

Abstract
It has been reported that radiolabelled agonist : antagonist binding affinity ratios can predict functional efficacy at several different receptors. This study investigates whether this prediction is true for recombinant and native tissue 5-HT1A receptors.
Saturation studies using [3H]-8-OH-DPAT and [3H]-MPPF revealed a single, high affinity site (KD∼1 nM) in HEK293 cells expressing human 5-HT1A receptors and rat cortex. In recombinant cells, [3H]-MPPF labelled 3–4 fold more sites than [3H]-8-OH-DPAT suggesting the presence of more than one affinity state of the receptor. [3H]-Spiperone labelled a single, lower affinity site in HEK293 cells expressing h5-HT1A receptors but did not bind to native tissue 5-HT1A receptors. These data suggest that, in transfected HEK293 cells, human 5-HT1A receptors exist in different affinity states but in native rat cortical tissue the majority of receptors appear to exist in the high agonist affinity state.
Receptor agonists inhibited [3H]-MPPF binding from recombinant 5-HT1A receptors in a biphasic manner, whereas antagonists and partial agonists gave monophasic inhibition curves. All compounds displaced [3H]-8-OH-DPAT and [3H]-spiperone binding in a monophasic manner. In rat cortex, all compounds displaced [3H]-MPPF and [3H]-8-OH-DPAT in a monophasic manner.
Functional evaluation of compounds, using [35S]-GTPγS binding, produced a range of intrinsic activities from full agonism, displayed by 5-HT and 5-CT to inverse agonism displayed by spiperone.
[3H]-8-OH-DPAT : [3H]-MPPF pKi difference correlated well with functional intrinsic activity (r=0.86) as did [3H]-8-OH-DPAT : [3H]-spiperone pKi difference with functional intrinsic activity (r=0.96).
Thus agonist : antagonist binding affinity differences may be used to predict functional efficacy at human 5-HT1A receptors expressed in HEK293 cells where both high and low agonist affinity states are present but not at native rat cortical 5-HT1A receptors in which only the high agonist affinity state was detectable.
Keywords: 5-HT1A receptor, [3H]-8-OH-DPAT, [3H]-MPPF, [3H]-spiperone, radioligand binding, intrinsic activity
Introduction
The classical ternary complex model assumes that there is a direct relationship between the ability of a drug to stabilize the active state of a receptor and its ability to elicit a response (De Lean et al., 1980). This concept is also used for more recent models such as the two-state model of agonist action, which suggests that ligands which bind with high affinity to the active form of a receptor are agonists whereas antagonists do not discriminate between the active or inactive state of the receptor. Ligands which bind with higher affinity to the inactive state of the receptor have been demonstrated to be inverse agonists (Sammama et al., 1993; Leff, 1995). These different agonist states have been shown to exist for several receptors such as serotonin, dopamine, muscarinic and β-adrenoreceptors (Clawges et al., 1997; Freedman et al., 1988; Garnier et al., 1998; Houston & Howlett, 1998; Lahti et al., 1992; Tayebati et al., 1999). It has previously been reported that the ratio of the affinity of a compound for high and low agonist affinity states of muscarinic receptors is a reasonable prediction of intrinsic activity (Freedman et al., 1988; Loudon et al., 1997; Tayebati et al., 1999). Similar studies have also shown that efficacy at cannabinoid CB1, D2 receptors and human 5-HT2A and 5-HT2C receptors also correlate well with high affinity agonist binding (Fitzgerald et al., 1999; Kearn et al., 1999; Lahti et al., 1992).
Human 5-HT1A receptors have also been shown to exist in high and low agonist affinity states in transfected cell lines (Clawges et al., 1997; Price et al., 1998). We have investigated the different states of the human recombinant 5-HT1A receptor expressed in HEK293 cells and native 5-HT1A receptors in rat cortex using [3H]-8-OH-DPAT as an agonist radioligand (Sijbesma et al., 1991) and [3H]-MPPF and [3H]-spiperone as antagonist radioligands (Kung et al., 1996; Sundaram et al., 1992). We have then compared compound-induced inhibition of binding of these radioligands with compound efficacy, measured by human recombinant 5-HT1A receptor stimulation of [35S]-GTPγS binding, to determine if a relationship exists between binding affinity differences and functional intrinsic activity.
