

Abstract
In rat isolated renal artery segments contracted with 0.1 μM phenylephrine and in the presence of the NO synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME), carbachol and acetylcholine produced endothelium-dependent relaxations. The mechanisms underlying these relaxations were studied.
These relaxations were not affected by ODQ (1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one) or indomethacin. In arteries contracted with 20–30 mM K+, L-NAME-resistant relaxations induced by carbachol and acetylcholine were virtually absent.
The Na+-K+ ATPase inhibitor ouabain reduced these relaxations in a concentration-dependent manner.
In K+-free media, addition of K+ (5 mM) produced 90.5±3.9% (n=3) relaxation of phenylephrine-induced tone. This relaxation was endothelium-independent and ouabain-sensitive.
Tetraethylammonium (TEA), charybdotoxin (ChTX) and iberiotoxin (IbTX) reduced the sensitivity of carbachol-induced relaxations, but did not change the maximal response. These relaxations were not altered by 4-aminopyridine (4-AP), glibenclamide or apamin. Acetylcholine (1 μM)-induced relaxation was reduced by ChTX, but not by TEA or IbTX.
The cytochrome P450 inhibitor miconazole, but not 17-octadecynoic acid, reduced the sensitivity of carbachol-induced relaxations, without changing the maximal response.
In conclusion, in rat isolated renal arteries, acetylcholine and carbachol produced a non-NO/non-PGI2 relaxation which is mediated by an endothelium-derived hyperpolarizing factor (EDHF). This factor does not appear to be a cytochrome P450 metabolite. The inhibition by ouabain of these relaxations suggests the possible involvement of Na+-K+ ATPase activation in EDHF responses, although other mechanisms cannot be totally ruled out.
Keywords: Acetylcholine, carbachol, EDRF, EDHF, Na+-K+ ATPase, ouabain, renal artery (rat)
Introduction
Vasodilatation produced by acetylcholine and other muscarinic agonists has been shown to be dependent on the presence of vascular endothelial cells (Furchgott & Zawadzki, 1980; Vanhoutte et al., 1986). Several endothelium-derived vasodilator factors have been identified. These include endothelium-derived relaxing factor (EDRF), prostaglandin I2 (PGI2, prostacyclin) and endothelium-derived hyperpolarizing factor (EDHF). EDRF is considered to be nitric oxide (NO), which is produced by NO synthase (NOS) from L-arginine and relaxes smooth muscle cells by increasing their intracellular cyclic GMP level (Moncada et al., 1991). PGI2 is a cyclo-oxygenase metabolite of arachidonic acid and evokes its relaxant effect by increasing intracellular cyclic AMP level (Siegel et al., 1989; Frolich, 1990). In many species, endothelium-dependent relaxations that were resistant to inhibitors of both NOS and cyclo-oxygenase have been demonstrated in a variety of blood vessels, and this response has been proposed to be mediated by EDHF (Garland et al., 1995; Kitagawa et al., 1994; Zygmunt et al., 1994; Kessler et al., 1996; Urakami-Harasawa et al., 1997). In contrast to NO and PGI2, the chemical identity of EDHF and the mechanisms by which it produces its biological activity are still uncertain (Mombouli & Vanhoutte, 1997; Quilley et al., 1997; Edwards & Weston, 1998).
Some recent studies suggested that EDHF may be arachidonic acid metabolites produced by cytochrome P450 epoxygenase, since some of these metabolites mimicked the biological activity of EDHF, and cytochrome P450 inhibitors inhibited EDHF-mediated responses (Hecker et al., 1994; Campbell et al., 1996; Chen & Cheung, 1996; Popp et al., 1996; Dong et al., 1997; Hayabuchi et al., 1998). However, some evidence directly against this hypothesis also has been reported (Zygmunt et al., 1996; Urakami-Harasawa et al., 1997). Two other mechanisms have been proposed to account for EDHF-produced relaxation and hyperpolarization; namely, activation of K+ channels (Edwards & Weston, 1998) and activation of Na+-K+ ATPase in the vascular smooth muscle cells (Kitagawa et al., 1994; Prieto et al., 1998). It appears that the extent to which each is involved may differ between species and tissues. Moreover, studies with various K+ channel blockers have led to controversial conclusions on the identity of the K+ channels involved in EDHF-mediated responses. Given the situation that there are considerable differences in the nature and biological activity of EDHF between blood vessels and species, some authors suggested that EDHF might not be a single factor, and its nature and cellular targets might differ between blood vessels and species (Triggle et al., 1999).
Evidence has shown that EDHF is an important regulator of vascular function in both physiological and pathophysiological conditions. For example, many studies suggest that in rat small resistance arteries (diameter of 100–300 μM), EDHF might be a major regulator of the vascular calibre under normal conditions, and therefore might be of primary importance in the regulation of vascular resistance (see Garland et al., 1995). Also, it has been reported that EDHF may contribute to the vascular adaptation to pregnancy in rats (Gerber et al., 1998). Under pathophysiological conditions, a decreased activity of EDHF has been observed in spontaneous hypertensive rats (Hayakawa et al., 1995), while in hypercholesterolemic rabbits, the EDHF-mediated component of endothelium-dependent vasorelaxations was enhanced (Brandes et al., 1997). Moreover, the significance of EDHF in the regulation of arterial functions in human beings has also been reported (Urakami-Harasawa et al., 1997). However, the importance of NO-independent factors (i.e. EDHF and PGI2) in endothelium-dependent relaxations in the rat renal arteries is still unclear (Nagao et al., 1992; Zygmunt et al., 1995).
The aims of the present study are (1) to determine if factors other than NO and PGI2 are involved in muscarinic agonists-induced endothelium-dependent relaxations of rat isolated renal arteries; and (2) to characterize the possible mechanisms underlying this response.
Methods
Isolated renal artery preparations and functional study
Adult Sprague-Dawley rats (300–400 g) of either sex were killed by CO2 inhalation followed by exsanguination. This procedure was approved by the RMIT Animal Experimentation Ethics Committee and is in compliance with the guidelines of the Australian National Health & Medical Research Council. Kidneys with the renal pedicle attached were removed and placed in a physiological salt solution (PSS) at room temperature in a Petri dish. The renal artery and its branches were isolated under a dissection microscope (Olympus SZ-30). The third branch of the renal artery (outer diameter of 350–450 μM) was cut into segments of around 1 mm in length for functional study. In some experiments, the endothelium was removed by rubbing the inner surface of the segment with a stainless steel wire.
