes mellitus. Diabet. Med. 1999;16:304–311. doi: 10.1046/j.1464-5491.1999.00049.x. [DOI] [PubMed] [Google Scholar]
- GEBREMEDHIN D., KOLTAI M.Z., POGATSA G., MAGYAR K., HADHAZY P. Influence of experimental diabetes on the mechanical responses of canine coronary arteries: role of endothelium. Cardiovasc. Res. 1988;22:537–544. doi: 10.1093/cvr/22.8.537. [DOI] [PubMed] [Google Scholar]
- GIUGLIANO D., CERIELLO A., PAOLISSO G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- HATTORI Y., KAWASAKI H., ABE K., KANNO M. Superoxide dismutase recovers altered endothelium-dependent relaxation in diabetic rat aorta. Am. J. Physiol. 1991;261:H1086–H1094. doi: 10.1152/ajpheart.1991.261.4.H1086. [DOI] [PubMed] [Google Scholar]
- HEAD R.J., LONGHURST P.A., PANEK R.L., STITZEL R.E. A contrasting effect of the diabetic state upon the contractile responses of aortic preparations from the rat and rabbit. Br. J. Pharmacol. 1987;91:275–286. doi: 10.1111/j.1476-5381.1987.tb10282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEYGATE K.M., LAWRENCE I.G., BENNETT M.A., THURSTON H. Impaired endothelium-dependent relaxation in isolated resistance arteries of spontaneously diabetic rats. Br. J. Pharmacol. 1995;116:3251–3259. doi: 10.1111/j.1476-5381.1995.tb15132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILL M.A., EGE E.A. Active and passive mechanical properties of isolated arterioles from STZ-induced diabetic rats. Effect of aminoguanidine treatment. Diabetes. 1994;43:1450–1456. doi: 10.2337/diab.43.12.1450. [DOI] [PubMed] [Google Scholar]
- HUIJBERTS M.S., WOLFFENBUTTEL B.H., STRUIJKER BOUDIER H.A., CRIJNS F.R., NIEWENHUIJZEN KRUSEMAN A.C., POITEVIN P., LÉVY B.I. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J. Clin. Invest. 1993;92:1407–1411. doi: 10.1172/JCI116716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSTONE M.T., CREAGER S.J., SCALES K.M., CUSCO J.A., LEE B.K., CREAGER M.A. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation. 1993;88:2510–2516. doi: 10.1161/01.cir.88.6.2510. [DOI] [PubMed] [Google Scholar]
- JORGENSEN R.G., RUSSO L., MATTIOLI L., MOORE W.V. Early detection of vascular dysfunction in type I diabetes. Diabetes. 1988;37:292–296. doi: 10.2337/diab.37.3.292. [DOI] [PubMed] [Google Scholar]
- KATUSIC Z.S., VANHOUTTE P.M. Superoxide anion is an endothelium-derived contracting factor. Am. J. Physiol. 1989;257:H33–H37. doi: 10.1152/ajpheart.1989.257.1.H33. [DOI] [PubMed] [Google Scholar]
- KHAN F., COHEN R.A., RUDERMAN N.B., CHIPKIN S.R., COFFMAN J.D. Vasodilator responses in the forearm skin of patients with insulin-dependent diabetes mellitus. Vasc. Med. 1996;1:187–193. doi: 10.1177/1358863X9600100303. [DOI] [PubMed] [Google Scholar]
- KEEGAN A., WALBANK H., COTTER M.A., CAMERON N.E. Chronic vitamin E treatment prevents defective endothelium-dependent relaxation in diabetic rat aorta. Diabetologia. 1995;38:1475–1478. doi: 10.1007/BF00400609. [DOI] [PubMed] [Google Scholar]
- KIFF R.J., GARDINER S.M., COMPTON A.M., BENNETT T. Selective impairment of hindquarters vasodilator responses to bradykinin in conscious wistar rats with streptozotocin-induced diabetes mellitus. Br. J. Pharmacol. 1991;103:1357–1362. doi: 10.1111/j.1476-5381.1991.tb09793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLTAI M.Z., HADHAZY P., POSA I., KOCSIS E., WINKLER G., RÖSEN P., POGATSA G. Characteristics of coronary endothelial dysfunction in experimental diabetes. Cardiovasc. Res. 1997;34:157–163. doi: 10.1016/s0008-6363(97)00050-3. [DOI] [PubMed] [Google Scholar]