Methods
Receptor binding
HEK (human embryonic kidney) 293 cells stably expressing human (h)5-HT1A receptors were homogenized and washed twice with ice-cold TRIS buffer (TRIS 50 mM MgCl2 10 mM, pH 7.4) (Watson et al., 1998). Rat cerebral cortex was excised, homogenized in TRIS buffer and incubated at 37°C for 15 min prior to washing twice. Both sets of membranes were stored at −80°C until required.
Membranes (from 5×105 cells or 10 mg original wet weight tissue per well) were incubated in TRIS buffer containing [3H]-8-OH-DPAT (2 nM), [3H]-MPPF (0.3 nM) or [3H]-spiperone (1 nM) with/without compounds at 37°C for 45 min. Non-specific binding was defined by buspirone (10 μM). In saturation studies increasing concentrations of radioligand were used : [3H]-8-OH-DPAT (0.1–40 nM), [3H]-MPPF (0.1–15 nM) and [3H]-spiperone (1.5–100 nM). Incubations were terminated by filtration over GF/B filters, followed by 6×1 ml washes with ice cold TRIS buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
[35S]-GTPγS binding
HEK293 cells stably expressing recombinant h5-HT1A receptors were prepared and assayed according to the method used by Watson et al., 1998. Briefly, membranes were pre-incubated in 20 mM HEPES containing 3 mM MgCl2, 100 mM NaCl, 0.2 mM ascorbate and 10 μM GDP. The reaction was started by the addition of 100 pM [35S]-GTPγS and incubated for a further 30 min. The reaction was terminated by rapid filtration over GF/B filters and bound radioactivity was measured by scintillation spectrometry. Intrinsic activity was calculated as per cent stimulation of basal specific [35S]-GTPγS binding and expressed as a fraction of the maximal 5-HT response.
Materials
Drugs and reagents were purchased from Sigma-Aldrich (Poole, U.K.), Calbiochem (Nottingham, U.K.), Bio-Rad (Hemel Hempstead, U.K.), Fisons Scientific Equipment (Loughborough, U.K.), Research Biochemicals International (Poole, U.K.), Tocris Cookson Ltd. (Bristol, U.K.) and GibcoBRL (Paisley, U.K.). [35S]-GTPγS, [3H]-8-OH-DPAT, [3H]-MPPF and [3H]-spiperone were supplied by Amersham International (Little Chalfont, U.K.).
Data analysis
Data from both radioligand binding studies and [35S]-GTPγS binding studies were fitted by a 4-parameter logistic equation using GRAFIT (Erithacus Software Ltd.) to yield IC50 and EC50 values respectively. In binding studies, pKi values were calculated from IC50 values using the correction for radioligand concentration described by Cheng & Prusoff (1973).
Results
Receptor binding studies
Saturation studies
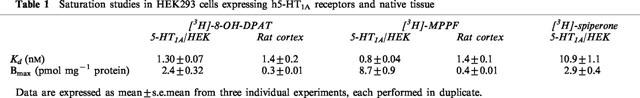
In HEK293 cells expressing h5-HT1A receptors, [3H]-8-OH-DPAT and [3H]-MPPF appeared to bind to a single population of sites, however [3H]-MPPF labelled over three times as many sites as [3H]-8-OH-DPAT (Table 1). [3H]-spiperone also appeared to bind a single component and labelled a similar number of sites as [3H]-8-OH-DPAT. In rat cortex, [3H]-8-OH-DPAT and [3H]-MPPF bound in a monophasic manner and labelled a similar number of binding sites. However, [3H]-spiperone failed to give a robust, specific binding signal in rat cortex (data not shown), presumably because of the low affinity that spiperone displays for this native receptor (see below).
Table 1.
Saturation studies in HEK293 cells expressing h5-HT1A receptors and native tissue
Inhibition of binding by agonists and antagonists
Cloned h5-HT1A receptors
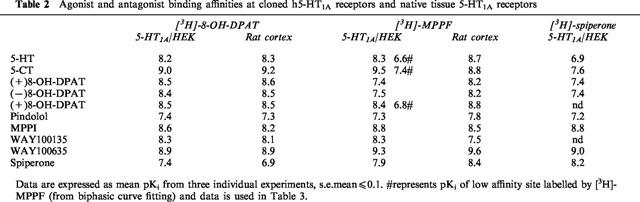
The agonists, 5-CT, 5-HT and 8-OH-DPAT displayed over 10 fold greater affinity at displacing [3H]-8-OH-DPAT binding than [3H]-MPPF or [3H]-spiperone (Figure 1a,c,e and Table 2). Monophasic inhibition curves were generated with agonists using both [3H]-8-OH-DPAT and [3H]-spiperone, however biphasic inhibition curves were observed with [3H]-MPPF. Data from 2-site curve fitting revealed that the high affinity agonist component represented 25% of specific binding which correlated well with the [3H]-8-OH-DPAT saturation studies, where the agonist radiolabel appeared to label only 0.27 of the total number of binding sites. The 5-HT1A receptor antagonists, WAY100635, MPPI and spiperone displayed monophasic inhibition curves with comparable potency, with all three radioligands (Table 2).