The mechanical activity of the segment was investigated using a wire myograph (Model 410A, J.P. Trading I/S, Denmark). Briefly, two parallel stainless steel wires of 50 μm diameter were threaded through the lumen of the segment: one wire was fixed to a displacement micrometer which controlled the distance between the two wires, the other one was connected to a tension transducer. The preparation was placed in a chamber containing 6 ml PSS, which was maintained at 36±1°C and gassed with 5% CO2 and 95% O2. The isometric tension of the vessel wall was displayed and recorded with a MacLab data recording system (MacLab/4, Model MK III, AD Instruments Pty. Ltd., Australia).
The PSS had the following composition (mM): NaCl 118, KCl 4.7, NaHCO3 25, MgSO4 0.45, KH2PO4 1.03, CaCl2 2.5, D-glucose 11.1, disodium edetate 0.067, and ascorbic acid 0.14. In some experiments, high-K+ PSS or K+-free PSS was used. The high-K+ PSS was prepared by replacing part of the NaCl in the normal PSS with an equal molar concentration of KCl. The final concentration of K+ was adjusted before use by mixing high-K+ PSS with normal PSS. The K+-free PSS was prepared by replacing KCl and KH2PO4 with NaCl and NaH2PO4, respectively.
Experimental protocols
After an equilibration period of 10 min, the tissue was normalized to 90% of the inner circumference that corresponds to a 100 mmHg blood pressure (Mulvany & Halpern, 1977), using a non-linear curve-fitting programme developed by McPherson (1992). This setting represents a resting tension of 2–3 mN under the present experimental condition. Vasodilator-induced relaxations were observed after the segment was contracted with 0.1 or 1 μM phenylephrine (as described in Results). After control responses were obtained, the segment was treated with one of various blocking drugs, and the relaxant responses were repeated in the presence of this drug. In studying NO-independent relaxations, the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) of 100 μM was added before the beginning of experimentation and was present throughout. During each experiment, the PSS was changed every 15–20 min.
Drugs
The following drugs were used: acetylcholine chloride (Sigma Chemical Co., St. Louis, MO, U.S.A.), 4-aminopyridine (4-AP, Sigma), apamin (Sigma), atropine sulphate (Sigma), carbachol (carbamylcholine chloride, Sigma), charybdotoxin (ChTX, Auspep, Melbourne, Australia), glibenclamide (Sigma), iberiotoxin (IbTX, Auspep), indomethacin (Merck Sharp & Dohme (Australia) Pty. Ltd.), (±) miconazole nitrate (Sigma), Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME, Sigma), 17-octadecynoic acid (17-ODYA, Sigma), ODQ (1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one) (Cayman Chemical Company, distributed by Sapphire Bioscience, NSW, Australia), ouabain (G-strophanthin, Sigma), L-phenylephrine hydrochloride (Sigma), sodium nitroprusside (SNP, Sigma), tetraethylammonium chloride (TEA, Sigma).
Stock solutions of various drugs were made by dissolving them in distilled water, except for miconazole, glibenclamide and ODQ, which were dissolved in 100% dimethylsulphoxide; 17-ODYA, which was dissolved in 100% ethanol; and indomethacin, which was dissolved in 5 mM Na2CO3. Stock solutions were added directly into the PSS during experimentation. The solvents had no effect in the final concentrations in which they were present in the PSS.
Data and statistical analysis
The tension of the vessel wall was measured in mN. Relaxations were expressed as percentage reductions of phenylephrine-produced contraction. EC50 values were calculated by linear regression using the linear portion of concentration-response curves. Data were presented as mean±standard error of the mean. The mean data were analysed with one-way analysis of variance (one-way ANOVA) followed by Student-Newman-Keuls Test. A value of P<0.05 was regarded as statistically significant.
Results
Phenylephrine-induced contractions
In the rat renal artery segments with intact endothelium, phenylephrine (10 nM–30 μM) produced concentration-dependent contractions, with an EC50 value of 0.34±0.044 μM (n=6). The maximal response (Rmax) produced by 30 μM phenylephrine was 14.7±1.7 mN (n=6). The contraction produced by 1 μM phenylephrine had a mean tension of 12.1±0.82 mN, n=24).
Carbachol and acetylcholine-induced relaxations
In segments contracted with 1 μM phenylephrine, carbachol (10 nM–10 μM) elicited concentration-dependent relaxations, which were blocked by atropine (1 μM) and were absent in segments denuded of endothelium (data not shown). The relaxant response was markedly reduced by the NOS inhibitor L-NAME (100 μM) and the inhibitor of NO-sensitive guanylate cyclase, ODQ (1 μM) (Figure 1). With L-NAME, the EC50 values of carbachol were shifted from 0.15±0.045 to 0.21±0.044 μM (n=6) and the Rmax was decreased from 88.5±2.2 to 48.3±9.6% (n=6). With ODQ, the EC50 values of carbachol was shifted from 0.15±0.026 to 0.36±0.11 μM (n=4) and the Rmax was decreased from 83.1±2.8 to 30.1±5.9% (n=4).
Figure 1.
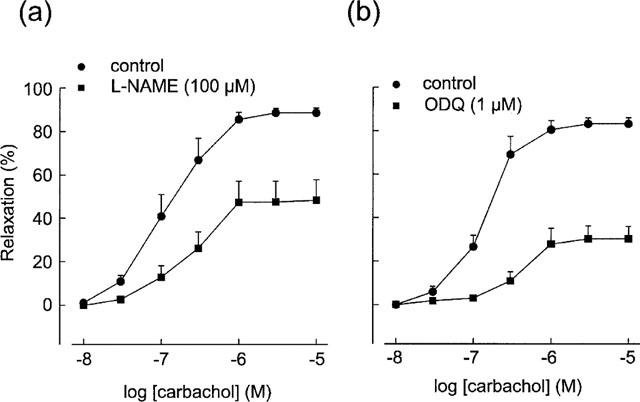
Effects of the nitric oxide synthase inhibitor L-NAME (n=6) and the NO-sensitive guanylate cyclase inhibitor ODQ (n=4) on carbachol-induced relaxations in the rat renal arteries contracted with phenylephrine (1 μM). Both L-NAME and ODQ significantly reduced the relaxations. Relaxations were expressed as percentage reductions of phenylephrine-induced tone and data are presented as mean±standard error of the mean (s.e.mean).