- KONTOS H.A. Oxygen radicals from arachidonate metabolism in abnormal vascular responses. Am. Rev. Respir. Dis. 1987;136:474–477. doi: 10.1164/ajrccm/136.2.474. [DOI] [PubMed] [Google Scholar]
- KOYA D., KING G.L. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- KUNISAKI M., BURSELL S.E., CLERMONT A.C., ISHII H., BALLAS L.M., JIROUSEK M.R., UMEDA F., NAWATA H., KING G.L. Vitamin E prevents diabetes-induced abnormal retinal blood flow via the diacylglycerol-protein kinase C pathway. Am. J. Physiol. 1995;269:E239–E246. doi: 10.1152/ajpendo.1995.269.2.E239. [DOI] [PubMed] [Google Scholar]
- LAMBERT J., AARSEN M., DONKER A.J., STEHOUWER C.D. Endothelium-dependent and independent vasodilation of large arteries in normoalbuminuric insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996;16:705–711. doi: 10.1161/01.atv.16.5.705. [DOI] [PubMed] [Google Scholar]
- LASH J.M., BOHLEN H.G. Structural and functional origins of suppressed acetylcholine vasodilation in diabetic rat arterioles. Circ. Res. 1991;69:1259–1268. doi: 10.1161/01.res.69.5.1259. [DOI] [PubMed] [Google Scholar]
- LEKAKIS J., PAPAMICHAEL C., ANASTASIOU H., ALEVIZAKI M., DESSES N., SOUVATZOGLOU A., STAMATELOPOULOS S., KOUTRAS D.A. Endothelial dysfunction of conduit arteries in insulin-dependent diabetes mellitus without microalbuminuria. Cardiovasc. Res. 1997;34:164–168. doi: 10.1016/s0008-6363(97)00015-1. [DOI] [PubMed] [Google Scholar]
- MACALLISTER R.J., CALVER A.L., COLLIER J., EDWARDS M.B., HERREROS B., NUSSEY S.S., VALLANCE P. Vascular and hormonal responses to arginine: provision of substrate for nitric oxide or non-specific effect. Clin. Sci. 1995;89:183–190. doi: 10.1042/cs0890183. [DOI] [PubMed] [Google Scholar]
- MATSUNAGA T., OKUMURA K., ISHIZAKA H., TSUNODA R., TAYAMA S., TABUCHI T., YASUE H. Impairment of coronary blood flow regulation by endothelium-derived nitric oxide in dogs with alloxan-induced diabetes. J. Cardiovasc. Pharmacol. 1996;28:60–67. doi: 10.1097/00005344-199607000-00010. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G. Superoxide dismutase partially restores impaired dilatation of the basilar artery during diabetes mellitus. Brain. Res. 1997;760:204–209. doi: 10.1016/s0006-8993(97)00282-5. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., FARACI F.M. Responses of cerebral arterioles in diabetic rats to activation of ATP-sensitive potassium channels. Am. J. Physiol. 1993;265:H152–H157. doi: 10.1152/ajpheart.1993.265.1.H152. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., PATEL K.P. Acute effects of glucose on reactivity of cerebral microcirculation: role of activation of protein kinase C. Am. J. Physiol. 1995;269:H1297–H1302. doi: 10.1152/ajpheart.1995.269.4.H1297. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., PATEL K.P. Treatment with dimethylthiourea prevents impaired dilation of the basilar artery during diabetes mellitus. Am. J. Physiol. 1998;274:H1895–H1901. doi: 10.1152/ajpheart.1998.274.6.H1895. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., PATEL K.P., SHARPE G.M. Effect of L-arginine on reactivity of hamster cheek pouch arterioles during diabetes mellitus. Int. J. Microcirc. 1997;17:107–112. doi: 10.1159/000179217. [DOI] [PubMed] [Google Scholar]
- MAYHAN W.G., SIMMONS L.K., SHARPE G.M. Mechanisms of impaired responses of cerebral arterioles during diabetes mellitus. Am. J. Physiol. 1991;260:H319–H326. doi: 10.1152/ajpheart.1991.260.2.H319. [DOI] [PubMed] [Google Scholar]