Figure 1.
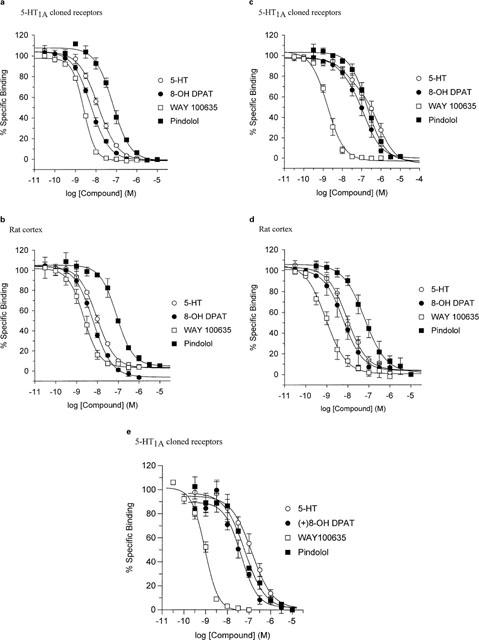
Displacement of agonist and antagonist radioligand binding by compounds with affinity for 5-HT1A receptors. (a and b) Shows inhibition of binding of [3H]-8-OH-DPAT from human 5-HT1A receptors expressed in HEK293 cells and rat cortex, respectively. (c and d) Shows inhibition of [3H]-MPPF binding from human 5-HT1A receptors expressed in HEK293 cells and rat cortex, respectively. (e) Shows inhibition of [3H]-spiperone binding from cloned 5-HT1A receptors. Binding to rat cortex could not be detected (data not shown). Data are expressed as the mean±s.e.mean from at least three individual experiments, each point performed in duplicate. All curves were fitted by a 4-parameter logistic equation.
Table 2.
Agonist and antagonist binding affinities at cloned h5-HT1A receptors and native tissue 5-HT1A receptors
Rat cortex
Agonists and antagonists inhibited [3H]-8-OH-DPAT and [3H]-MPPF binding with similar affinity and gave monophasic inhibition curves (Figure 1b,d and Table 2).
Functional studies – [35S]-GTPγS binding studies
The full agonist 5-HT, stimulated [35S]-GTPγS binding by approximately 60% above basal with a pEC50=8.4. Other compounds displayed a range of intrinsic activities when their response (per cent stimulation) was expressed as a fraction of the maximal 5-HT response (Figure 2a,b and Table 3). Inverse agonist activities were expressed relative to spiperone which was designated as a full inverse agonist.
Figure 2.
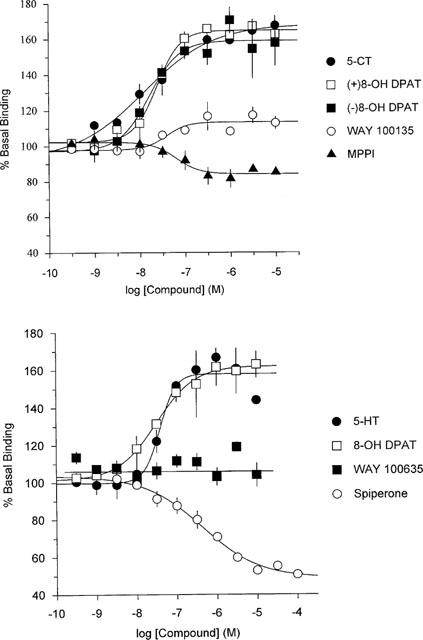
[35S]-GTPγS binding to HEK293 cells expressing human 5-HT1A receptors. Data are expressed as per cent of basal [35S]-GTPγS binding and are the mean±s.e.mean from three individual experiments, each performed in duplicate. All curves were fitted by a 4-parameter logistic equation.
