

Abstract
The systemic administration of Δ9-tetrahydrocannabinol (2.5–7.5 mg kg−1) reduced hippocampal extracellular acetylcholine concentration and impaired working memory in rats.
Both effects were antagonized not only by the CB1 cannabinoid receptor antagonist SR141716A (0.5 mg kg−1, i.p.) but also unexpectedly by the D2 dopamine receptor antagonist S(−)-sulpiride (5, 10 and 25 mg kg−1, i.p.). Conversely, Δ9-tetrahydrocannabinol-induced memory impairment and inhibition of hippocampal extracellular acetylcholine concentration were potentiated by the subcutaneous administration of the D2 dopamine receptor agonist (−)-quinpirole (25 and 500 μg kg−1). The inhibition of hippocampal extracellular acetylcholine concentration and working memory produced by the combination of (−)-quinpirole and Δ9-tetrahydrocannabinol was suppressed by either SR141716A or S(−)-sulpiride.
Our findings suggest that impairment of working memory and inhibition of hippocampal extracellular acetylcholine concentration are mediated by the concomitant activation of D2 dopamine and CB1 cannabinoid receptors, and that D2 dopamine receptor antagonists may be useful in the treatment of the cognitive deficits induced by marijuana.
Keywords: Δ9-Tetrahydrocannabinol, D2 dopamine receptors, working memory, acetylcholine
Introduction
The effect of Cannabis sativa on cognitive functions is one of the most controversial debated questions pertaining to the consequences of marijuana use on health (Hall & Solowij, 1998; Thomas, 1993). While there is no question concerning the cognitive deficits present during marijuana intoxication, there is some doubt whether or not permanent memory deficits occur after chronic use (Thomas, 1993). Indeed, it has been argued that the cognitive deficits observed in heavy chronic marijuana users may represent antecedents, concomitants, or consequences from the chronic exposure to the drug (Pope et al., 1995). Investigation about the influence of cannabis sativa on cognitive processes may help not only to resolve the contention of marijuana effects on cognition, but also to elucidate the physiological and pathophysiological role that endogenous cannabinoids play in cognitive functions.
Animal studies have shown that memory processes are impaired by the main active principle of cannabis sativa, Δ9-tetrahydrocannabinol, by different synthetic CB1 cannabinoid receptor agonists, as well as by the endogenous cannabinoids (Brodkin & Moerschbaecher, 1997; Collins et al., 1995; Lichtman et al., 1995; Lichtman & Martin, 1996; Mallet & Benninger, 1996; 1998; Stella et al., 1997). In particular, Lichtman & Martin (1996) have shown that cannabinoid-induced memory impairment is specifically reversed by the CB1 cannabinoid receptor antagonist SR141716A [N-(piperidine-1-yl) -5 -(4-chlorophenyl) -1- (2,4-dichloro-phenyl) -4-methyl -1H-pyrazole-3-carboxamide hydrochloride], suggesting that this effect is mediated by cannabinoid receptors. Additional studies have shown that cannabinoids impair working memory after intrahippocampal administration (Lichtman et al., 1995), inhibit hippocampal long-term potentiation (Collins et al., 1995) and reduce hippocampal extracellular acetylcholine concentration (Gifford & Ashby, 1996; Gifford et al., 1997; Gessa et al., 1997; Carta et al., 1998). Moreover, further evidence has suggested that the negative effects of cannabinoids on working memory are caused from dopaminergic hyperactivity in the prefrontal cortex (Jentsch et al., 1997). In particular, Jentsch et al. (1997) have shown that both Δ9-tetrahydrocannabinol-induced increase in dopamine turnover in the prefrontal cortex and