- MCFARLANE R., MCCREDIE R.J., BONNEY M., MOLYNEAUX L., ZILKENS R., CELERMAJER D.S., YUE D.K. Angiotensin converting enzyme inhibition and arterial endothelial function in adults with type 1 diabetes mellitus. Diabet. Med. 1999;16:62–66. doi: 10.1046/j.1464-5491.1999.00021.x. [DOI] [PubMed] [Google Scholar]
- MCNALLY P.G., WATT P.A., RIMMER T., BURDEN A.C., HEARNSHAW J.R., THURSTON H. Impaired contraction and endothelium-dependent relaxation in isolated resistance vessels from patients with insulin-dependent diabetes mellitus. Clin. Sci. 1994;87:31–36. doi: 10.1042/cs0870031. [DOI] [PubMed] [Google Scholar]
- MCVEIGH G.E., BRENNAN G.M., JOHNSTON G.D., MCDERMOTT B.J., MCGRATH L.T., HENRY W.R., ANDREWS J.W. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1992;35:771–776. doi: 10.1007/BF00429099. [DOI] [PubMed] [Google Scholar]
- MCVEIGH G.E., BRENNAN G.M., JOHNSTON G.D., MCDERMOTT B.J., MCGRATH L.T., HENRY W.R., ANDREWS J.W., HAYES J.R. Dietary fish oil augments nitric oxide production or release in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1993;36:33–38. doi: 10.1007/BF00399090. [DOI] [PubMed] [Google Scholar]
- MIYATA T., VAN YPERSELE DE STRIHOU C., KUROKAWA K., BAYNES J.W. Alterations in nonenzymatic biochemistry in uremia: origin and significance of ‘carbonyl stress' in long-term uremic complications. Kidney Int. 1999;55:389–399. doi: 10.1046/j.1523-1755.1999.00302.x. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MULHERN M., DOCHERTY J.R. Effects of experimental diabetes on the responsiveness of rat aorta. Br. J. Pharmacol. 1989;97:1007–1012. doi: 10.1111/j.1476-5381.1989.tb12555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLEN M.J., CLARKSON P., DONALD A.E., THOMSON H., THORNE S.A., POWE A.J., FURUNO T., BULL T., DEANFIELD J.E. Effect of enalapril on endothelial function in young insulin-dependent diabetic patients: a randomized double-blind study. J. Am. Coll. Cardiol. 1998;31:1330–1335. doi: 10.1016/s0735-1097(98)00099-0. [DOI] [PubMed] [Google Scholar]
- NITENBERG A., VALENSI P., SACHS R., DALI M., APTECAR E., ATTALI J. Impairment of coronary vascular reserve and ACh-induced coronary vasodilation in diabetic patients with angiographically normal coronary arteries and normal left ventricular systolic function. Diabetes. 1993;42:1017–1025. doi: 10.2337/diab.42.7.1017. [DOI] [PubMed] [Google Scholar]
- O'DRISCOLL G., GREEN D., MAIORANA A., STANTON K., COLREAVY F., TAYLOR R. Improvement in endothelial function by angiotensin converting enzyme inhibition in non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1999;33:1506–1511. doi: 10.1016/s0735-1097(99)00065-0. [DOI] [PubMed] [Google Scholar]
- O'DRISCOLL G., GREEN D., RANKIN J., STANTON K., TAYLOR R. Improvement in endothelial function by angiotensin converting enzyme inhibition in insulin-dependent diabetes mellitus. J. Clin. Invest. 1997;100:678–684. doi: 10.1172/JCI119580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OTTER D.J., CHESS-WILLIAMS R. The effects of aldose reductase inhibition with ponalrestat on changes in vascular function in streptozotocin diabetic rats. Br. J. Pharmacol. 1994;113:576–580. doi: 10.1111/j.1476-5381.1994.tb17028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OYAMA Y., KAWASAKI H., HATTORI Y., KANNO M. Attenuation of endothelium-dependent relaxation in aorta from diabetic rats. Eur. J. Pharmacol. 1986;131:75–78. doi: 10.1016/0014-2999(86)90013-0. [DOI] [PubMed] [Google Scholar]
- PALMER A.M., GOPAUL N., DHIR S., THOMAS C.R., POSTON L., TRIBE R.M. Endothelial dysfunction in streptozotocin-diabetic rats is not reversed by dietary probucol or simvastatin supplementation. Diabetologia. 1998b;41:157–164. doi: 10.1007/s001250050884. [DOI] [PubMed] [Google Scholar]