Acetylcholine (0.1–10 μM) produced similar concentration-dependent relaxations which were endothelium-dependent and blocked by atropine (1 μM) (data not shown). L-NAME (100 μM) reduced the maximal acetylcholine-induced relaxation from 79.4±4.9 to 22.9±7.7% (P<0.001, n=5).
Effect of the phenylephrine concentration on L-NAME resistant relaxations to carbachol and acetylcholine
The residual relaxations produced by carbachol and acetylcholine in the presence of 100 μM L-NAME and 1 μM phenylephrine were difficult to study because of their small size. However, the L-NAME-resistant relaxations were more pronounced when the concentration of phenylephrine was lowered. Phenylephrine at 0.1 μM in the presence of L-NAME produced a mean contraction of 7.6±1.1 mN (n=10), and carbachol and acetylcholine then produced prominent relaxations. Typical tracings comparing the magnitude of L-NAME resistant relaxations to carbachol in the presence of 1 and 0.1 μM phenylephrine are illustrated in Figure 2.
Figure 2.
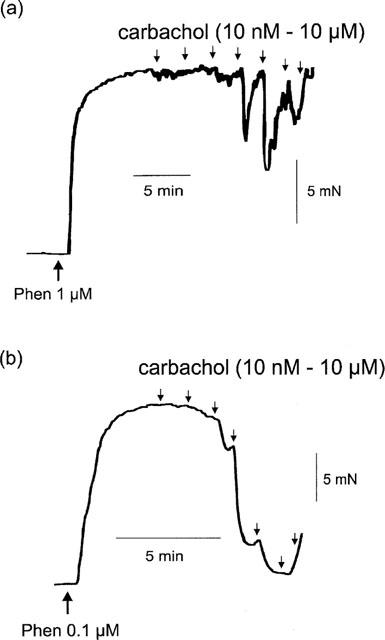
Typical tracings showing the different magnitude of relaxations induced by carbachol (10 nM–10 μM) in artery segments incubated with L-NAME (100 μM) and contracted with (a) 1 and (b) 0.1 μM phenylephrine. Carbachol was added cumulatively as indicated by the downward arrows.
Lack of effect of ODQ and indomethacin on L-NAME resistant relaxations
In the presence of L-NAME (100 μM) and 0.1 μM phenylephrine, carbachol (10 nM–10 μM) produced consistent endothelium-dependent and atropine-sensitive relaxations. These relaxations were not significantly affected by ODQ (1 μM) (Figure 3a), whereas the response to SNP (10 nM), which produced nearly full relaxation under the same conditions, was abolished (data not shown). The EC50 values of carbachol before and after ODQ treatment were 0.17±0.016 and 0.20±0.055 μM, respectively (P>0.05, n=5); the Rmax values were 86.8±3.6 and 79.5±9.1%, respectively (P>0.05, n=5).
Figure 3.
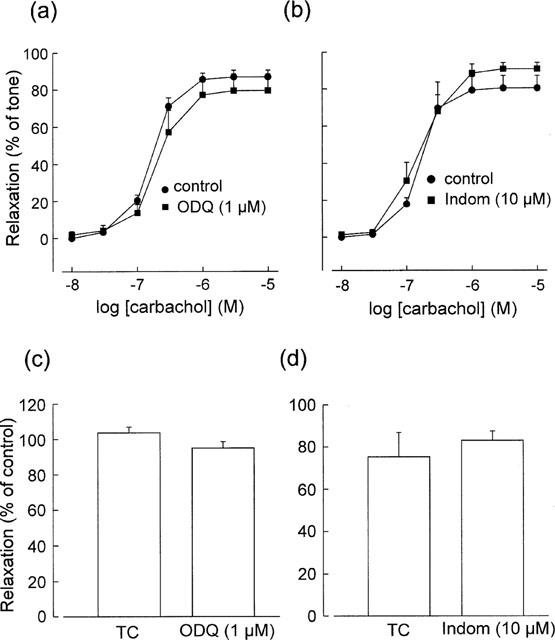
Effects of ODQ and the cyclo-oxygenase inhibitor indomethacin (Indom) on L-NAME-resistant relaxations induced by carbachol (a,b) and acetylcholine (1 μM) (c,d) in artery segments contracted with phenylephrine (0.1 μM). Acetylcholine-induced relaxations have been converted to percentages of the initial control responses and compared with parallel time-control (TC) responses. Data are means±s.e.mean (n=3–5).
The cyclo-oxygenase inhibitor indomethacin (10 μM) did not significantly affect carbachol-induced relaxations (Figure 3b). The EC50 values for carbachol before and after indomethacin treatment were 0.16±0.014 and 0.19±0.056 μM, respectively (P>0.05, n=4); the Rmax values were 80.3±6.7 and 90.7±3.5%, respectively (P>0.05, n=4).
In the presence of L-NAME and 0.1 μM phenylephrine, acetylcholine (0.1–1 μM) also produced concentration-dependent relaxations. However, responses to acetylcholine were less consistent with time than were those to carbachol; therefore, acetylcholine was added at only one concentration (1 μM) and the responses were converted to percentages of the initial control responses and compared with parallel time controls. Acetylcholine-induced relaxations were not affected by ODQ (1 μM, n=4) or indomethacin (10 μM, n=3) (Figure 3c,d).
Effects of K+ channel blockers on L-NAME-resistant relaxations
The non-selective K+ channel blocker TEA (1 mM) and the large-conductance Ca2+-activated K+ channel blockers ChTX (200 nM) and IbTX (100 nM) produced statistically significant 2.5–6.3 fold rightward shifts of the concentration-response curve of carbachol-induced relaxations in segments contracted with phenylephrine (0.1 μM), (Figure 4a–c). The effects of TEA, ChTX and IbTX on the EC50 of carbachol and the Rmax of the relaxations are summarized in Table 1.
Figure 4.