memory impairment are prevented by HA966 [3-amino-1-hydroxy-2-pyrrolidone], an inhibitor of dopamine neuronal activity (Morrow et al., 1997). It is widely accepted that dopaminergic modulation of neural activity in the prefrontal cortex is essential for working memory (Durstewitz et al., 1999; Levy & Goldman-Rakic; 1999; Seamans et al., 1998; Cai & Arnsten, 1997; Zahrt et al., 1997; Watanabe et al., 1997; Desimone, 1995; Williams & Goldman-Rakic, 1995; Arnsten et al., 1994; Sawaguchi & Goldman-Rakic, 1991). Indeed, recent evidence has demonstrated that working memory is impaired not only when prefrontal dopamine levels or D1 dopamine receptors activity are below normal (Sawaguchi & Goldman-Rakic, 1991), but also when they are above an optimal range (Zahrt et al., 1997). In accord with this theory several studies involving both rats and monkeys have shown that stress-induced memory impairment could be prevented by the D1 dopamine antagonist SCH23390 [R(+)-7-Chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine] (Arnsten et al., 1998) and by the D2 dopamine antagonist haloperidol (Arnsten et al., 1998) and that these stress effects on working memory could be mimicked by the administration of high doses of the D1 dopamine agonist A77636 [(−)-(1R,3S)-3-adamanty-1-(aminomethyl)-3,4-dihydro-5,6-dihydroxy-1H-2-benzopyran hydrochloride] or SKF81297 [R(+)-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzaze-pine hydrobromide] (Cai & Arnsten, 1997). Since cannabinoids activate mesocortical dopamine neurons (Diana et al., 1998; Gessa et al., 1998b), increase dopamine release (Chen et al., 1990a) and turnover in the prefrontal cortex (Jentsch et al., 1997) and since HA966 (Jentsch et al., 1997), a compound that inhibits the activity of dopaminergic neurons (Morrow et al., 1997), antagonizes the Δ9-tetrahydrocannabinol-induced memory deficits (Jentsch et al, 1997), we studied the role that dopamine plays on negative effects induced by Δ9-tetrahydrocannabinol. Specifically, the aim of this study was to examine the role of dopamine and acetylcholine during Δ9-tetrahydrocannabinol-induced inhibition of working memory. In particular, we set out to investigate whether the inhibitory effect of Δ9-tetrahydrocannabinol on extracellular acetylcholine concentration and working memory was modified by the blockade or the stimulation of D2 dopamine receptors.
Methods
Animals
Sprague Dawley rats (200–250 g; Charles River, Como, Italy) were housed individually in a Plexiglass chamber (height 15, length 40 and width 15 cm) at 22±1°C with 55% humidity. Food and water were freely available and the animals were maintained under an artificial 12 h/12 h light/dark schedule with the light cycle ranging from 0800 h to 2000 h. Experiments were conducted between 0900 h and 1700 h.
Experimental procedure
The effect of Δ9-tetrahydrocannabinol on hippocampal acetylcholine release was studied on freely moving rats by means of microdialysis, while the effect on working memory was studied in separate groups of unoperated animals trained to correctly perform a delayed alternation task in the T-maze.
Surgery and microdialysis
Implantation of the microdialysis probes was performed under general anaesthesia as previously described (Imperato et al., 1992). Briefly, rats were implanted with a trasversal dialysis probe (AN 69-HF, tube outer diameter 320 μm; Hospal-Dasco, Bologna, Italy), passing the hippocampi bilaterally (A=−3.2 and V=−3.6; A and V being referred to bregma and skull, respectively). Coordinates were chosen according to the atlas of Paxinos & Watson (1986). The dialysis probe had an active dialysis length of 1 cm.
