

Abstract
The A1-adenosine receptor (A1AdoR) reserve for N6-cyclopentyladenosine (CPA) mediated inhibition of (−)isoprenaline stimulated cyclic AMP accumulation and stimulation of [35S]-guanosine-5′-O-(thiotriphosphate) (GTPγS) binding, a measure of guanine nucleotide binding protein (G-protein) activation, was determined in DDT1 MF-2 cells.
Inactivation of the A1AdoRs with the chemoreactive ligand 8-cyclopentyl-3-[3-[[4-(fluorosulphonyl)benzoyl]oxy]propyl]-1-propylxanthine (FSCPX) caused a progressive rightward shift of the concentration-response curves for CPA to inhibit cyclic AMP accumulation, with a maximum of 10 fold increase in the EC50 value. In contrast, inactivation of A1AdoR's caused only a 1.7 fold rightward shift in the CPA concentration-response for stimulation of [35S]-GTPγS binding.
The A1AdoR occupancy-response relationship for CPA inhibition of cyclic AMP accumulation was hyperbolic with 43% receptor occupancy required to elicit the maximal response, i.e. a 57% A1AdoR reserve. In contrast, the A1AdoR occupancy-response relationship for CPA mediated stimulation of [35S]-GTPγS binding was linear indicating little or no receptor reserve for G-protein activation. The relationship between CPA stimulation of [35S]-GTPγS binding and cyclic AMP inhibition was also hyperbolic with 44% G-protein activation sufficient to cause maximal inhibition.
The data suggest that the A1AdoR reserve for CPA mediated inhibition of cyclic AMP accumulation occurs at the level of G-protein interaction with adenylyl cyclase. However, each A1AdoR appears to activate a constant fraction of the total G-protein population suggesting signal amplification at the receptor-G-protein level which may also contribute to the receptor reserve for CPA.
Keywords: A1-adenosine receptor, receptor reserve, DDT1 MF-2 cells, irreversible antagonist, 8-cyclopentyl-3-[3-[[4-(fluorosulphonyl)benzoyl]oxy]propyl]-1-propylxanthine (FSCPX), cyclic AMP, G-protein, N6-cyclopentyladenosine
Introduction
The extent of receptor reserve, a measure of the receptor effector coupling efficency, is an important determinant of cellular responsiveness to receptor agonists. The presence of a receptor reserve indicates signal amplification and the magnitude of the reserve is dependent upon several factors including the agonist, the cell and/or tissue preparation and the response (Kenakin, 1993). Although not well studied, the receptor reserve for a given agonist appears to be a dynamic property involved in regulating tissue responsiveness (Nyborg, 1991; Brown et al., 1992). Several experimental approaches have been employed to determine the presence and magnitude of receptor reserve. For example partial receptor inactivation using an irreversible antagonist had been used to estimate agonist affinity, which is subsequently used to construct the relationship between receptor occupancy and response (Furchgott & Bursztyn, 1967). Another approach has been to directly relate agonist-mediated responses with receptor number by varying the latter over a wide range with an irreversible antagonist (Baker & Deyrup, 1994). The Ki/EC50 ratio for an agonist has also been used to indicate the absence or presence of receptor reserve (Adham et al., 1993; Traynor & Nahorshi, 1995; Breivogel et al., 1997).
Using irreversible antagonists, the receptor reserve for a number of guanine nucleotide binding protein (G-protein) coupled receptors such as the α and β-adrenoceptor (Ruffolo & Yaden, 1984; Jasper et al., 1988), muscarinic (Brown & Goldstein, 1986), dopamine (Meller et al., 1987), and serotonin receptors (Adham et al., 1993) has been reported. Recently, irreversible antagonists for the G-protein coupled A1-adenosine receptor (A1AdoR) have been synthesized, pharmacologically characterized and thse ligands have been used to estimate the receptor reserve for A1-mediated cardiac electrophysiological responses (Jacobson et al., 1989; Dennis et al, 1992; Scammells et al., 1994; Srinivas et al., 1997). The importance of a receptor reserve for the A1AdoR is underscored by its contribution to the differential potency of adenosine to activate the inward rectifying K+ current (IKAdo) and inhibition of isoprenaline-stimulate L-type Ca2+ current (ICa,L) in guinea-pig atrial myocytes (Srinivas et al., 1997). However, the majority of studies on receptor reserve for G-protein coupled receptors, including those for the A1AdoR, have been limited in defining the relationship between receptor occupancy and a response that is one or more steps downstream from the initial receptor occupancy and a response that is one or more steps downstream from the initial receptor-induced activation of a G-protein. Recently, agonist-dependent binding of the hydrolysis resistant guanine nucleotide [35S]-guanosine-5′-O-(-thiotriphosphate) ([35S]-GTPγS) has been used to investigate receptor activation of G-proteins in some cell and tissue membrane preparations (Hilf et al., 1989; Gierschik et al., 1991; Lorenzen et al., 1993; Traynor et al., 1995). In several studies, it was found that significant amplification exists in that one receptor is capable of activating several G-protein molecules (Gierschik et al., 1991; Sim et al., 1996). Results of these studies suggest that the measurement of agonist-dependent [35S]-GTPγS binding would be useful to ascertain if receptor activation of G-protein contributes to the overall receptor reserve.
In the present study, the relationship between A1AdoR occupancy and the effect of the full A1AdoR agonist N6-cyclopentyladenosine (CPA) to inhibit cyclic AMP accumulation and to stimulate [35S]-GTPγS binding to G-proteins was determined. This was accomplished by constructing concentration-response curves for CPA inhibition of (−)isoprenaline-stimulated cyclic AMP accumulation and stimulation of [35S]-GTPγS binding before and after irreversible A1AdoR inactivation with the chemoreactive ligand 8-cyclopentyl-3-[3-[[ 4 - (fluorosulphonyl) benzoyl]oxy] propyl] - 1 - propylxanthine (FSCPX) in DDT1 MF-2 (DDT) cells. These cells were found to be a particularly useful model system for this study because they express a relatively high density of A1AdoRs which are coupled to the inhibition of adenylyl cyclase activity through a mechanism requiring pertussis toxin sensitive G-proteins (Gerwins et al., 1990; Ramkumar et al., 1990).
Methods
Cell culture
DDT cells were grown as monolayers in 150-mm petri dishes using Dulbecco's Modified Eagle's Medium (DMEM) containing 2.5 μg ml−1 amphotericin B, 100 u ml−1 penicillin G, 0.1 mg ml−1 streptomycin sulphate and 5% foetal bovine serum in a humidified atmosphere of 95% air and 5% CO2. Cells were subcultured twice weekly by dispersion in Hank's Balanced Salt Solution (HBSS) without the divalent cations and containing 1 mM EDTA. The cells were then seeded in growth medium at a density of 1.2×125 cells per plate and experiments were peformed 4 days later at approximately 1 day preconfluence.