Table 3.
Comparison of functional activity and agonist : antagonist binding difference in h5-HT1A receptor clones
Correlation between functional intrinsic activity (I.A.) and agonist : antagonist pKi difference at h5-HT1A receptors
[3H]-8-OH-DPAT : [3H]-MPPF pKi difference for standard compounds correlated well with functional I.A. resulting in a correlation coefficient (r) of 0.86 (Figure 3a). [3H]-8-OH-DPAT : [3H]-spiperone pKi difference for standards showed a linear relationship with functional I.A. resulting in r=0.96 (Figure 3b). The pKi difference between [3H]-8-OH-DPAT and either antagonist radioligands, for standard compounds, correlated well with r=0.96 (data not shown).
Figure 3.
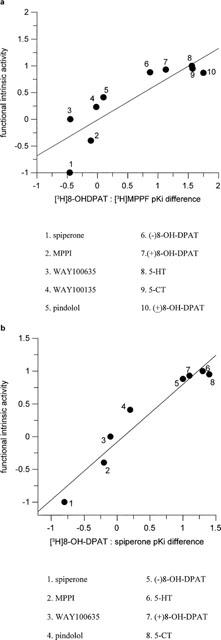
Relationship between agonist: antagonist binding affinity difference and functional intrinsic activity (I.A.), in HEK293 cells expressing human 5-HT1A receptors. Functional efficacy was determined by [35S]-GTPγS binding assays. (a) Shows the relationship between [3H]-8-OH-DPAT : [3H]-MPPF pKi difference and I.A. The correlation between [3H]-8-OH-DPAT : [3H]-spiperone pKi difference and I.A. is shown in (b).
Discussion
Evaluation of agonist activity at receptors has been based, primarily, on functional tests performed in cell lines expressing cloned receptors and/or native tissue preparations. In most cases, especially for native tissue, functional assessment can be time consuming and complicated to perform. The report of a correlation between [3H]-N-methyl scopolamine (muscarinic antagonist) and [3H]-Oxotremorine-M (muscarinic agonist) binding affinity ratio and functional assessment of muscarinic drugs in rat cortex, which was presumed to be primarily of the M1 receptor sub-type was, therefore, of interest (Freedman et al., 1988). These studies resulted in the development of a simple radioligand binding assay, the results of which could estimate the relative intrinsic activity of muscarinic compounds at M1 receptors. Later studies also confirmed this correlation (Loudon et al., 1997). These studies were possible since, in rat cortex, muscarinic receptors existed in high and low agonist affinity states (Birdsall et al., 1978; 1980), a phenomenon common to several G protein coupled receptors (Clawges et al., 1997; De Lean et al., 1980; Garnier et al., 1998; Houston & Howlett, 1998; Lahti et al., 1992). The studies of muscarinic receptors alluded to above were limited to the analysis of the profile of compounds at M1 receptors but recent reports have shown that antagonist : agonist radioligand binding affinity ratios can predict the intrinsic activity of compounds at native tissue cannabinoid and M2 receptors and recombinant 5-HT2 receptors (Fitzgerald et al., 1999; Kearn et al., 1999; Tayebati et al., 1999).
It has been reported that human 5-HT1A receptors, expressed in cultured cells, can also exist in both high and low agonist affinity states (Clawges et al., 1997; Price et al., 1998). The aim of our study was, therefore, to determine if the agonist : antagonist binding affinity difference for a selection of compounds predicted functional intrinsic activity at recombinant human 5-HT1A receptors. Similar studies in rat cortex were carried out in parallel. From saturation analysis, the 5-HT1A receptor agonist radioligand, [3H]-8-OH-DPAT (Sijbesma et al., 1991), appeared to label a single, high agonist affinity site in both HEK293 cells expressing h5-HT1A receptors and rat cortex. The density of sites in the recombinant system was 8 fold greater than that of the native preparation, a common feature of many cloned receptors. The 5-HT1A antagonist radioligand, [3H]-MPPF (Kung et al., 1996), also labelled recombinant and native receptors with high affinity but in the recombinant system, it labelled 3–4 fold more sites than [3H]-8-OH-DPAT. Data from agonist inhibition curves suggested that h5-HT1A receptors, expressed in HEK293 cells, exist in both high and low agonist affinity states, the majority of which are in the low agonist affinity state. The antagonist radioligand, [3H]-spiperone, appeared to selectively label the low agonist affinity site in the HEK cells expressing h5-HT1A receptors but did not bind to native 5-HT1A receptors (data not shown).