Upon completion of the experiments, each rat was sacrificed and the location of the probe verified histologically. Only data from rats with a proper location of the probe were used. Microdialysis perfusion was performed 24 h after probe implantation. The probe was perfused at a constant rate of 2 μl min−1 with a Ringer solution containing (mM): KCl, 4.0; NaCl, 147; CaCl2, 1.5; pH 6.5. Neostigmine bromide was added at a final concentration of 10−7 mM in order to recover detectable concentrations of dialysate acetylcholine. Samples were collected every 20 min, corresponding to a volume of 40 μl, and were injected in a high-performance liquid chromatography (HPLC) system with electrochemical detection according to the technique described by Damsma & Westernik (1991). The detection limit for acetylcholine was 0.05 fmol per 1 μl of the sample. The average concentration of acetylcholine in the last three pre-drug samples was taken as 100% and all subsequent post-treatment values are expressed as a per cent (mean±s.e.mean) of basal values.
Delayed alternation task in the T-maze
Working memory was evaluated in a standard T-maze made of black plexiglass and consisting of a central stem (height 10, length 40 and width 20 cm) with a start compartment (the first 20 cm of the stem) and two arms (height 10, length 60 and width 20 cm) with a wire mesh floor. The start compartment and each goal arm were separated from the distal part of the central stem by a guillotine door. Complete entrance into the goal arm was necessary in order to reach the food cup. The T-maze was located in a silent and dimly illuminated room and a weak light (25 W) was located 30 cm over the right-hand food cup, to distinguish the right from the left arm by brightness.
Training and testing of the animals were performed according to the method described previously by Murphy et al. (1996).
The animals were housed individually for 7 days before starting the training session in a plexiglass chamber (height 15, length 40 and width 25 cm) at 22±1°C and 55% humidity. To increase their motivation for food, animals were maintained at 85% of their pre-experimental body weight by feeding them a limited amount of chow following the daily session. No rat experienced weight loss during the experiment. Water was always available in the home cage.
Each training session consisted of 11 consecutive trials in which the rats had to alternate between the right and left arm of the maze in order to obtain their reward consisting of a shelled sunflower seed. During the first trial of each session, access to one of the two arms was blocked forcing the rat to enter the opposite arm. The direction of the forced trial was alternated daily. In each of the consecutive 10 trials, the food was placed in the opposite arm to that visited in the previous successful trial, with both arms being unblocked (free-choice trials). A correct trial ended with the rat eating the food. An incorrect trial ended with the rat reaching the empty food cup. If the rat did not enter an arm within 2 min, the trial was not counted and the rat was given another attempt. After each trial, the rat was removed from the goal arm and kept in the start compartment for a delay period after which the door of central stem was opened. The criteria defining a correct daily training session was nine successful trials out of 10 choice trials for 3 consecutive days. Three different delay periods were used, ranging at 2, 8, and 16 s. In the delay period of 2 s the rats' performance was stabilized at 10 correct entries (or 0 incorrect entries) in the T-maze on 10 trials, while during the delay period of 8 and 16 s performance was stabilized at nine and seven correct entries (or one and three incorrect entries) in the T-maze on 10 trials, respectively. All drugs were injected after animals had attained the above criteria. Data collected from each session were analysed in terms of number of correct and incorrect entries in the T-maze on 10 trials.
In the working memory experiments we used a cross over design. In particular, in order to reduce inter-group variables, for each different time we used the same group of animals tested after a washout period of 1 week during which they received no drug and re-established their optimal performance.
Drugs
Δ9-Tetrahydrocannabinol (RBI, Italy) solutions were prepared from vials containing 10 mg of the drug in 1 ml of absolute ethanol. Vials were evaporated under nitrogen and the residue dissolved in two drops of Tween 80 and then diluted in saline. The specific CB1 cannabinoid receptor antagonist SR 141617A (Sanofi Recherche, Montpellier, France) was dissolved in two drops of Tween 80 and then diluted in saline.
Δ9-Tetrahydrocannabinol, SR 141617A and S(−)-sulpiride were administered intraperitoneally (i.p.) in a volume of 3 ml kg−1, while (−)-quinpirole hydrochloride was given subcutaneously (s.c.) in a volume of 2 ml kg−1. Control rats were treated with the vehicle used to dissolve the active ingredient.