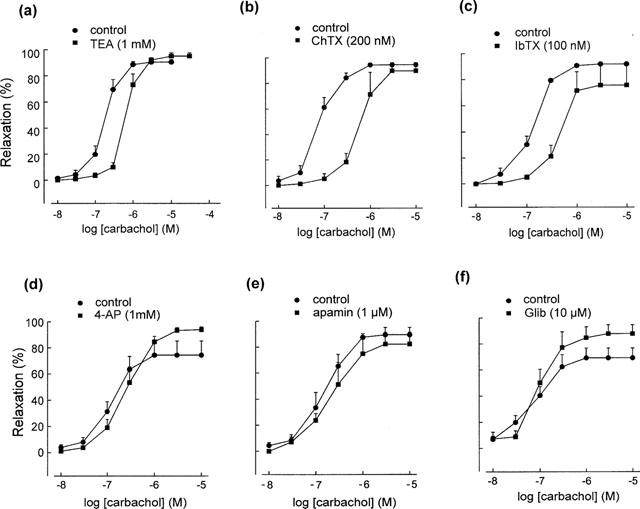
Effects of various K+ channel blockers on carbachol-induced, L-NAME-resistant relaxations. The relaxations were significantly reduced by tetraethylammonium (TEA), charybdotoxin (ChTX) and iberiotoxin (IbTX), but were not affected by 4-aminopyridine (4-AP), apamin or glibenclamide (Glib). Relaxations are expressed as % reduction of 0.1 μM phenylephrine-induced tone. Data are means±s.e.mean (n=3–6).
Table 1.
Effects of TEA, ChTX, IbTX, ouabain, miconazole and K+ on the EC50 and Rmax of L-NAME resistant relaxations induced by carbachol
The relaxant responses were not significantly altered by the voltage-sensitive K+ channel blocker 4-AP (1 mM), the small-conductance Ca2+-activated K+ channel blocker apamin (1 μM) or the ATP-sensitive K+ channel blocker glibenclamide (10 μM) (Figure 4d–f).
The relaxation induced by acetylcholine (1 μM) was reduced by ChTX (200 nM), the mean data in time control and ChTX-treated tissues being 84.7±11.3 and 29.5±5.9% (expressed as % of the initial control response, P<0.005, n=5–6), respectively. Considerably variable results were obtained with TEA (1 and 10 mM) and IbTX (150 nM), and there was no significant difference between the mean data (not shown).
Effect of ouabain on L-NAME resistant relaxations
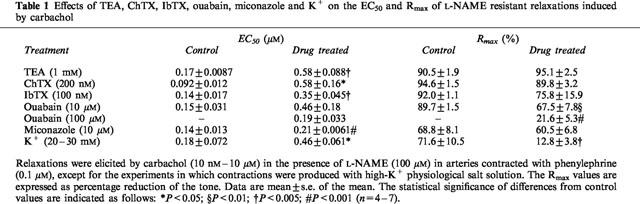
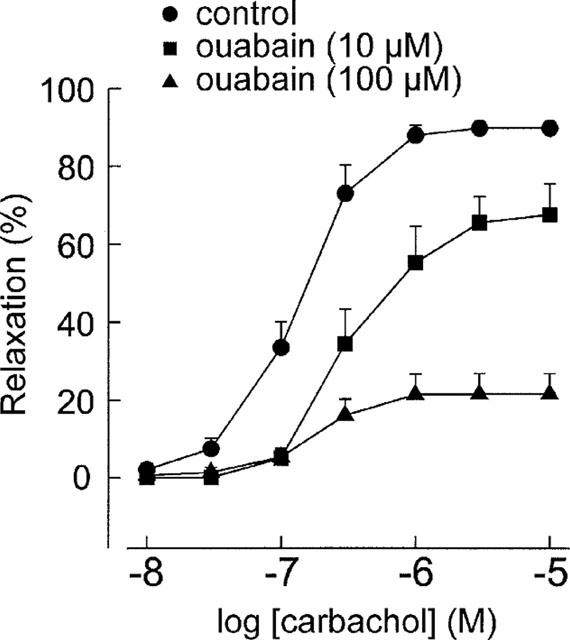
In L-NAME-treated segments, the Na+-K+ adenosine triphosphatase (Na+-K+ ATPase) inhibitor ouabain (10 and 100 μM) produced slowly-developing but transient contractions in five of the nine preparations used with the maximal developed tension of 0.6–7.5 mN, which appeared about 5 min after the addition of ouabain. Then the tension tended to decline. Carbachol experiments were started 20 min after ouabain treatment, when the tension had nearly returned to the baseline. Ouabain did not significantly change the final tone elicited by 0.1 μM phenylephrine. Ouabain (10 and 100 μM) reduced carbachol-induced relaxations in a concentration-dependent manner (Figure 5). The EC50 values of carbachol and the Rmax of carbachol-induced relaxations before and after ouabain were shown in Table 1. Typical tracings showing the effect of 100 μM ouabain on carbachol-induced relaxations were shown in Figure 6.
Figure 5.
Effect of the Na+-K+ adenosine triphosphatase (Na+-K+ ATPase) inhibitor ouabain on L-NAME-resistant relaxations induced by carbachol in artery segments contracted with 0.1 μM phenylephrine. Ouabain significantly reduced the relaxations in a concentration-dependent manner. Data are means±s.e.mean (n=5).
Figure 6.
Typical tracings showing the effects of the Na+-K+ ATPase inhibitor ouabain on L-NAME-resistant relaxations induced by carbachol. Carbachol was added cumulatively as indicated by the downward arrows.
Ouabain (100 μM) also significantly reduced relaxations induced by acetylcholine (1 μM). The mean data of the relaxation in time control and ouabain-treated tissues were 98.6±4.9% (expressed as % of the initial response) and 51.9±15.0% (P<0.05, n=4), respectively.
Ouabain at 100 μM had no effect on the relaxations induced by SNP (1–30 nM) under the same condition (data not shown).
Effects of high-K+ PSS on L-NAME-resistant relaxations
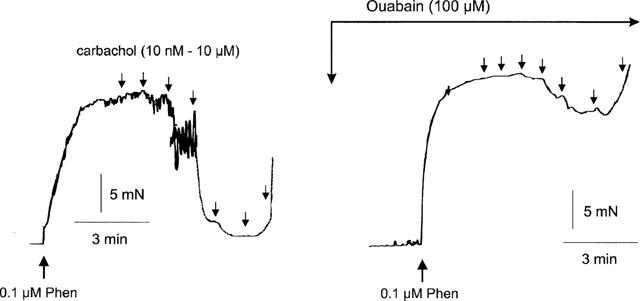
In segments treated with L-NAME, PSS containing 20–30 mM K+ produced contractions. The concentration of K+ in each experiment was adjusted to produce a contraction matching that to 0.1 μM phenylephrine, thus eliminating possible effects of differences in contractile tone. Under these conditions, carbachol-induced relaxations were virtually absent (Figure 7). The EC50 of carbachol and the Rmax values in control and high-K+ PSS-contracted tissues were given in Table 1.
Figure 7.