In recombinant cells and native tissue, a range of compounds with affinity for 5-HT1A receptors inhibited [3H]-8-OH-DPAT binding in a monophasic manner. However, agonists inhibited [3H]-MPPF binding to human receptors in a biphasic manner, whereas antagonist inhibition curves were monophasic. This supports the above finding that h5-HT1A receptors exist in two agonist affinity states in HEK293 cells. [3H]-MPPF labelled both of these states with similar affinity but agonists, and to a lesser extent partial agonists, discriminated between the two states.
Both saturation and competition studies in rat cortex suggested that the 5-HT1A receptor high affinity state predominates in this preparation. Both agonist and antagonist radioligands labelled a similar number of receptors and agonists produced monophasic inhibition curves with high affinity, with either ligand. This phenomenon has also been reported for native 5-HT1B receptors (Selkirk et al., 1997).
In HEK cells expressing h5-HT1A receptors, both agonists and antagonists displaced [3H]-spiperone binding in a monophasic manner but the binding affinity for agonists was lower than those obtained when displacing [3H]-8-OH-DPAT binding or [3H]-MPPF from the high affinity site. There was a good correlation between agonist affinity measured by [3H]-spiperone and the ‘low affinity' component of [3H]-MPPF binding. From the studies using [3H]-8-OH-DPAT it was evident that the high agonist affinity state exists in native tissue and so the fact that [3H]-spiperone did not bind to rat cortex and the aforementioned data, together suggest that [3H]-spiperone only binds to the low agonist affinity state. An observation which is consistent with its inverse agonist activity (Sammama et al., 1993; Leff, 1995).
[35S]-GTPγS binding studies were carried out in HEK293 cells expressing h5-HT1A receptors to measure the functional intrinsic activity of the compounds evaluated in radioligand binding studies. A range of intrinsic activities was observed, from full agonism (5-HT, 5-CT) to inverse agonism (spiperone) and the correlation between these efficacy values and binding affinity difference was assessed. The data showed a linear relationship between functional intrinsic activity and [3H]-8-OH-DPAT : [3H]-MPPF and [3H]-8-OH-DPAT : [3H]-spiperone binding affinity differences with correlation coefficients ranging from 0.86–0.96. The results also revealed a good correlation between [3H]-8-OH-DPAT : [3H]-MPPF and [3H]-8-OH-DPAT : [3H]-spiperone binding affinity differences suggesting that either [3H]-MPPF or [3H]-spiperone could be used as an antagonist radioligand for these particular studies.
It has been suggested that receptor reserve may influence the relationship we are describing in this study (Fitzgerald et al., 1999) in that, for systems with high reserve, the I.A. for a partial agonist may increase, with little effect on the high and low-affinity binding values. This certainly appears to be the case in our study. Weak partial agonists such as pindolol and WAY100135, which in many native tissue studies fail to display any intrinsic activity, demonstrate significant efficacy in the [35S]-GTPγS assay. Interestingly, these compounds show very little difference in their displacement of agonist and antagonist radioligands, consistent with very low efficacy/antagonism. This suggests that the binding affinity ratio is giving a better approximation of tau in a system with high 5-HT1A receptor reserve. The very high receptor expression in this recombinant system is presumably responsible for this receptor reserve. Paradoxically, it is probably this same receptor reserve which allows expression of the low agonist affinity state. A finite number of G proteins exist in recombinant cells and so, assuming receptor : G protein interaction is 1 : 1, if the number of receptors present exceed G protein levels then a proportion of receptors will not couple to G proteins and hence exist in a low agonist affinity state. Many other factors determine which agonist state a receptor can exist in such as Na+, Mg++ concentration, cofactors, guanine nucleotides (Houston & Howlett, 1998; Nanoff et al., 1995; Stiles, 1988; Vickroy et al., 1983) and these will dictate whether or not the studies described can be used to estimate functional activity. Basal constitutive activity may also hinder these types of studies as I.A. may be reduced in cell lines with high basal coupling present.