Cell pretreatments
Cell monolayers were rinsed twice with HBSS (2×10 ml) and incubated in 20 ml of HBSS per plate containing the indicated concentrations of FSCPX for 20 min at 37°C. At the end of the incubation, the HBSS was aspirated and drug free HBSS (10 ml) was added. After 5 min, the solution was aspirated and fresh drug free HBSS was added again. This cell washing procedure was repeated 15 times to remove unbound FSCPX. In some experiments, cells were preincubated with 100 nM FSCPX two or three times as above with three wash cycles between each drug exposure and 15 wash cycles after the last exposure.
Membrane preparations
Attached cells were washed twice with HBSS (2×10 ml), scraped free of plate with the aid of a rubber policeman in 5 ml of 50 mM Tris-HCl buffer pH 7.4 at 4°C and the suspension homogenized with a Tekmar homogenizer at setting 4 for 10 s. The suspension was then centrifuged at 27,000 ×g for 10 min. The pellet was resuspended in homogenization buffer by vortexing and centrifuged as described above. The final pellet was resuspended in one volume of 50 mM Tris-HCl buffer pH 7.4 containing 5 mM MgCl2 for A1AdoR assays. For the [35S]-GTPγS binding assay the final pellet was resuspended in 50 mM Tris-HCl pH 7.4 containing (mM): MgCl2 5, NaCl 100, and dithiothreitol 1. This membrane suspension was then placed in liquid N2 for 10 min, thawed and used for assays. The protein content was determined by the method of Bradford (1976) using bovine serum albumin as standard.
Receptor binding assay
The A1AdoR number in DDT cell membranes was obtained by determining the specific [3H]-8-cyclopentyl-1,3-dipropylxanthine ([3H]-CPX) binding (Scammells et al., 1994). Briefly, membranes (0.05–0.2 mg protein) were incubated in a total volume of 0.25 ml containing Tris-HCl buffer (50 mM) pH 7.4, MgCl2 (5 mM), 2 units ml−1 adenosine deaminase, [3H]-CPX (0.06–5 nM), and with and without 1 μM cyclopentyltheophylline (CPT) for 90 min at 25°C. At the end of the incubation, the suspensions were diluted with 3 ml of ice-cold incubation buffer. The suspension was then filtered under reduced pressure using a Brandell cell harvester. The filters were washed with a 6 ml of ice-cold buffer, placed in a vial with 3 ml of Scinti-Verse BD, and the radioactivity determined in a scintillation counter. Specific radioligand binding to the A1AdoR was calculated as the difference between total binding in the absence of CPT and the nonspecific binding determined in the presence of CPT.
[35S]-GTPγS binding assays
A1-agonist stimulated [35S]-GTPγS binding was determined by a modification of the method described by Gierschik et al. (1991) and Lorenzen et al. (1993). Membranes (30–50 μg protein) were incubated in a volume of 0.1 ml containing (mM): Tris-HCl buffer 50, pH 7.4, MgCl2 5, NaCl 100, dithiothreitol 1, 0.2 units ml−1 adenosine deaminase, 0.5% BSA, EDTA 1, 10 μM guanosine diphosphate (GDP), [35S]-GTPγS (0.3–0.4 nM) and with or without varying concentrations of CPA for 90 min at 30°C. Nonspecific binding was determined by the addition of 10 μM GTPγS. Agonist stimulated binding was determined as the difference between total binding in the presence of CPA and basal binding determined in the absence of CPA. In saturation experiments, 0.4 nM [35S]-GTPγS was incubated with 0.5–10 nM cold GTPγS. At the end of the incubation, each suspension was filtered and the retained radioactivity determined as described above.
Although the binding of [35S]-GTPγS to purified G-proteins has been shown to be irreversible (Bokoch et al., 1984), in several recent studies, agonist stimulated binding of this radioligand to G-proteins in membranes has been reported to be largely reversible (Breivogel et al., 1998; Yang & Lanier, 1999). This suggests that in membranes the interaction of [35S]-GTPγS with G-proteins is an equilibration process allowing the data to be analysed by standard radioligand binding methods. However, it should be pointed out that agonist stimulated [35S]-GTPγS binding is complex partly because of the requirement for the presence of GDP. Therefore, the calculated parameters of [35S]-GTPγS binding (Bmax and KD) are to be considered apparent and are used for relative comparisons.
Cyclic AMP determinations
Monolayers of DDT cells were rinsed twice with HBSS (2×10 ml) and detached from the plate in 5 ml of HBSS by gentle scraping with rubber cell scraper. The suspension was centrifuged at 500 ×g for 5 min and the cell pellet was gently resuspended in one volume of HBSS. Aliquots of the cell suspension were then incubated in HBSS (0.5 ml total volume) containing 50 μM rolipram, 1 μM (−)isoprenaline and varying concentrations of CPA for 10 min at 37°C. At the end of the incubation, the suspensions were placed in a boiling water bath for 5 min, cooled to room temperature and centrifuged at 13,000 ×g for 5 min. The cyclic AMP content of the supernatant was determined by radioimmunoassay similar to that described by Harper & Brooker (1975). An aliquot of the supernatant (0.05 ml) was added to 0.05 ml of HBSS containing adenosine 3′,5′-cyclic phosphoric acid 2′-O-succinyl[125I]-iodotyrosine methyl ester (10,000 c.p.m.) followed by the addition of 0.05 ml of H2O containing 0.1% BSA and anti-cyclic AMP antibody (1:2000 dilution). The samples were incubated at 4°C for 18 h. At the end of the incubation, 70 μl of a 50% (v v−1) hydroxyapatite suspension in H2O was added followed by a further incubation for 10 min at 4°C. The antibody/radioligand complex adsorbed to the hydroxyapatite was retained on filters using the Brandel cell harvester. The filters were washed with 6 ml of ice-cold 10 mM Tris-HCl buffer pH 7.0 and the radioactivity determined in a gamma counter. Nonspecific binding of the radioligand was determined in parallel assays that contained 0.1 μM cyclic AMP, and was subtracted from the total binding. The amount of cyclic AMP accumulated was calculated from a standard curve using known amounts of cyclic AMP.
Data analysis
The concentration of agonists that inhibited (−)isoprenaline stimulated cyclic AMP accumulation by 50% or stimulated [35S]-GTPγS binding by 50% (EC50) were determined from nonlinear regression analysis of the curves using the GraphPad Prism program (GraphPad Software Inc., San Diego, CA, U.S.A.). The KD and maximal binding (Bmax) values for the radioligands were determined from nonlinear regression analysis (GraphPad Prism) of saturation binding data and presented in the form of Scatchard (1949) plots. The receptor occupancy-response relationships were fitted to a rectangular hyperbola equation using nonlinear regression (GraphPad Prism) and the receptor reserve for the maximal response, defined as 90% of the maximum, was calculated from the standard curve. Statistical analysis of the data was performed using the Student's t-test and differences were considered significant if P<0.05.