- PALMER A.M., THOMAS C.R., GOPAUL N., DHIR S., ÄNGGARD E.E., POSTON L., TRIBE R.M. Dietary antioxidant supplementation reduces lipid peroxidation but impairs vascular function in small mesenteric arteries of the streptozotocin-diabetic rat. Diabetologia. 1998a;41:148–156. doi: 10.1007/s001250050883. [DOI] [PubMed] [Google Scholar]
- PELLIGRINO D.A., KOENIG H.M., WANG Q., ALBRECHT R.F. Protein kinase C suppresses receptor-mediated pial arteriolar relaxation in the diabetic rat. NeuroReport. 1994;5:417–420. doi: 10.1097/00001756-199401120-00011. [DOI] [PubMed] [Google Scholar]
- PERICO N., REMUZZI G. Diabetic nephropathy: animal models of human type 2 diabetes Nephropathy in type 2 diabetes 1999New York: Oxford University Press; 47–57.ed. Ritz, E. & Rychlík, I. pp [Google Scholar]
- PFEIFER M.A., SCHUMER M.P., GELBER D.A. Aldose reductase inhibitors: the end of an era or the need for different trial designs. Diabetes. 1997;46 Suppl. 2:S82–S89. doi: 10.2337/diab.46.2.s82. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., ADAMS M.B., ROZA A.M. Pancreatic transplantation reverses endothelial dysfunction in experimental diabetes mellitus. Surgery. 1998a;123:89–95. [PubMed] [Google Scholar]
- PIEPER G.M., GROSS G.J. Oxygen free radicals abolish endothelium-dependent relaxation in diabetic aorta. Am. J. Physiol. 1988;255:H825–H833. doi: 10.1152/ajpheart.1988.255.4.H825. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., LANGENSTROER P., SIEBENEICH W. Diabetic-induced endothelial dysfunction in rat aorta: role of hydroxyl radicals. Cardiovasc. Res. 1997;34:145–156. doi: 10.1016/s0008-6363(96)00237-4. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., MEI D.A., LANGENSTROER P., O'ROURKE S.T. Bioassay of endothelium-derived relaxing factor in diabetic rat aorta. Am. J. Physiol. 1992;263:H676–H680. doi: 10.1152/ajpheart.1992.263.3.H676. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., MOORE-HILTON G., ROZA A.M. Evaluation of the mechanism of endothelial dysfunction in the genetically-diabetic BB rat. Life Sci. 1996;58:147–152. doi: 10.1016/0024-3205(95)02360-7. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., PELTIER B.A. Amelioration by L-arginine of a dysfunctional arginine/nitric oxide pathway in diabetic endothelium. J. Cardiovasc. Pharmacol. 1995;25:397–403. doi: 10.1097/00005344-199503000-00008. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M., SIEBENEICH W. Oral administration of the antioxidant, N-acetylcysteine, abrogates diabetes-induced endothelial dysfunction. J. Cardiovasc. Pharmacol. 1998b;32:101–105. doi: 10.1097/00005344-199807000-00016. [DOI] [PubMed] [Google Scholar]
- QUILLEY J., MCGIFF J.C., MIEYAL P., RAPACON M., FULTON D. NO-independent coronary vasodilation to bradykinin in diabetes. Hypertension. 1996;28:P178. [Google Scholar]
- RÖSEN P., BALLHAUSEN T., STOCKKLAUSER K. Impairment of endothelium dependent relaxation in the diabetic rat heart: mechanisms and implications. Diabetes Res. Clin. Pr. 1996;31:S143–S155. doi: 10.1016/0168-8227(96)01242-9. [DOI] [PubMed] [Google Scholar]
- RUBANYI G.M., VANHOUTTE P.M. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am. J. Physiol. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822. [DOI] [PubMed] [Google Scholar]
- SAKAMOTO S., MINAMI K., NIWA Y., OHNAKA M., NAKAYA Y., MIZUNO A., KUWAJIMA M., SHIMA K. Effect of exercise training and food restriction on endothelium-dependent relaxation in the Otsuka Long-Evans Tokushima Fatty rat, a model of spontaneous NIDDM. Diabetes. 1998;47:82–86. doi: 10.2337/diab.47.1.82. [DOI] [PubMed] [Google Scholar]