We have shown that human 5-HT1A receptors expressed in HEK293 cells exist in high and low agonist affinity states, whereas 5-HT1A receptors in rat cortex predominately exist in the high agonist affinity state. Functional evaluation of h5-HT1A receptors expressed in HEK293 cells, using [35S]-GTPγS binding, revealed a correlation between functional efficacy and agonist : antagonist binding affinity difference for a range of compounds. Therefore, the results of this study suggest that radioligand binding techniques may be a useful tool for predicting the functional activity of compounds at h5-HT1A receptors expressed in HEK293 cells, where both the high and low agonist affinity state exists. The predominance of the high agonist affinity state in rat cortex does not allow this relationship to extrapolate to native tissue 5-HT1A receptors in our studies but a recent report has shown that when rat hippocampal 5-HT1A receptors were manipulated to display both agonist affinity states, then a similar correlation was observed (Assie et al., 1999). In this report, antagonist radioligand binding experiments were carried out in the presence of non-hydrolazable GTP analogues to ensure 5-HT1A receptors existed in a low agonist affinity state. Similar studies have been performed in cerebellum membranes to manipulate agonist affinity states of cannabinoid receptors (Kearn et al., 1999). Our studies did not include these analogues but future studies are required to investigate their effect on 5-HT1A receptor affinity states in rat cortex.
Abbreviations
- h
human
- HEK
human embryonic kidney
- 8-OH-DPAT
8-Hydroxy-dipropylaminotetralin hydrobromide
References
- ASSIE M.-B., COSI C., KOEK W. Correlation between low/high affinity ratios for 5-HT1A receptors and intrinsic activity. E. J. Pharmacol. 1999;386:97–103. doi: 10.1016/s0014-2999(99)00738-4. [DOI] [PubMed] [Google Scholar]
- BIRDSALL N.J.M., BURGEN A.S.V., HUME E.C. The binding of agonists to brain muscarinic receptors. Mol. Pharmacol. 1978;14:723–739. [PubMed] [Google Scholar]
- BIRDSALL N.J.M., BURGEN A.S.V., HUME E.C. The character of the muscarinic receptors in different regions of the rat brain. Proc. R. Soc. 1980;207:1–12. doi: 10.1098/rspb.1980.0011. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSSOF W.H. Relationship between inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;92:881–894. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLAWGES H.M., DEPREE K.M., PARKER E.M., GRABER S.G. Human 5-HT1 receptor subtypes exhibit distinct G protein coupling behaviours in membranes from Sf9 cells. Biochem. 1997;36:12930–12938. doi: 10.1021/bi970112b. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., STADEL J.M., LEFKOWITZ R.J. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J. Biol. Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- FITZGERALD L.W., CONKLIN D.S., KRAUSE C.M., MARSHALL A.P., PATTERSON J.P., TRAN D.P., IYER G., KOSTICH W.A., LARGENT B.L., HARTIG P.R. High-affinity agonist binding correlates with efficacy (intrinsic activity) at the human serotonin 5-HT2A and 5-HT2C receptors: evidence favouring the ternary complex and two-state models of agonist action. J. Neurochem. 1999;72:2127–2134. doi: 10.1046/j.1471-4159.1999.0722127.x. [DOI] [PubMed] [Google Scholar]
- FREEDMAN S.B., HARLEY E.A., IVERSEN L.L. Relative affinities of drugs acting at cholinoceptors in displacing agonist and antagonist radioligands: the NMS/Oxo-M ratio as an index of efficacy at cortical muscarinic receptors. Br. J. Pharmacol. 1988;93:437–445. doi: 10.1111/j.1476-5381.1988.tb11451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARNIER V., ZINI R., MORIN D., TILLEMENT J.-P. Evidence for the stabilisation of the high-affinity state of β-adrenoreceptors by an endogenous factor in rat brain. Pharmacol. Res. 1998;37:365–373. doi: 10.1006/phrs.1998.0305. [DOI] [PubMed] [Google Scholar]