Materials
The radiochemicals [3H]-CPX (88–120 Ci mmol−1) and [35S]-GTPγS (1100–1300 Ci mmol−1) were purchased from Dupont NEN (Boston, MA, U.S.A.). CPA and CPX were from Research Biochemicals (Natick, MA, U.S.A.). DMEM, HBSS and foetal bovine serum were obtained from GIBCO (Grand Island, NY, U.S.A.). DDT cells were from Americal Type Culture Collection (Rockville, MD, U.S.A.). Rolipram was a gift from Berlex Laboratories (Cedar Knolls, NJ, U.S.A.). FSCPX was synthesized as described by Scammells et al. (1994) and stock solutions (10 mM) were prepared in DMSO. Adenosine 3′,5′-cyclic phosphoric acid 2′-O-succinyl[125I]-iodotyrosine methyl ester was prepared by the method described by Patel & Linden (1988). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Results
Characteristics of CPA stimulated [35S]-GTPγS binding in DDT cell membranes
Previous studies have shown that basal and agonist stimulated [35S]-GTPγS binding is dependent upon the presence GDP (Hilf et al., 1989; Gierschik et al., 1991; Lorenzen et al., 1993; Traynor & Nahorski, 1995; Breivogel et al., 1998). In DDT cell membranes, GDP reduced the basal level of [35S]-GTPγS binding (0.3 nM) from 1424±9 fmol mg−1 protein in the absence of GDP to 11±3 fmol mg−1 protein in the presence of 100 μM GDP. Furthermore, there was little or no (<10%) CPA (1 μM)-mediated increase in [35S]-GTPγS binding above the basal level in the absence of GDP (data not shown). As depicted in Figure 1, there is a GDP concentration-dependent increase in CPA stimulated [35S]-GTPγS binding with a maximum stimulation at 5–10 μM of the guanine nucleotide. At GDP concentrations higher than 10 μM, the CPA stimulation of [35S]-GTPγS binding decreased. The less than maximal stimulation of [35S]-GTPγS binding at the lower GDP concentrations (0.1–0.5 μM) may be underestimated if maximal GDP binding required more than the 90 min incubation used. Figure 2 shows the time course of CPA dependent [35S]-GTPγS binding. Basal [35S]-GTPγS binding increased over the 180 min incubation period and this time-dependent increase was potentiated in the presence of CPA (1 μM). The CPA stimulated component of [35S]-GTPγS binding increased during the initial 80–90 min of incubation after which a binding plateau was attained.
Figure 1.
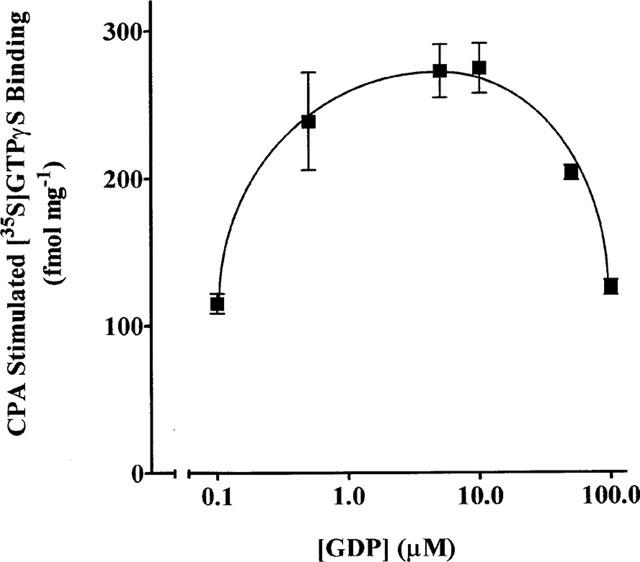
Effect of GDP on the CPA-stimulated component of [35S]-GTPγS binding. DDT cell membranes were incubated with 0.3 nM [35S]-GTPγS, 1 μM CPA and in the absence and presence of the indicated concentrations of GDP for 90 min at 30°C. Each point on the graph is the mean±s.d. of triplicate determinations and is representative of three separate experiments.
Figure 2.
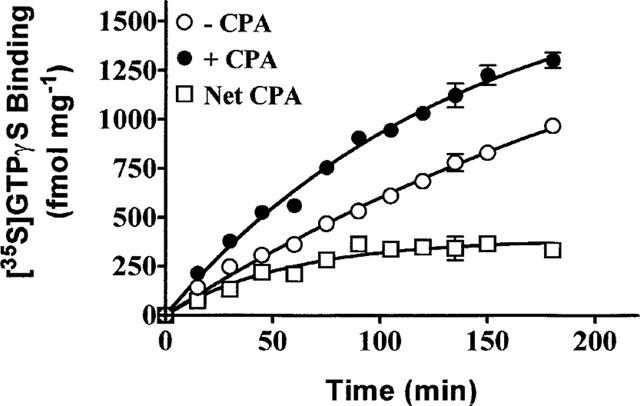
Time course of [35S]-GTPγS binding in the absence (−) and presence (+) of CPA. DDT cell membranes were incubated with 0.3 nM [35S]-GTPγS, 10 μM GDP and in the absence and presence of 1 μM CPA at 30°C for the times indicated. The net CPA stimulated-component of [35S]-GTPγS binding is the difference between the binding in the presence and absence of CPA. Each point on the graph is the mean±s.d. of triplicate determinations and is representative of three experiments.
Effect of FSCPX on A1AdoRs in DDT cells
Table 1 summarizes the Bmax and KD values for [3H]-CPX binding to the A1AdoR in DDT cell membranes before and after pretreatment of cells with FSCPX followed by cell washing. FSCPX caused a concentration-dependent decrease in the number of A1AdoRs with a 39, 65, 78 and 83% reduction after pretreatment with 0.5, 5, 50 and 100 nM FSCPX, respectively. After three consecutive pretreatments with 100 nM FSCPX the A1AdoR number was decreased by 91.5%. The KD value for [3H]-CPX binding to the A1AdoR did not change with any of the FSCPX pretreatments. This data indicates that FSCPX is an irreversible ligand for the A1AdoR in DDT cells. Pretreatment of DDT cells with concentrations of FSCPX up to 1 μM had no effect on (−)isoprenaline (1 μM) or forskolin (1 μM) stimulated cyclic AMP accumulation. Furthermore, the inclusion of 20 μM CPT during pretreatment of DDT cells with 100 nM FSCPX prevented the loss of A1AdoR's and did not change the concentration-response for CPA to inhibit (−)isoprenaline stimulated cyclic AMP accumulation or CPA-mediated [35S]-GTPγS binding (data not shown).