When contractions of arteries were induced by high K+ PSS, carbachol-induced L-NAME-resistant relaxations were almost absent, in contrast to those when contractions were induced with 0.1 μM phenylephrine (control).
The relaxation-induced by acetylcholine (1 μM) was also significantly suppressed in tissues contracted with 20–30 mM K+. The mean data of acetylcholine-induced relaxations in control and high-K+ PSS-contracted tissues were 59.4±16.4 and 17.2±3.0% (% of tone, P<0.05, n=5), respectively.
Effect of ouabain on the relaxations induced by K+
In segments treated with L-NAME, changing to K+-free PSS increased the basal tone, the maximal tension produced being 10.7±1.3 mN (n=3). Then, after phenylephrine (0.1 μM), addition of K+ (5 mM) produced a 90.5±3.9% (n=3) relaxation of the induced tone. In the presence of ouabain (100 μM), the K+-induced relaxations were significantly decreased compared with those in control tissues, the mean relaxation being 28.4±5.1% (P<0.001, n=3). In segments denuded of endothelium, the K+-induced relaxations were not significantly changed from those in endothelium-intact segments, the mean relaxation being 83.0±5.4% (P>0.05 vs control, n=3). Tracings illustrating the relaxations induced by addition of K+ in control, ouabain-treated and endothelium-denuded segments are shown in Figure 8.
Figure 8.
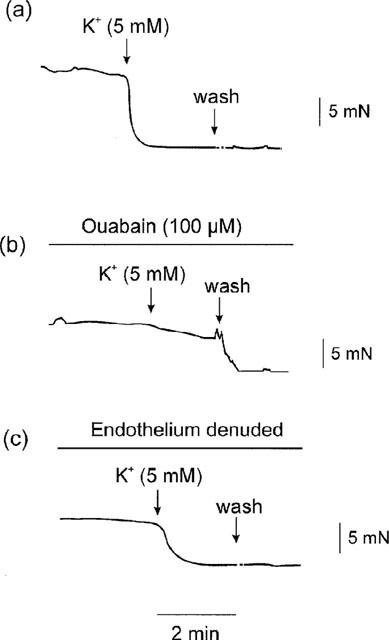
Tracings showing: (a) in arteries in K+-free PSS plus phenylephrine (0.1 μM) in the presence of L-NAME, addition of K+ (5 mM) to the K+-free PSS produced relaxations; (b) the K+-induced relaxation was significantly suppressed after ouabain (100 μM); (c) the K+-induced relaxation was still present after endothelium-denudation.
Effects of cytochrome P450 inhibitors on L-NAME resistant relaxations
Treatment with the cytochrome P450 inhibitor miconazole (10 μM) produced a statistically significant 1.5 fold rightward shift of the concentration-response curve for carbachol-induced relaxations in the presence of L-NAME (Figure 9a), but the Rmax value was not significantly changed (Table 1). Another P450 inhibitor, 17-ODYA (3 and 10 μM), did not significantly affect carbachol-induced relaxations (Figure 9b).
Figure 9.
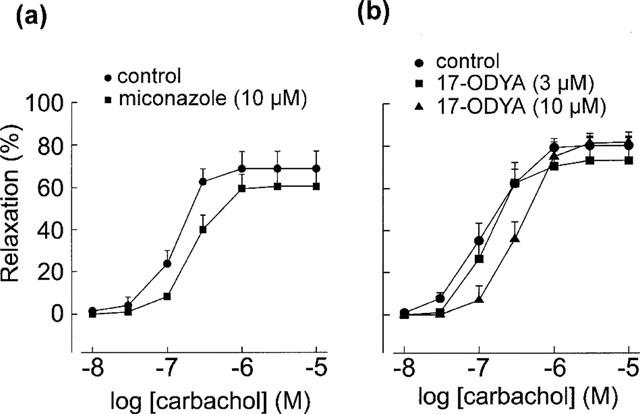
Effects of cytochrome P450 inhibitor miconazole (n=6) and 17-octadecynoic acid (17-ODYA) (n=5) on L-NAME-resistant relaxations induced by carbachol. The relaxation was slightly yet significantly reduced by miconazole but was not significantly changed by 17-ODYA.
Discussion
The muscarinic agonists carbachol and acetylcholine produced endothelium-dependent relaxations in segments of rat renal arteries contracted with 1 μM phenylephrine. The relaxations were blocked by atropine, indicating that the response was mediated by endothelial muscarinic receptors. Inhibition of NOS with L-NAME or of NO-sensitive guanylate cyclase with ODQ reduced but did not abolish the relaxations. This suggests that one or more endothelium-derived mediator(s) other than EDNO is likely to be involved in mediating the smooth muscle relaxation. Similar observations have been made with the rabbit carotid artery (Dong et al., 1997) and the rat aorta (Chen et al., 1988).
The NO-independent, carbachol-induced relaxation was more prominent when the artery segment was contracted with 0.1 μM phenylephrine than it was when contracted with 1 μM phenylephrine. Cheung et al. (1999) found that the ability of hyperpolarization to induce relaxation is limited within a certain range of membrane potential, and hyperpolarization is ineffective in producing relaxation when the contraction is mediated by membrane potential-independent mechanisms. Therefore, a possible explanation for the present finding is that different mechanisms might be involved in the contractions produced by phenylephrine at low and high concentrations.
Carbachol-induced relaxations after contraction with 0.1 μM phenylephrine in the presence of 100 μM L-NAME were not further decreased by ODQ (which completely abolished relaxations induced by the NO donor SNP), or the cyclo-oxygenase inhibitor indomethacin, indicating that under these conditions, the relaxation was totally independent of the NO/cyclic GMP pathway and was unlikely to be mediated by vasorelaxant prostanoids such as PGI2 (see Vane et al., 1982; Zygmunt et al., 1998). On the other hand, the relaxations were virtually absent when contraction was induced by depolarizing the smooth muscle with high-K+ PSS, suggesting that the putative EDHF might be involved in mediating this response (Adeagbo & Triggle, 1993; Zygmunt et al., 1994; Petersson et al., 1998).