Statistical analysis
Between-group comparisons were assessed by a two-way analysis of variance (ANOVA) for repeated measures. Post-hoc comparisons were performed by Student-Newman-Keuls tests. Statistical significance was reached at P<0.05.
Results
Hippocampal extracellular acetylcholine concentration
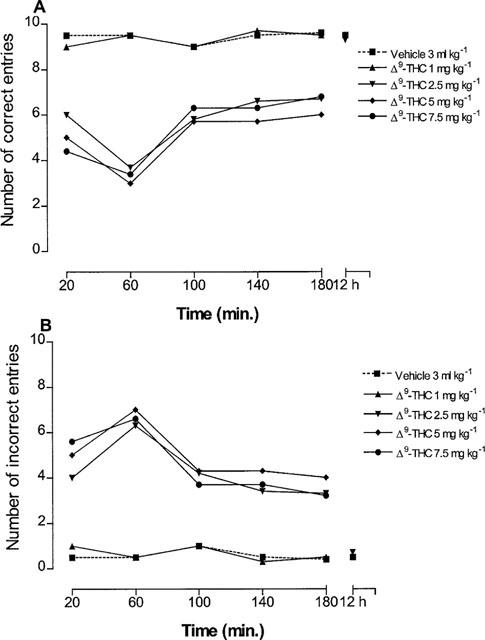
In agreement with previous results (Carta et al., 1998; Gessa et al., 1997; 1998a), the administration of Δ9-tetrahydrocannabinol at the dose of 2.5 and 5 mg kg−1 reduced, in a dose-related manner, hippocampal extracellular acetylcholine concentration (Figure 1). A significant inhibition appeared at 80 min after treatment, was maximal at 120 and 180 min, and was no longer present at 12 h. A higher dose of 7.5 mg kg−1 produced no further reduction (Figure 1). A Δ9-tetrahydrocannabinol dose of 1 mg kg−1 had no significant effect (Figure 1).
Figure 1.
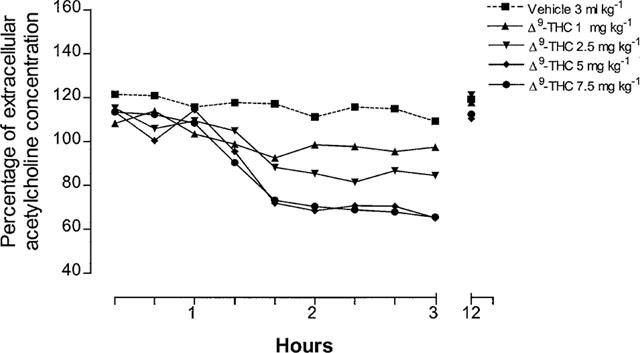
Time-course for the reduction of hippocampal extracellular acetylcholine concentration after Δ9-tetrahydrocannabinol administration (ANOVA main effect Δ9-THC2.5 mg kg−1 F1.16=20.84, P<0.0001; ANOVA main effect of repeated measures Δ9-THC2.5 mg kg−1 F8.36=0.82, P=0.59; ANOVA main effect Δ9-THC5 mg kg−1 F1.16=20, P<0.0001; ANOVA main effect of repeated measures Δ9-THC5 mg kg−1 F8.36=3.36, P<0.01; ANOVA main effect Δ9-THC7.5 mg kg−1 F1.16=19.25, P<0.0001; ANOVA main effect of repeated measures Δ9-THC7.5 mg kg−1 F8.36=3.54, P<0.01). P<0.05 vs controls (Student-Newman-Keuls test) 80 min after Δ9-tetrahydrocannabinol. Data are expressed as percentage (mean±s.e.m.; n=5) of the baseline concentration. Basal values of extracellular acetylcholine concentration, prior to drug administration, were: 1.38±0.13 fmol μl−1 for control group and 1.27±0.24, 1.34±0.18, 1.16±0.12 and 1.42±0.32 fmol μl−1 for the different groups treated with Δ9-THC at the doses of 1, 2.5, 5 and 7.5 mg kg−1, respectively. A Δ9-tetrahydrocannabinol dose of 1 mg kg−1 had no significant effect on hippocampal acetylcholine concentration. s.e. values were not more than ±17.86%.