Table 1.
Effect of FSCPX pretreatment on maximal specific binding (Bmax) and KD of [3H]-CPX binding to A1-adenosine receptors in DDT1 MF-2 cell membranes

Effect of reducing A1AdoR content on CPA-mediated cyclic AMP inhibition and stimulation of [35S]-GTPγS binding
A representative concentration-response for CPA mediated inhibition of (−)isoprenaline stimulated cyclic AMP accumulation and stimulation of [35S]-GTPγS binding before and after FSCPX pretreatment of DDT cells is depicted in Figure 3a,b, respectively. From the concentration-response curves, the pEC50's (EC50's) and maximal responses for CPA were determined, and are presented in Table 2. With increasing concentrations of FSCPX, the concentration-responses for CPA mediated inhibition of cyclic AMP accumulation were shifted progressively to the right. A maximal shift of approximately 10 fold was achieved after pretreatment with 100 or 3×100 nM FSCPX. The maximal inhibition of (−)isoprenaline-stimulated cyclic AMP accumulation was not changed in cells pretreated with 0.5 nM FSCPX but was reduced by 10 and 16% in cells pretreated with 5 and 50 nM FSCPX, respectively. The maximal effect of CPA was reduced by 31 and 69% after pretreatment with 100 nM and 3×100 nM of the irreversible antagonist, respectively. The pEC50 for CPA-stimulated [35S]-GTPγS binding in control cells was not changed after pretreatment with 0.5 or 5 nM FSCPX. However, after pretreatment with 100 nM FSCPX there was a small decrease in the pEC50 for CPA (1.7 fold increase in EC50). The maximal CPA-mediated stimulation of [35S]-GTPγS binding was decreased by 39, 65 and 78% after pretreatment with 0.5, 5 and 100 nM FSCPX, respectively. The CPA (0.1 μM) mediated inhibition of (−)isoprenaline (1 μM) stimulated cyclic AMP accumulation and stimulation [35S]-GTPγS binding was completely prevented by the addition of 1 μM CPX (data not shown).
Figure 3.

Effect of FSCPX on CPA-mediated inhibition of (−)isoprenaline-stimulated cyclic AMP accumulation (a) and CPA-stimulated [35S]-GTPγS binding (b). (a) DDT cells were incubated in the absence (control) and presence of 0.5, 5, 50, 100 or 3×100 nM FSCPX for 20 min at 37°C followed by 15 cell washes. The cells were then incubated for 10 min in the presence of 50 μM rolipram, 1 μM (−)isoprenaline and the indicated concentrations of CPA for 10 min at 37°C. At the end of the incubation, the cyclic AMP accumulated was determined. Basal cyclic AMP accumulation in control cells was 13±4 pmol mg−1 (mean±s.d., n=3). Data are the mean of triplicate determinations and are representative of 4–10 separate experiments. (b) DDT cells were incubated in the absence (control) and presence of 0.5, 5 or 100 nM FSCPX for 20 min at 37°C. After 15 cell washes, membranes were prepared and the concentration-response for CPA-stimulated [35S]-GTPγS binding (0.3 nM) was determined. Basal [35S]-GTPγS binding in control membranes was 146±8 fmol mg−1 protein (mean±s.d., n=3). Data are the mean of triplicate determinations and are representative of 3–10 experiments.
Table 2.
pEC50 values and maximal response for CPA inhibition of cylic AMP accumulation and stimulation of [35S]-GTPγS binding in FSCPX pretreated cells
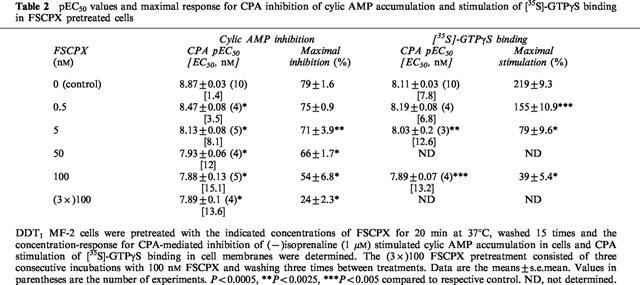
Figure 4a shows the concentration-dependence of GTPγS binding in DDT membranes after pretreatment with 5 nM FSCPX. These experiments were performed using a constant concentration of [35S]-GTPγS (0.4 nM) and varying concentrations of unlabelled ligand. In control membranes, basal GTPγS binding increased as the concentration of ligand was increased from 0.4 to 10.2 nM and this increase was potentiated in the presence of 1 μM CPA. After pretreatment with FSCPX, basal GTPγS binding was not altered but the binding in the presence of CPA was reduced. Figure 4b shows a representative Scatchard plot of the CPA-stimulated component of GTPγS binding in DDT cell membranes before and after pretreatment of the intact cells with FSCPX. Under the assay conditions used, the Bmax was 4.3±0.2 (n=3) pmol mg−1 protein in control membranes. After preincubation of DDT cells with 5 nM FSCPX for 20 min followed by repeated cell washing, the Bmax was reduced by 51% to 2.1±0.2 pmol mg−1 protein (P<0.005). There was no change in the apparent KD value for the CPA-stimulated component of [35S]-GTPγS binding (control=0.63±0.03 nM, FSCPX pretreatment=0.72±0.07 nM). Based on the Bmax values for [3H]-CPX and [35S]-GTPγS binding the estimated ratio of A1AdoR to G-proteins before and after pretreatment with 5 nM FSCPX are 10.3 and 13.3, respectively.
Figure 4.
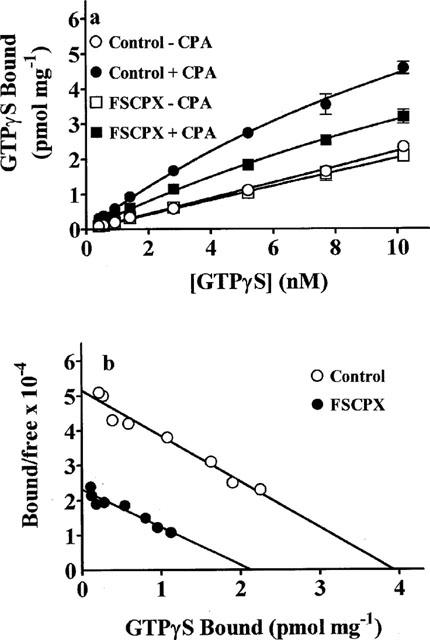
Representative [35S]-GTPγS binding (a) and Scatchard plot (b) of the CPA-stimulated [35S]-GTPγS binding component after pretreatment with FSCPX. DDT cells were incubated in the absence (control) or presence of 5 nM FSCPX for 20 min at 37°C followed by 15 cell washes. Cell membranes were then prepared and the concentration-dependence of [35S]-GTPγS binding, using cold GTPγS with 0.4 nM [35S]-GTPγS, in the absence (basal) and presence of CPA (5 μM) was determined. Points on the graph are means±s.d. of triplicate determinations and are representative of three separate experiments. In (b), the net CPA-stimulated [35S]-GTPγS binding is shown as a Scatchard plot.