- SCHMIDT A.M., YAN S.D., WAUTIER J.L., STERN D. Activation of receptor for advanced glycation end products. A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999;84:489–497. doi: 10.1161/01.res.84.5.489. [DOI] [PubMed] [Google Scholar]
- SHIBANO T., VANHOUTTE P.M. Involvement of 5-HT2 receptors in chronic endothelial dysfunction after balloon injury of porcine coronary arteries. Circulation. 1994;89:1776–1785. doi: 10.1161/01.cir.89.4.1776. [DOI] [PubMed] [Google Scholar]
- SHIMIZU K., MURAMATSU M., KAKEGAWA Y., ASANO H., TOKI Y., MIYAZAKI Y., OKUMURA K., HASHIMOTO H., ITO T. Role of prostaglandin H2 as an endothelial-derived contracting factor in diabetic state. Diabetes. 1993;42:1246–1252. doi: 10.2337/diab.42.9.1246. [DOI] [PubMed] [Google Scholar]
- SMITS P., KAPMA J., JACOBS M., LUTTERMAN J., THIEN T. Endothelium-dependent vascular relaxation in patients with type I diabetes. Diabetes. 1993;42:148–153. doi: 10.2337/diab.42.1.148. [DOI] [PubMed] [Google Scholar]
- SOBREVIA L., MANN G.E. Dysfunction of the endothelial nitric oxide signalling pathway in diabetes and hyperglycaemia. Exp. Physiol. 1997;82:423–452. doi: 10.1113/expphysiol.1997.sp004038. [DOI] [PubMed] [Google Scholar]
- TAYLOR P.D., GRAVES J.E., POSTON L. Selective impairment of acetylcholine-mediated endothelium-dependent relaxation in isolated resistance arteries of streptozotocin-induced diabetic rat. Clin. Sci. 1995;88:519–524. doi: 10.1042/cs0880519. [DOI] [PubMed] [Google Scholar]
- TAYLOR P.D., MCCARTHY A.L., THOMAS C.R., POSTON L. Endothelium-dependent relaxation and noradrenaline sensitivity in mesenteric resistance arteries of streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1992;107:393–399. doi: 10.1111/j.1476-5381.1992.tb12757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR P.D., OON B.B., THOMAS C.R., POSTON L. Prevention by insulin treatment of endothelial dysfunction but not enhanced noradrenaline-induced contractility in mesenteric resistance arteries from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1994a;111:35–41. doi: 10.1111/j.1476-5381.1994.tb14020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR P.D., WICKENDEN A.D., MIRRLEES D.J., POSTON L. Endothelial function in the isolated perfused mesentery and aortae of rats with streptozotocin-induced diabetes: effect of treatment with the aldose reductase inhibitor, ponalrestat. Br. J. Pharmacol. 1994b;111:42–48. doi: 10.1111/j.1476-5381.1994.tb14021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., BROWN M.L., COHEN R.A. Elevated glucose impairs endothelium-dependent relaxation by activating protein kinase C. J. Clin. Invest. 1991;87:1643–1648. doi: 10.1172/JCI115179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., BROWN M.L., DEYKIN D., COHEN R.A. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J. Clin. Invest. 1990;85:929–932. doi: 10.1172/JCI114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESFAMARIAM B., COHEN R.A. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am. J. Physiol. 1992;263:H321–H326. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- TESFAMARIAM B., JAKUBOWSKI J.A., COHEN R.A. Contraction of diabetic rabbit aorta caused by endothelium-derived PGH2-TxA2. Am. J. Physiol. 1989;257:H1327–H1333. doi: 10.1152/ajpheart.1989.257.5.H1327. [DOI] [PubMed] [Google Scholar]