The mechanism(s) underlying EDHF-induced smooth muscle hyperpolarization and relaxation has not been fully elucidated. Many pharmacological and electrophysiological studies in various tissues and species suggested that EDHF produces its biological effect through the opening of K+ channels on vascular smooth muscle cells (Popp et al., 1996; Zygmunt & Hogestatt, 1996; McCulloch et al., 1997; Petersson et al., 1997; Yamakawa et al., 1997; Zygmunt et al., 1997; Hayabuchi et al., 1998; Nishiyama et al., 1998; Yamanaka et al., 1998), although the types of K+ channels identified are heterogeneous. In the present study, the non-selective K+ channel blocker TEA and the large-conductance Ca2+-activated K+ channel blockers ChTX and IbTX reduced the NO-independent relaxations induced by low concentrations of carbachol but did not change the maximal response. Similarly, ChTX reduced acetylcholine-induced relaxations. Although TEA and IbTX had inconsistent effects on acetylcholine-induced relaxations, these results indicate that activation of K+ channels is at least partly involved in acetylcholine and carbachol-induced EDHF responses. However, the site of actions of these blockers is not clear. The inhibitory effects of the K+ channel blockers might be 2 fold: blockade of the K+ channels in the smooth muscle that prevents K+ efflux and subsequent hyperpolarization; and/or blockade of K+ channels in the endothelial cells thus preventing the release of EDHF (Edwards & Weston, 1998). On the other hand, these data suggest that other K+-channel independent mechanisms may also be involved.
A major finding of the present study is that the NO-independent relaxations were reduced in a concentration-dependent manner by the Na+-K+ ATPase inhibitor ouabain, which is consistent with the observations by Kitagawa et al. (1994) in the rabbit renal artery. The involvement of Na+-K+ ATPase in endothelium-dependent hyperpolarization and NO-independent relaxations induced by acetylcholine was also reported in dog coronary arteries and horse penile small arteries (Feletou & Vanhoutte, 1988; Prieto et al., 1998), while opposite results were also reported (Suzuki, 1988; Chen et al., 1989; Zygmunt & Hogestatt, 1996). It has been established that Na+-K+ ATPase activity is a electrogenic Na+-K+ pump, which extrudes Na+ and brings in K+ across the plasma membrane in a ratio of approximately 3:2, resulting in a net cumulation of intracellular negative charge (hyperpolarization) (see Fleming, 1980). The inhibitory effect of ouabain suggests that the relaxation produced by EDHF may be through Na+-K+ ATPase activation. Addition of K+ (5 mM) to the K+-free PSS produced nearly full relaxation of the K+-free PSS plus phenylephrine-induced tone, and the K+-induced relaxation was also observed in endothelium-denuded segments, suggesting the functional presence of Na+-K+ ATPase activity in the renal arterial smooth muscle cells (Hendrickx & Casteels, 1974; Webb & Bohr, 1978). Moreover, the inhibitory effect of ouabain on K+-induced relaxations indicates that ouabain at 100 μM has completely blocked the Na+-K+ ATPase activity. However, other possibilities underlying the effect of ouabain cannot be ruled out. For example, ouabain may depolarize the smooth muscle cell membrane as a consequence of Na+-K+ ATPase inhibition, which in turn offsets the effect of EDHF. In addition, the depolarization of endothelial cells may inhibit the release of endothelium-derived factors (Luckhoff & Busse, 1990). On the other hand, activation of Na+-K+ ATPase did not appear to be effective in counteracting contraction caused by high K+-induced depolarization, since in segments depolarized with the PSS containing 20–30 mM K+, carbachol- and acetylcholine-induced relaxations were nearly abolished. This might be because that Na+-K+ ATPase plays a major functional role in vasorelaxation only when the total extracellular K+ concentration is within a relatively low range (e.g. <5 mM) (McCarron & Halpern, 1990).
Evidence has shown that ouabain at high concentrations may release noradrenaline from perivascular nerves and thereby produce contractions (Toda, 1980). Although arteries contracted with the higher concentration of phenylephrine (1 μM) relaxed poorly in the presence of L-NAME, this mechanism does not appear to play a major role in the ouabain-induced inhibition of the L-NAME resistant relaxations, since if release of noradrenaline contributed appreciably to the inhibitory effect, ouabain should have produced contractions similar to those induced by phenylephrine. In the present study, however, ouabain only produced slight and transient contractions in some of the preparations studied. Moreover, ouabain did not change the relaxations induced by the NO donor SNP, suggesting that a non-specific antagonism was unlikely. The tension developed in K+-free PSS might be explained by depolarization resulting from K+ depletion caused by inhibition of Na+-K+ ATPase (Hendrickx & Casteels, 1974).
The chemical identity of EDHF is still controversial. In the present study, carbachol-induced NO-independent relaxations in rat renal arteries were not affected or only slightly reduced by the cytochrome P450 inhibitors 17-ODYA and miconazole. This finding does not support the view that EDHF might be an endothelium-derived cytochrome P450 metabolite (Hecker et al., 1994; Campbell et al., 1996; Chen & Cheung, 1996; Popp et al., 1996; Dong et al., 1997; Hayabuchi et al., 1998). Similar results indicating that EDHF is unlikely to be a cytochrome P450 metabolite were also obtained in other blood vessels from various species (Zygmunt et al., 1996; Urakami-Harasawa et al., 1997; Van de Voorde & Vanheel, 1997; Vanheel & Van de Voorde, 1997; Petersson et al., 1998; Yamanaka et al., 1998). It should be noted that in contrast to other P450 inhibitors, 17-ODYA is not associated with major non-specific effects, therefore the marginal effect of miconazole in the present study might be due to other non-specific activities (such as inhibition of certain types of K+ channels) of this class of compounds rather than inhibition of the P450 enzyme (Zygmunt et al., 1996; Edwards et al., 1996; Vanheel & Van de Voorde, 1997).
In the rat hepatic artery Edwards et al. (1998) demonstrated recently that EDHF released by acetylcholine might be the K+ ion per se that effluxes through ChTX- and apamin-sensitive K+ channels on endothelial cells, and the hyperpolarization and relaxation produced by the increase in myoendothelial K+ concentration involve the activation of both K+ channels and ouabain-sensitive Na+-K+ ATPase in the smooth muscle cells. It was also demonstrated that acetylcholine at 10 μM can raise the K+ concentration in the myoendothelial space by 5.9±1.0 mM, a concentration that matches the K+ concentration used in the present study that elicited nearly full relaxation. This hypothesis is also supported by the findings of Prior et al. (1998), using rabbit small renal arteries, in which addition of K+ induced relaxation mainly through the activation of Na+-K+ ATPase in the smooth muscle membrane. The present study did not provide direct evidence showing K+ per se as being responsible for the NO-independent relaxations induced by muscarinic agonists; however, the postulation that EDHF in the rat renal artery is K+ would provide a plausible explanation for the findings.