Receptor occupancy-response relationships for CPA to inhibit cyclic AMP accumulation and stimulate [35S]-GTPγS binding
The relationship between receptor number and CPA-mediated inhibition of (−)isoprenaline-stimulated cyclic AMP accumulation and CPA-stimulated [35S]-GTPγS binding is depicted in Figure 5. DDT cells were pretreated with varying concentrations of FSCPX (0.1–3×100 nM) and washed to remove unbound ligand. The maximal CPA (5 μM) inhibition of cyclic AMP accumulation was determined in intact cells and the A1AdoR content and maximal CPA-stimulated [35S]-GTPγS binding was determined in cell membranes. As illustrated in Figure 5, the relationship between A1AdoR occupancy and inhibition of cyclic AMP accumulation is curvilinear, indicating that to achieve the maximal response less than full receptor occupancy by the agonist is required. The calculated receptor reserve based upon 90% of the maximal response is 57%. On the other hand, the receptor occupancy-response relationship for CPA-mediated stimulation of [35S]-GTPγS binding is linear (linear regression analysis, r2=0.98) indicating that full receptor occupancy is required to achieve maximal agonist-mediated [35S]-GTPγS binding (Figure 5). Similar to the occupancy-response relationship for the A1AdoR and cyclic AMP inhibition, the relationship between the CPA mediated stimulation of [35S]-GTPγS binding and cyclic AMP inhibition is also curvilinear (Figure 6). The maximal inhibition of cyclic AMP accumulation, defined as 90% of maximum, is achieved when CPA-stimulated [35S]-GTPγS binding reached 44% of the maximum.
Figure 5.
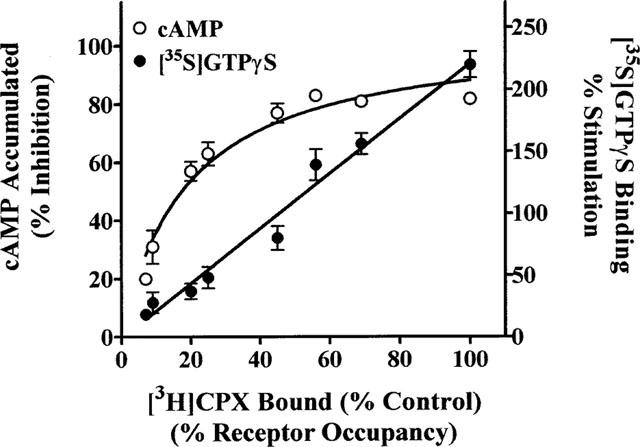
Relationship between A1AdoR number (per cent receptor occupancy) and maximal CPA-mediated inhibition of cyclic AMP accumulation and maximal CPA-stimulated [35S]-GTPγS binding. DDT cells were pretreated in the absence and presence of 0.5, 1, 5, 50, 100, 2×100 or 3×100 nM FSCPX for 20 min at 37°C followed by 15 cell washings. The maximal A1AdoR-mediated inhibition of cyclic AMP accumulation was determined by incubation of the cells with 50 μM rolipram, 1 μM (−)isoprenaline and in the presence or absence of CPA (5 μM). Cell membranes were prepared from additional cells and the maximal CPA (5 μM)-stimulated G-protein activation was determined by [35S]-GTPγS (0.3 nM) binding. The A1AdoR content of the cell membranes was determined by specific [3H]-CPX (5 nM) binding. Each point on the graph is the mean±s.e.mean (n=4–12).
Figure 6.
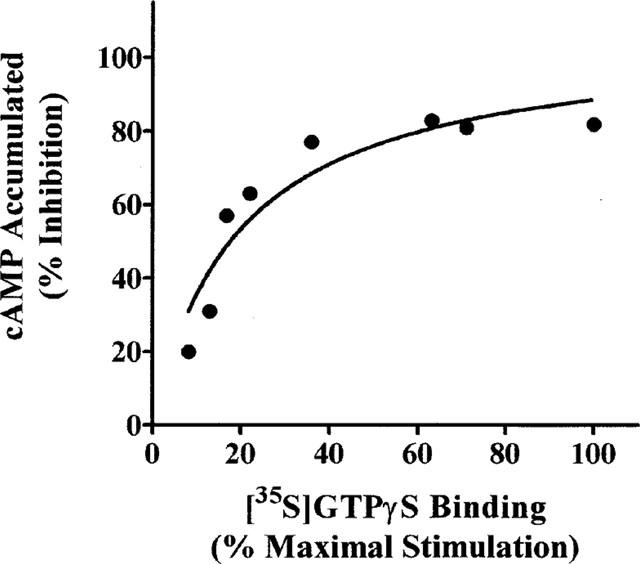
Relationship between maximal CPA-stimulated [35S]-GTPγS binding and maximal CPA-mediated inhibition of (−)isoprenaline-stimulation of cyclic AMP accumulation. Data were taken from Figure 5.
Discussion
Results of the present study show a receptor reserve for CPA-mediated inhibition of cyclic AMP accumulation but little or no receptor reserve for CPA-stimulation of [35S]-GTPγS binding in DDT cells. The experimental approach used was to inactivate variable fractions of receptors using the irreversible A1AdoR antagonist FSCPX (Scammells et al., 1994; Srinivas et al., 1996). Thus, pretreatment of DDT cells with FSCPX, followed by extensive washing, produced a concentration-dependent decrease in maximal [3H]-CPX binding without a change in affinity of the radioligand for the non-inactivated A1AdoR's, i.e. the remaining receptors. This indicates that FSCPX acts as an irreversible antagonist for the A1AdoR in DDT cells, a finding that is consistent with the irreversible nature of this antagonist as previously reported using brain and cardiac membranes (Srinivas et al., 1996; 1997; Morey et al., 1998). The lack of effect of FSCPX on forskolin or (−)isoprenaline-stimulated cyclic AMP accumulation indicates that this chemoreactive ligand had little or no nonspecific effects on the β2-adrenoceptor, stimulatory Gs protein or adenylyl cyclase. Furthermore, pretreatment of DDT cells with FSCPX in the presence of an excess of the A1AdoR antagonist CPT, prevented the irreversible inactivation of the receptor and did not affect the subsequent effect of CPA to inhibit cyclic AMP accumulation or basal and CPA-stimulated [35S]-GTPγS binding. This indicates that FSCPX did not affect the coupling or function of G-proteins associated with the A1AdoR.