- HOUSTON D.B., HOWLETT A.C. Differential receptor-G-protein coupling evoked by dissimilar cannabinoid receptor agonists. Cell. Signal. 1998;10:667–674. doi: 10.1016/s0898-6568(98)00013-8. [DOI] [PubMed] [Google Scholar]
- KEARN C.S., GREENBERG M.J., DICAMELLI R., KURZAWA K., HILLARD C.J. Relationship between ligand affinities for the cerebellar cannabinoid receptor CB1 and the induction of GDP/GTP exchange. J. Neurochem. 1999;72:2379–2387. doi: 10.1046/j.1471-4159.1999.0722379.x. [DOI] [PubMed] [Google Scholar]
- KUNG H.F. New 5-HT1A receptor antagonist: [3H]p-MPPF. Synpase. 1996;23:344–346. doi: 10.1002/(SICI)1098-2396(199608)23:4<344::AID-SYN13>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- LAHTI R.A., FIGUR L.M., PIERCEY M.F., RUPPEL P.L., EVANS D.L. Intrinsic activity determinations at the dopamine D2 guanine nucleotide-binding protein-coupled receptor: utilisation of receptor state binding affinities. Mol. Pharmacol. 1992;42:432–438. [PubMed] [Google Scholar]
- LEFF P. The two-state model of receptor activation. Trends. Pharmacol. Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- LOUDON J.M., BROMIDGE S.M., BROWN F., CLARK M.S.G., HATCHER J.P., HAWKINS J., RILEY G.J., NOY G., ORLEK B.S. SB 202026 : a novel muscarinic partial agonist with functional selectivity for M1 receptors. J. Pharmacol. Exp. Ther. 1997;283:1059–1068. [PubMed] [Google Scholar]
- NANOFF C., MITTERAUER T., ROKA F., HOHENEGGER M., FREISSMUTH M. Species differences in A1 adenosine receptor/G protein coupling: identification of a membrane protein that stabilises the association of the receptor/G protein complex. Mol. Pharmacol. 1995;48:806–817. [PubMed] [Google Scholar]
- PRICE G.W., HO M., SCOTT C., SELKIRK J.V., BROWN A.M. 5-HT receptor agonist-antagonist binding affinity ratios as a measure of intrinsic activity using [3H]8-OH-DPAT and [3H]MPPF as radioligands. Br. J. Pharmacol. 1998;125:58P. doi: 10.1038/sj.bjp.0703394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMMAMA P., COTECCHIA S., COSTA T., LEFKOWITZ R.J. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- SELKIRK J.V., SCOTT C., JERMAN J.C., PRICE G.W. [3H] GR125743 labels high and low affinity states of 5-HT1B and 5-HT1D receptors. Br. J. Pharmacol. 1997;120:295P. [Google Scholar]
- SIJBESMA H. Species differences in the distribution of central 5-HT1 binding sites: a comparative autoradiographic study between rat and guinea-pig. Brain Res. 1991;555:295–304. doi: 10.1016/0006-8993(91)90355-y. [DOI] [PubMed] [Google Scholar]
- STILES G.L. A1 adenosine receptor-G protein coupling in bovine brain membranes:effects of guanine nucleotides, salt and solubilisation. J. Neurochem. 1988;51:1592–1598. doi: 10.1111/j.1471-4159.1988.tb01129.x. [DOI] [PubMed] [Google Scholar]
- SUNDARAM H., NEWMAN-TANCREDI A., STRANGE P.G. Pharmacological characterisation of the 5-HT1A serotonin receptor using the agonist [3H]8-OH-DPAT, and the antagonist [3H]spiperone. Biochem. Soc. Trans. 1992;20:145S. doi: 10.1042/bst020145s. [DOI] [PubMed] [Google Scholar]
- TAYEBATI S.K., PIERGENTILI A., NATALE D., AMENTA F. Evaluation of an agonist index: affinity ratio for compounds active on muscarinic cholinergic M2 receptors. J. Auton. Pharmacol. 1999;19:77–84. doi: 10.1046/j.1365-2680.1999.00118.x. [DOI] [PubMed] [Google Scholar]
- VICKROY T.W., YAMAMURA H.I., ROESKE W.R. Differential regulation of high-affinity agonist binding to muscarinic sites in the rat heart, cerebellum and cerebral cortex. Biochem. Biophys. Res. Comm. 1983;116:284–290. doi: 10.1016/0006-291x(83)90412-6. [DOI] [PubMed] [Google Scholar]
- WATSON J., BROUGH S., COLDWELL M.C., GAGER T., HO M., HUNTER A.J., JERMAN J., MIDDLEMISS D.N., RILEY G.J., BROWN A.M. Functional effects of the muscarinic receptor agonist, xanomeline, at 5-HT1 and 5-HT2 receptors. Br. J. Pharmacol. 1998;125:1413–1420. doi: 10.1038/sj.bjp.0702201. [DOI] [PMC free article] [PubMed] [Google Scholar]