In conclusion, the present study demonstrated that in rat isolated renal arteries with intact endothelium, muscarinic agonists produce relaxations that are mediated by EDNO and another non-NO/non-PGI2 hyperpolarizing factor (EDHF). The inhibition by ouabain of NO-independent relaxations suggests the possible involvement of Na+-K+ ATPase activation in EDHF responses, although other mechanisms cannot be totally ruled out. This hyperpolarizing factor does not appear to be a cytochrome P450 metabolite but it could be K+ itself released from endothelium through ChTX-sensitive K+ channels, resulting in the Na+-K+ ATPase-dependent hyperpolarization and relaxation of smooth muscle cells. This study also supports the view that EDHF might not be a single factor, and its nature and cellular targets might differ between vascular regions and species (Triggle et al., 1999).
Acknowledgments
This research was supported by a grant from the National Health and Medical Research Council and a grant from the Australian Smoking and Health Foundation, which also awarded a postgraduate scholarship to F. Jiang.
Abbreviations
- 4-AP
4-aminopyridine
- AMP
adenosine monophosphate
- ChTX
charybdotoxin
- EC50
concentration producing 50% of maximal effect
- EDHF
endothelium-derived hyperpolarizing factor
- EDNO
endothelium-derived nitric oxide
- EDRF
endothelium-derived relaxing factor
- GMP
guanosine monophosphate
- IbTX
iberiotoxin
- L-NAME
Nω-nitro-L-arginine methyl ester
- NO
nitric oxide
- NOS
nitric oxide synthase
- ODQ
1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one
- 17-ODYA
17-octadecynoic acid
- PGI2
prostacyclin
- PSS
physiological salt solution
- Rmax
maximal response
- SNP
sodium nitroprusside
- TEA
tetraethylammonium
References
- ADEAGBO A.S., TRIGGLE C.R. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- BRANDES R.P., BEHRA A., LEBHERZ C., BOGER R.H., BODE-BOGER S.M., PHIVTHONG-NGAM L., MUGGE A. NG-nitro-L-arginine- and indomethacin-resistant endothelium-dependent relaxation in the rabbit renal artery: effect of hypercholesterolemia. Atherosclerosis. 1997;135:49–55. doi: 10.1016/s0021-9150(97)00145-7. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GERBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHEN G., CHEUNG D.W. Modulation of endothelium-dependent hyperpolarization and relaxation to acetylcholine in rat mesenteric artery by cytochrome P450 enzyme activity. Circ. Res. 1996;79:827–833. doi: 10.1161/01.res.79.4.827. [DOI] [PubMed] [Google Scholar]
- CHEN G., HASHITANI H., SUZUKI H. Endothelium-dependent relaxation and hyperpolarization of canine coronary artery smooth muscles in relation to the electrogenic Na-K pump. Br. J. Pharmacol. 1989;98:950–956. doi: 10.1111/j.1476-5381.1989.tb14625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEUNG D.W., CHEN G., MACKAY M.J., BURNETTE E. Regulation of vascular tone by endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 1999;26:172–175. doi: 10.1046/j.1440-1681.1999.03008.x. [DOI] [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., GALIPEAU D., COLE W.C., TRIGGLE C.R. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br. J. Pharmacol. 1997;120:695–701. doi: 10.1038/sj.bjp.0700945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Endothelium-derived hyperpolarizing factor–a critical appraisal. Prog. Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., ZYGMUNT P.M., HOGESTATT E.D., WESTON A.H. Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. Br. J. Pharmacol. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEMING W.W. The electrogenic Na+, K+-pump in smooth muscle: physiologic and pharmacologic significance. Annu. Rev. Pharmacol. Toxicol. 1980;20:129–149. doi: 10.1146/annurev.pa.20.040180.001021. [DOI] [PubMed] [Google Scholar]
- FROLICH J.C. Prostacyclin in hypertension. J. Hypertens. Suppl. 1990;8:S73–S78. [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GERBER R.T., ANWAR M.A., POSTON L. Enhanced acetylcholine induced relaxation in small mesenteric arteries from pregnant rats: an important role for endothelium-derived hyperpolarizing factor (EDHF) Br. J. Pharmacol. 1998;125:455–460. doi: 10.1038/sj.bjp.0702099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYABUCHI Y., NAKAYA Y., MATSUOKA S., KURODA Y. Endothelium-derived hyperpolarizing factor activates Ca2+-activated K+ channels in porcine coronary artery smooth muscle cells. J. Cardiovasc. Pharmacol. 1998;32:642–649. doi: 10.1097/00005344-199810000-00018. [DOI] [PubMed] [Google Scholar]
- HAYAKAWA H., HIRATA Y., SUZUKI E., KAKOKI M., KIKUCHI K., NAGANO T., HIROBE M., OMATA M. Endothelium-derived relaxing factor in the kidney of spontaneously hypertensive rats. Life Sci. 1995;56:PL401–408. doi: 10.1016/0024-3205(95)00157-2. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolic in mammals. J. Physiol. (Lond) 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENDERICKX H., CASTEELS R. ELectrogenic sodium pump in arterial smooth muscle cells. Pflugers Arch. 1974;346:299–306. doi: 10.1007/BF00596185. [DOI] [PubMed] [Google Scholar]
- KESSLER P., LISCHKE V., HECKER M. Etomidate and thiopental inhibit the release of endothelium-derived hyperpolarizing factor in the human renal artery. Anesthesiology. 1996;84:1485–1488. doi: 10.1097/00000542-199606000-00025. [DOI] [PubMed] [Google Scholar]
- KITAGAWA S., YAMAGUCHI Y., KUNITOMO M., SAMESHIMA E., FUJIWARA M. NG-nitro-L-arginine-resistant endothelium-dependent relaxation induced by acetylcholine in the rabbit renal artery. Life Sci. 1994;55:491–498. doi: 10.1016/0024-3205(94)00741-1. [DOI] [PubMed] [Google Scholar]