Pretreatment of DDT cells with relatively low concentrations of FSCPX had no effect on the maximal CPA-mediated inhibition of (−)isoprenaline-stimulated cyclic AMP accumulation although the concentration-response curves were rightward shifted. With higher concentration of FSCPX the maximal CPA mediated response was also reduced. This pattern of effect using an irreversible antagonist is consistent with the presence of a receptor reserve. Indeed, the relationship between receptor number and maximal response for CPA was curvilinear with 43% receptor occupancy necessary to achieve the maximal response. Thus, for the agonist CPA, there is a 57% receptor reserve for the maximal response in DDT cells. This receptor reserve was similar (64%) to that recently reported for an irreversible A1AdoR agonist mediating an inhibition of cyclic AMP accumulation in the same cells (Zhang et al., 1997). In contrast to cyclic AMP inhibition, the relationship between receptor number and CPA stimulated [35S]-GTPγS binding was linear indicating that full receptor occupancy is required to achieve maximal G-protein activation, i.e. there is little or no receptor reserve. This indicates that the observed receptor reserve for the CPA-mediated inhibition of cyclic AMP accumulation in DDT cells occurs at the level of G-protein (or its subunits) interaction with adenylyl cyclase rather than at the level of the interaction between the receptor and the G-protein. This conclusion is supported by the observation that the maximal inhibition of cyclic AMP accumulation requires 44% of maximal G-protein activation. Furthermore, based upon the Bmax values for [3H]-CPX binding to the A1AdoR (0.417 pmol mg−1 protein) and CPA-stimulated [35S]-GTPγS binding (4.3 pmol mg−1 protein), one A1AdoR appears to activate an estimated 10 molecules of a G-protein (amplification factor). This amplification of G-protein activation by each A1AdoR could increase the number of G-protein-adenylyl cyclase interactions such that a maximal inhibition of cyclic AMP can occur at submaximal receptor occupancy. Our data therefore support the original conjecture of Keen & Nahorski (1988) that the receptor reserve for inhibition of adenylyl cyclase is actually a G-protein reserve.
Similar to the data in the present report, a relatively high amplification factor for G-protein activation has been reported for the chemotactic peptide receptor (1:20) in HL-60 granulocytes (Gierschik et al., 1991) and for the μ-opioid (1:17) and δ-opioid (1:22) receptors in the rat striatum (Sim et al., 1996). On the other hand, a relatively low amplification factor for G-protein activation has been reported for the μ-opioid receptor (1:2) in SH-SY5Y neuroblastoma cells (Traynor & Nahorshi, 1995) and for the muscarinic receptor (1:2 to 3) in porcine atrial membranes (Hilf et al., 1989). In several studies, the agonist Ki/EC50 ratio has been used to indicate the presence or absence of a receptor reserve for G-protein activation as determined by stimulation of [35S]-GTPγS binding. Alt et al. (1998) reported that the Ki's for several mu-opioid receptor agonists closely matched their EC50's for stimulation of [35S]-GTPγS binding in rat C6 glioma cells suggesting no receptor reserve. In contrast, the EC50 value for [D-Ala2,MePhe4,Gly-ol5]enkephalin to stimulate [35S]-GTPγS binding was 100 fold smaller than its Ki value for the μ-opioid receptor in SH-SY5Y cells indicating a large receptor reserve for G-protein activation (Traynor & Nahorski, 1995). Although in the above studies the Ki and EC50 values were determined under similar conditions, there are two or more agonist affinity states for most G-protein coupled receptors and it is unclear which state is relevant to the stimulation of [35S]-GTPγS binding. For example, Lorenzen et al. (1993) first reported that the agonist high affinity binding state of the A1AdoR was identical to the EC50 for stimulation of [35S]-GTPγS binding. However, in a more recent study, the agonist low affinity binding state of the A1AdoR more closely correlated with G-protein activation (Lorenzen et al., 1996). Therefore, it remains to be established which affinity state of the receptor is responsible for the stimulation of [35S]-GTPγS binding. In the present study, the receptor reserve was determined by varying the fraction of active receptors using an irreversible antagonist. This experimental approach does not require knowledge of the Ki of an agonist or which affinity state of the receptor is coupled to a response, and thereby allows for a direct determination of the presence and magnitude of a receptor reserve.
As indicated by the linear relationship between the A1AdoR number and stimulation of [35S]-GTPγS binding, the ratio of receptor and activated G-proteins remains relatively constant as the receptor number is varied. This observation is consistent with the A1AdoR and/or G-protein having restricted mobility within the cell membrane. Although early experimental models of G-protein coupled receptor activation mechanisms suggested that these proteins and their effectors are freely mobile within the membrane, increasing evidence now suggests that the G-protein and receptors have highly organized and restricted interactions (Neubig, 1994). In the case of the A1AdoR, it has been reported that in some tissues there is a tight interaction between the receptor and G-protein which is resistant to uncoupling by guanine nucleotides and the complex of the two proteins is retained upon protein solubilization (Klotz et al., 1986; Stiles, 1988). More recently, a membrane protein distinct from both the A1AdoR and G-protein has been shown to stabilize the receptor G-protein complex and may regulate signal amplification (Nanoff et al., 1995). These observations suggest that the A1AdoR and/or its coupled G-proteins have restrictions on their interactions which may help to explain the ratio of one A1AdoR activating a relatively constant number of G-proteins in DDT cells.
Potential limitations
It should be pointed out that the calculated ratio between A1AdoRs and activated G-proteins is an estimate (∼1:10) that is likely to be affected by several factors. First, the analysis assumes that all A1AdoRs in DDT cells are coupled to the cyclic AMP inhibitory response. However, it is known that activation of A1AdoRs in these cells also mediate phosphoinositide metabolism (Gerwins & Fredholm, 1992). Although to our knowledge there is no evidence for fractions of this receptor exclusively coupling to either cyclic AMP inhibition or another response, if this were the case, then the calculated ratio of 10–13 would be an underestimation. Second, DDT cells have been shown to express Gαi2 and Gαi3 which may mediate cyclic AMP inhibition (Gerwins & Fredholm, 1991). If the CPA-stimulated [35S]-GTPγS binding involves G-proteins not coupled to the inhibition of cyclic AMP then this may result in an overestimation of the ratio between receptor and G-protein. Thirdly, the cyclic AMP response was determined in intact cells whereas the [35S]-GTPγS binding assay was performed in membranes. The loss of cell integrity with isolated membranes may restrict receptor-G-protein interactions such that the ratio is underestimated. Alternatively, the process of membrane preparation may remove mobility constraints on receptors and/or G-proteins increasing the probability of their interaction and leading to an overestimation of the ratio. Finally, receptor mediated amplification of G-protein activation may be higher under physiological conditions. In intact cells, GTP binds to and activates a G-protein followed by inactivation due to hydrolysis to GDP (Gilman, 1987). This cycling between active and inactive states may allow a single receptor to repetitively activate a single G-protein and thus further enhance the amplification of the signal. On the other hand, GTPγS is poorly hydrolyzable and slowly dissociates from G-proteins (Pfeuffer & Helmreich, 1975; Breivogel et al., 1998) which may prevent or slow the cycling between active and inactive states of the G-protein. Thus, it is not clear to what extent agonist stimulated [35S]-GTPγS binding accurately and quantitatively reflects receptor activation of the G-protein population under physiological conditions. Regardless of these limitations however, our results indicate that each A1AdoR can activate more than one molecule of a G-protein and that this amplification remains relatively constant over a wide range of active receptor expression.