- TESFAMARIAM B., PALACINO J.J., WEISBROD R.M., COHEN R.A. Aldose reductase inhibition restores endothelial cell function in diabetic rabbit aorta. J. Cardiovasc. Pharmacol. 1993;21:205–211. doi: 10.1097/00005344-199302000-00004. [DOI] [PubMed] [Google Scholar]
- THE DIABETES CONTROL AND COMPLICATIONS TRIAL RESEARCH GROUP/EPIDEMIOLOGY OF DIABETES INTERVENTIONS AND COMPLICATIONS RESEARCH GROUP Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N. Eng. J. Med. 2000;342:381–389. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TILTON R.G., CHANG K., HASAN K.S., SMITH S.R., PETRASH J.M., MISKO T.P., MOORE W.M., CURRIE M.G., CORBETT J.A., MCDANIEL M.L., WILLIAMSON J.R. Prevention of diabetic vascular dysfunction by guanidines. Inhibition of nitric oxide synthase versus advanced glycation end-product formation. Diabetes. 1993;42:221–232. doi: 10.2337/diab.42.2.221. [DOI] [PubMed] [Google Scholar]
- TING H.H., TIMIMI F.K., BOLES K.S., CREAGER S.J., GANZ P., CREAGER M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1996;97:22–28. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USUI M., MATSUOKA H., MIYAZAKI H., UEDA S., OKUDA S., IMAIZUMI T. Endothelial dysfunction by acute hyperhomocyst(e)inaemia: restoration by folic acid. Clin. Sci. 1999;96:235–239. [PubMed] [Google Scholar]
- VANHOUTTE P.M., BOULANGER C.M., MOMBOULI J.V. Endothelium-derived relaxing factors and converting enzyme inhibition. Am. J. Cardiol. 1995;76:3E–12E. [PubMed] [Google Scholar]
- VERHAAR M.C., WEVER R.M., KASTELEIN J.J., VAN LOON D., MILSTIEN S., KOOMANS H.A., RABELINK T.J. Effects of oral folic acid supplementation on endothelial function in familial hypercholesterolemia: a randomized placebo-controlled trial. Circulation. 1999;100:335–338. doi: 10.1161/01.cir.100.4.335. [DOI] [PubMed] [Google Scholar]
- WAKABAYASHI I., HATAKE K., KIMURA N., KAKISHITA E., NAGAI K. Modulation of vascular tonus by the endothelium in experimental diabetes. Life Sci. 1987;40:643–648. doi: 10.1016/0024-3205(87)90265-7. [DOI] [PubMed] [Google Scholar]
- WANG Y., BROOKS D.P., EDWARDS R.M. Attenuated glomerular cGMP production and renal vasodilation in streptozotocin-induced diabetic rats. Am. J. Physiol. 1993;264:R952–R956. doi: 10.1152/ajpregu.1993.264.5.R952. [DOI] [PubMed] [Google Scholar]
- WHITE R.E., CARRIER G.O. Supersensitivity and endothelium dependency of histamine-induced relaxation in mesenteric arteries of diabetic rats. Pharmacology. 1986;33:34–38. doi: 10.1159/000138197. [DOI] [PubMed] [Google Scholar]
- WILLIAMS S.B., CUSCO J.A., RODDY M., JOHNSTONE M.T., CREAGER M.A. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 1996;27:567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- WOLFF S.P., DEAN R.T. Glucose autoxidation and protein modification. The potential role of oxidative glycosylation in diabetes. Biochem. J. 1987;245:243–250. doi: 10.1042/bj2450243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN S.D., SCHMIDT A.M., ANDERSON G.M., ZHANG J., BRETT J., ZOU Y.S., PINSKY D., STERN D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Cell. Biol. 1994;269:9889–9897. [PubMed] [Google Scholar]
- ZENERE B.M., ARCARO G., SAGGIANI F., ROSSI L., MUGGEO M., LECHI A. Noninvasive detection of functional alterations of the arterial wall in IDDM patients with and without microalbuminuria. Diabetes Care. 1995;18:975–982. doi: 10.2337/diacare.18.7.975. [DOI] [PubMed] [Google Scholar]
- ZENON G.J., ABOBO C.V., CARTER B.L., BALL D.W. Potential use of aldose reductase inhibitors to prevent diabetic complications. Clin. Pharm. 1990;9:446–457. [PubMed] [Google Scholar]