The reduction of hippocampal extracellular acetylcholine concentration induced by Δ9-tetrahydrocannabinol (5 mg kg−1) was antagonized not only, as expected, by the CB1 cannabinoid receptor antagonist SR141716A (0.5 mg kg−1), but also, dose-dependently, by the D2 dopamine receptor antagonist S(−)-sulpiride (5, 10 and 25 mg kg−1), (Figure 2). The latter also suppressed the inhibition of extracellular acetylcholine concentration induced by the effective Δ9-tetrahydrocannabinol dose of 7.5 mg kg−1, indicating a non competitive type of antagonism (Figure 3). Given alone, neither SR141716A (0.5 mg kg−1) nor S(−)-sulpiride (25 mg kg−1) modified hippocampal extracellular acetylcholine concentration (Figure 2). Conversely, administration of the D2 dopamine receptor agonist (−)-quinpirole (25 μg kg−1) markedly potentiated the effect of the ineffective Δ9-tetrahydrocannabinol dose of 1 mg kg−1 (Figure 4). A higher dose of (−)-quinpirole (500 μg kg−1) produced similar results (Figure 4). (−)-Quinpirole (25 and 500 μg kg−1) given alone failed to modify extracellular acetylcholine concentration (Figure 4). Inhibition of hippocampal extracellular acetylcholine concentration induced by the combination of (−)-quinpirole (25 and 500 μg kg−1) and Δ9-tetrahydrocannabinol (1 mg kg−1), was reversed by either SR 141716A (0.5 mg kg−1) or S(−)-sulpiride (25 mg kg−1) (Figure 4).
Figure 2.
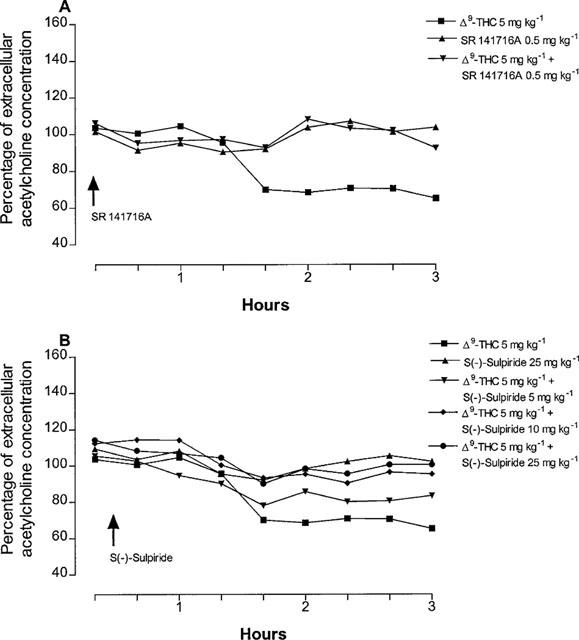
Time-course for the antagonism by SR141716A (A) and S(−)-sulpiride (B) of Δ9-tetrahydrocannabinol-induced reduction of hippocampal extracellular acetylcholine concentration. (ANOVA main effect Δ9-THC+SR 141716A F1.16=5.35, P<0.05; ANOVA main effect Δ9-THC+S(−)-sulpiride 5 mg kg−1 F1.16=0.42, P=0.52; ANOVA main effect Δ9-THC+S(−)-sulpiride 10 mg kg−1 F1.16=5.83, P<0.05; ANOVA main effect Δ9-THC+S(−)-sulpiride 25 mg kg−1 F1.16=6.97, P<0.05). P<0.05 vs Δ9-THC 5 mg kg−1 (Student-Newman-Keuls test). Data are expressed as percentage (mean±s.e.mean; n=5) of the baseline concentration. Basal values of extracellular acetylcholine concentration, prior to drug administration, were: 1.22±0.11, 1.32±0.18 and 1.41±0.23 for Δ9-THC, SR 141716A and S(−)-sulpiride groups, respectively and 1.25±0.21, 1.23±0.23, 1.45±0.22 fmol μl−1 for the different groups treated with Δ9-THC+S(−)-sulpiride at the doses of 5, 10 and 5 mg kg−1, respectively. SR141716A and S(−)-sulpiride were given 20 and 30 min after Δ9-tetrahydrocannabinol, respectively. SR141716A and S(−)-sulpiride given alone had no effect on acetylcholine release. s.e. values were not more than ±19.32%.