In summary, the data from the present study show that there is a relatively large A1AdoR reserve for CPA-mediated inhibition of cyclic AMP accumulation in DDT cells. Although little or no receptor reserve was found between A1AdoR occupancy and G-protein activation, each A1AdoR appear to be able to activate a number of G-protein molecules. This amplification of G-protein activation appeared independent of A1AdoR number and is likely to contribute to the receptor reserve between the A1AdoR and inhibition of cyclic AMP. It is well known that changes in the expression of receptors and G-proteins can occur under a variety of conditions. Therefore, the present data suggest that the receptor:G-protein activation ratio, rather than independent measurements of receptors and G-proteins, may prove to be important in further understanding the regulation of cell responsiveness to agonists.
Acknowledgments
The authors thank Deborah Otero and Jackie Ruble for technical assistance. We also thank Dr Donn Dennis for helpful discussions of the data analysis. This work was supported by a grant from the American Heart Association, Florida Affiliate and by National Institutes of Health Grant HL56785.
Abbreviations
- A1AdoR
A1-adenosine receptor
- CPA
cyclopentyladenosine
- CPT
cyclopentyltheophylline
- CPX
cyclopentyl-1,3-dipropylxanthine
- DDT
DDT1 MF-2
- FSCPX
8-cyclopentyl-3-[3-[[4-(fluorosulphonyl)benzoyl]oxy]propyl]-1-propylxanthine
- GDP
guanosine diphosphate
- G-protein
guanine nucleotide binding protein
- GTPγS
guanosine-5′-O-(thiotriphosphate)
References
- ADHAM N., ELLERBROOK B., HARTIG P., WEINSHANK R.L., BRANCHEK T. Receptor reserve masks partial agonist activity of drugs in a cloned 5-hydroxytryptamine1B receptor expression system. Mol. Pharmacol. 1993;43:427–433. [PubMed] [Google Scholar]
- ALT A., MANSOUR A., AKIL H., MEDZIHRADSKY F., TRAYNOR J.R., WOODS J.H. Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding by endogenous opioids acting at a cloned Mu receptor. J. Pharmacol. Exp. Ther. 1998;286:282–288. [PubMed] [Google Scholar]
- BAKER S.B., DEYRUP M.D. Development of novel irreversible antagonists. Neuroprotocols. 1994;4:66–75. [Google Scholar]
- BOKOCH G.M., KATADA T., NORTHUP J.K., UI M., GILMAN A.G. Purification and properties of the inhibitory guanine nucleotide-binding regulatory component of adenylate cyclase. J. Biol. Chem. 1984;259:3560–3567. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quanties of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SELLEY D.E., CHILDERS S.R. Cannabinoid receptor agonist efficacy for stimulating [35S]GTPγS binding to rat cerebellar membranes correlates with agonist-induced decreases in GDP affinity. J. Biol. Chem. 1998;273:16865–16873. doi: 10.1074/jbc.273.27.16865. [DOI] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SIM L.J., CHILDERS S.R. Regional differences in cannabinoid receptor/G-protein coupling in rat brain. J. Pharmacol. Exp. Ther. 1997;282:1632–1642. [PubMed] [Google Scholar]
- BROWN L., DEIGHTON N.M., BALS S., SOHLMANN W., ZERKOWSKI H.-R., MICHEL M.C., BRODDE O.-E. Spare receptors for β-adrenoceptor-mediated positive inotropic effects of catecholamines in the human heart. J. Cardiovasc. Pharmacol. 1992;19:222–232. doi: 10.1097/00005344-199202000-00011. [DOI] [PubMed] [Google Scholar]
- BROWN J.H., GOLDSTEIN D. Differences in muscarinic receptor reserve for inhibition of adenylate cyclase and stimulation of phosphoinositide hydrolysis in chick heart cells. Mol. Pharmacol. 1986;30:566–570. [PubMed] [Google Scholar]
- DENNIS D., JACOBSON K., BELARDINELLI L. Evidence of spare A1-adenosine receptors in guinea pig atrioventricular node. Am. J. Physiol. 1992;262:H661–H671. doi: 10.1152/ajpheart.1992.262.3.H661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F., BURSZTYN P. Comparison of dissociation constants and of relative efficacies of selected agonists acting on parasympathetic receptors. Ann. New York Acad. Sci. 1967;144:882–899. [Google Scholar]
- GERWINS P., FREDHOLM B.B. Glucocorticoid receptor activation leads to up-regulation of adenosine A1 receptors and down-regulation of adenosine A2 responses in DDT1 MF-2 smooth muscle cells. Mol. Pharmacol. 1991;40:149–155. [PubMed] [Google Scholar]
- GERWINS P., FREDHOLM B.B. Stimulation of adenosine A1 receptors and bradykinin receptors, which act via different G proteins, synergistically raise inositol 1,4,5-trisphosphate and intracellular free calcium in DDT1 MF-2 smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7330–7334. doi: 10.1073/pnas.89.16.7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERWINS P., NORDSTEDT C., FREDHOLM B.B. Characterization of adenosine A1 receptors in intact DDT1 MF-2 smooth muscle cells. Mol. Pharmacol. 1990;38:660–666. [PubMed] [Google Scholar]
- GIERSCHIK P., MOGHTADER R., STRAUB C., DIETERICH K., JAKOBS K.H. Signal amplification in HL-60 granulocytes. Evidence that the chemotactic peptide receptor catalytically activates guanine-nucleotide-binding regulatory proteins in native plasma membranes. Eur. J. Biochem. 1991;197:725–732. doi: 10.1111/j.1432-1033.1991.tb15964.x. [DOI] [PubMed] [Google Scholar]
- GILMAN A.G. G proteins: transducers of receptor-generated signals. Ann. Rev. Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- HARPER J.F., BROOKER G. Femtomole sensitive radioimmunoassay for cyclic AMP and cyclic GMP after 2′O acetylation by acetic anhydride in aqueous solution. J. Cyclic Nucleotide Res. 1975;1:207–218. [PubMed] [Google Scholar]