- LUCKHOFF A., BUSSE R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- MCCARRON J.G., HALPERN W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am. J. Physiol. 1990;259:H902–H908. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH A.I., BOTTRILL F.E., RANDALL M.D., HILEY C.R. Characterization and modulation of EDHF-mediated relaxations in the rat isolated superior mesenteric arterial bed. Br. J. Pharmacol. 1997;120:1431–1438. doi: 10.1038/sj.bjp.0701066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCPHERSON G.A. Optimal conditions for assessing vascular reactivity in small resistance arteries in small vessel myograph. Clin. Exp. Pharmacol. Physiol. 1992;19:815–825. doi: 10.1111/j.1440-1681.1992.tb00420.x. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends Pharmacol. Sci. 1997;18:252–256. [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NAGAO T., ILLIANO S., VANHOUTTE P.M. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am. J. Physiol. 1992;263:H1090–H1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- NISHIYAMA M., HASHITANI H., FUKUTA H., YAMAMOTO Y., SUZUKI H. Potassium channels activated in the endothelium-dependent hyperpolarization in guinea-pig coronary artery. J. Physiol. (Lond) 1998;510:455–465. doi: 10.1111/j.1469-7793.1998.455bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., HOGESTATT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.M., JONSSON P., HOGESTATT E.D. Characterization of endothelium-dependent relaxation in guinea pig basilar artery–effect of hypoxia and role of cytochrome P450 mono-oxygenase. J. Vasc. Res. 1998;35:285–294. doi: 10.1159/000025595. [DOI] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. (Lond) 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIETO D., SIMONSEN U., HERNANDEZ M., GARCIA-SACRISTAN A. Contribution of K+ channels and ouabain-sensitive mechanism to the endothelium-dependent relaxations of horse penile small arteries. Br. J. Pharmacol. 1998;123:1609–1620. doi: 10.1038/sj.bjp.0701780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIOR H.M., WEBSTER N., QUINN K., BEECH D.J., YATES M.S. K+-induce dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovasc. Res. 1998;37:780–790. doi: 10.1016/s0008-6363(97)00237-x. [DOI] [PubMed] [Google Scholar]
- QUILLEY J., FULTON D., MCGIFF J.C. Hyperpolarizing factors. Biochem. Pharmacol. 1997;54:1059–1070. doi: 10.1016/s0006-2952(97)00039-7. [DOI] [PubMed] [Google Scholar]
- SIEGEL G., SCHNALKE F., STOCK G., GROTE J. Prostacyclin, endothelium-derived relaxing factor and vasodilatation. Adv. Prostaglandin Thromboxane Leukot. Res. 1989;19:267–270. [PubMed] [Google Scholar]
- SUZUKI H. The electrogenic Na-K pump does not contribute to endothelium-dependent hyperpolarization in the rabbit ear artery. Eur. J. Pharmacol. 1988;156:295–297. doi: 10.1016/0014-2999(88)90337-8. [DOI] [PubMed] [Google Scholar]
- TODA N. Mechanisms of ouabain-induced arterial muscle contraction. Am. J. Physiol. 1980;239:H199–H205. doi: 10.1152/ajpheart.1980.239.2.H199. [DOI] [PubMed] [Google Scholar]
- TRIGGLE C.R., DONG H., WALDRON G.J., COLE W.C. Endothelium-derived hyperpolarizing factor(s): species and tissue heterogeneity. Clin. Exp. Pharmacol. Physiol. 1999;26:176–179. doi: 10.1046/j.1440-1681.1999.03007.x. [DOI] [PubMed] [Google Scholar]
- URAKAMI-HARASAWA L., SHIMOKAWA H., NAKASHIMA M., EGASHIRA K., TAKESHITA A. Importance of endothelium-derived hyperpolarizing factor in human arteries. J. Clin. Invest. 1997;100:2793–2799. doi: 10.1172/JCI119826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DE VOORDE J., VANHEEL B. Influence of cytochrome P-450 inhibitors on endothelium-dependent nitro-L-arginine-resistant relaxation and cromakalim-induced relaxation in rat mesenteric arteries. J. Cardiovasc. Pharmacol. 1997;29:827–832. doi: 10.1097/00005344-199706000-00018. [DOI] [PubMed] [Google Scholar]
- VANE J.R., BUNTING S., MONCADA S. Prostacyclin in physiology and pathophysiology. Int. Rev. Exp. Pathol. 1982;23:161–207. [PubMed] [Google Scholar]
- VANHEEL B., VAN DE VOORDE J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. J. Physiol. (Lond) 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANHOUTTE P.M., RUBANYI G.M., MILLER V.M., HOUSTON D.S. Modulation of vascular smooth muscle contraction by the endothelium. Annu. Rev. Physiol. 1986;48:307–320. doi: 10.1146/annurev.ph.48.030186.001515. [DOI] [PubMed] [Google Scholar]
- WEBB R.C., BOHR D.F. Potassium-induced relaxation as an indicator of Na+-K+ ATPase activity in vascular smooth muscle. Blood Vessels. 1978;15:198–207. doi: 10.1159/000158166. [DOI] [PubMed] [Google Scholar]
- YAMAKAWA N., OHHASHI M., WAGA S., ITOH T. Role of endothelium in regulation of smooth muscle membrane potential and tone in the rabbit middle cerebral artery. Br. J. Pharmacol. 1997;121:1315–1322. doi: 10.1038/sj.bjp.0701285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMANAKA A., ISHIKAWA T., GOTO K. Characterization of endothelium-dependent relaxation independent of NO and prostaglandins in guinea pig coronary artery. J. Pharmacol. Exp. Ther. 1998;285:480–489. [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS S.C., HOGESTATT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., LARSSON B., HOGESTATT E.D. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997;121:141–149. doi: 10.1038/sj.bjp.0701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HOGESTATT E.D. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996;117:1600–1606. doi: 10.1111/j.1476-5381.1996.tb15327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., PLANE F., PAULSSON M., GARLAND C.J., HOGESTATT E.D. Interactions between endothelium-derived relaxing factors in the rat hepatic artery: focus on regulation of EDHF. Br. J. Pharmacol. 1998;124:992–1000. doi: 10.1038/sj.bjp.0701893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., RYMAN T., HOGESTATT E.D. Regional differences in endothelium-dependent relaxation in the rat: contribution of nitric oxide and nitric oxide-independent mechanisms. Acta Physiol. Scand. 1995;155:257–266. doi: 10.1111/j.1748-1716.1995.tb09972.x. [DOI] [PubMed] [Google Scholar]
- ZYGMUNT P.M., WALDECK K., HOGESTATT E.D. The endothelium mediates a nitric oxide-independent hyperpolarization and relaxation in the rat hepatic artery. Acta Physiol. Scand. 1994;152:375–384. doi: 10.1111/j.1748-1716.1994.tb09819.x. [DOI] [PubMed] [Google Scholar]