- HILF G., GIERSCHIK P., JAKOBS K.H. Muscarinic acetylcholine receptor-stimulated binding of guanosine 5′-O-(3-thiophosphate) to guanine-nucleotide-binding proteins in cardiac membranes. Eur. J. Biochem. 1989;186:725–731. doi: 10.1111/j.1432-1033.1989.tb15266.x. [DOI] [PubMed] [Google Scholar]
- JACOBSON K.A., BARONE S., KAMMULA U., STILES G. Electrophilic derivatives of purines as irreversible inhibitors of A1 adenosine receptors. J. Med. Chem. 1989;32:1043–1051. doi: 10.1021/jm00125a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JASPER J.R.M., MOTULSKY H.J., INSEL P.A. Characterization of a bromoacetylated derivative of pindolol as a high affinity, irreversible beta adrenergic antagonist in cultured cells. J. Pharmacol. Exp. Ther. 1988;244:820–824. [PubMed] [Google Scholar]
- KEEN M., NAHORSKI S.R. Muscarinic acetylcholine receptors linked to the inhibition of adenylate cyclase activity in membranes from the rat striatum and myocardium can be distinguished on the basis of agonist efficacy. Mol. Pharmacol. 1988;34:769–778. [PubMed] [Google Scholar]
- KENAKIN T. New York: Raven Press; 1993. Pharmacologic Analysis of Drug-Receptor Interaction; pp. 44–57. [Google Scholar]
- KLOTZ K.-N., LOHSE M.J., SCHWABE U. Characterization of the solubilized A1 adenosine receptor from rat brain membranes. J. Neurochem. 1986;46:1528–1534. doi: 10.1111/j.1471-4159.1986.tb01772.x. [DOI] [PubMed] [Google Scholar]
- LORENZEN A., FUSS M., VOGT H., SCHWABE U. Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding. Mol. Pharmacol. 1993;44:115–123. [PubMed] [Google Scholar]
- LORENZEN A., GUERRA L., VOGT H., SCHWABE U. Interaction of full and partial agonists of the A1 adenosine receptor with receptor/G protein complexes in rat brain membranes. Mol. Pharmacol. 1996;49:915–926. [PubMed] [Google Scholar]
- MELLER E., BOHMAKER K., NAMBA Y., FRIEDHOFF A.J., GOLDSTEIN M. Relationship between receptor occupancy and response at striatal dopamine autoreceptors. Mol. Pharmacol. 1987;31:592–598. [PubMed] [Google Scholar]
- MOREY T.E., BELARDINELLI L., DENNIS D.M. Validation of Furchgott's method to determine agonist-dependent A1-adenosine receptor reserve in guinea-pig atrium. Br. J. Pharmacol. 1998;123:1425–1433. doi: 10.1038/sj.bjp.0701747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NANOFF C., MITERAUER T., ROKA F., HOHENEGGER M., FREISSMUTH M. Species differences in A1 adenosine receptor/G protein coupling: identification of a membrane protein that stabilizes the association of the receptor/G protein complex. Mol. Pharmacol. 1995;48:806–817. [PubMed] [Google Scholar]
- NEUBIG R.R. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- NYBORG N.C.B. Ageing is associated with increased 5-HT2-receptor affinity and decreased receptor reserve in rat isolated coronary arteries. Br. J. Pharmacol. 1991;102:282–286. doi: 10.1111/j.1476-5381.1991.tb12167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL A., LINDEN J. Purification of 125I-labeled succinyl cyclic nucleotide tyrosine methyl esters by high-performance liquid chromatography. Anal. Biochem. 1988;168:417–420. doi: 10.1016/0003-2697(88)90338-7. [DOI] [PubMed] [Google Scholar]
- PFEUFFER T., HELMREICH E.J.M. Activation of pigeon erythrocyte membrane adenylate cyclase by guanyl nucleotide analogues and separation of a nucleotide binding protein. J. Biol. Chem. 1975;250:867–876. [PubMed] [Google Scholar]
- RAMKUMAR V., BARRINGTON W.W., JACOBSON K.A., STILES G.L. Demonstration of both A1 and A2 adenosine receptors in DDT1 MF-2 smooth muscle cells. Mol. Pharmacol. 1990;37:149–156. [PMC free article] [PubMed] [Google Scholar]
- RUFFOLO R.R., Jr, YADEN E.L. Existence of spare α1-adrenoceptors, but not α2-adrenoceptors, for respective vasopressor effects of cirazoline and B-HT 933 in the pithed rat. J. Cardiovasc. Pharmacol. 1984;6:1011–1019. [PubMed] [Google Scholar]
- SCAMMELLS P.J., BAKER S.P., BELARDINELLI L., OLSSON R.A. Substituted 1,3-dipropylxanthines as irreversible antagonists of A1 adenosine receptors. J. Med. Chem. 1994;37:2704–2712. doi: 10.1021/jm00043a010. [DOI] [PubMed] [Google Scholar]
- SCATCHARD G. The attractions of proteins for small molecules and ions. Ann. NY Acad. Sci. 1949;51:660–672. [Google Scholar]
- SIM L.J., SELLEY D.E., XIAO R., CHILDERS S.R. Differences in G-protein activation by μ- and δ-opioid, and cannabinoid receptors in rat striatum. Eur. J. Pharmacol. 1996;307:97–105. doi: 10.1016/0014-2999(96)00211-7. [DOI] [PubMed] [Google Scholar]
- SRINIVAS M., SHYROCK J.C., DENNIS D.M., BAKER S.P., BELARDINELLI L. Differential A1 adenosine receptor reserve for two actions of adenosine on guinea pig atrial myocytes. Mol. Pharmacol. 1997;52:683–691. doi: 10.1124/mol.52.4.683. [DOI] [PubMed] [Google Scholar]
- SRINIVAS M., SHRYOCK J.C., SCAMMELS P.J., RUBLE J., BAKER S.P., BELARDINELLI L. A novel irreversible antagonist of the A1-adenosine receptor. Mol. Pharmacol. 1996;50:196–205. [PubMed] [Google Scholar]
- STILES G.L. A1 adenosine receptor-G protein coupling in bovine brain membranes: effects of guanine nucleotides, salt and solubilization. J. Neurochem. 1988;51:1592–1598. doi: 10.1111/j.1471-4159.1988.tb01129.x. [DOI] [PubMed] [Google Scholar]
- TRAYNOR J.R., NAHORSKI S.R. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- YANG Q., LANIER S.M. Influence of G protein type on agonist efficacy. Mol. Pharmacol. 1999;56:651–656. doi: 10.1124/mol.56.3.651. [DOI] [PubMed] [Google Scholar]
- ZHANG J., BELARDINELLI L., JACOBSON K.A., OTERO D.H., BAKER S.P. Persistent activation by and receptor reserve for an irreversible A1-adenosine receptor agonist in DDT1 MF-2 cells and in guinea pig heart. Mol. Pharmacol. 1997;52:491–498. doi: 10.1124/mol.52.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]