Figure 3.
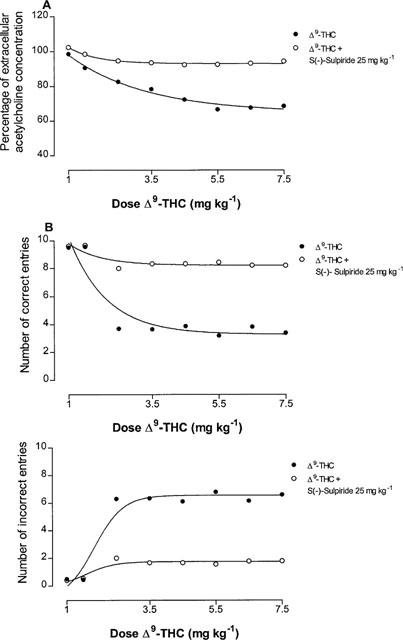
Effects on percentage of hippocampal extracellular acetylcholine concentration (A) (120 min after Δ9-tetrahydrocannabinol administration) and on correct and incorrect entries in T-maze (B) (60 min after Δ9-tetrahydrocannabinol administration) as a function of dose of Δ9-tetrahydrocannabinol alone or in combination with S(−)-sulpiride (25 mg kg−1). Each point represents the mean±s.e.mean of five animals. s.e. values were not more than ±8.40%.
Figure 4.
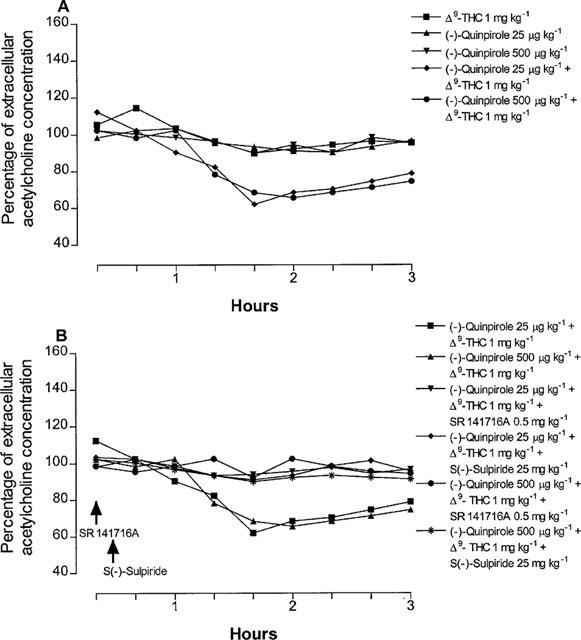
Time course for the potentiation by (−)-quinpirole on Δ9-tetrahydrocannabinol-effect on hippocampal acetylcholine release (A) and reversal by SR141716A or S(−)-sulpiride (B). (ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC F1.16=7.23, P<0.05; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC F1.16=9.44, P<0.01; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+SR141716A F1.16=6.65, P<0.05; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+S(−)-sulpiride F1.16=9.32, P<0.01; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+SR141716A F1.16=20.57, P<0.0001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+S(−)-sulpiride F1.16=6.63, P<0.01). In (A) P<0.05 vs Δ9-THC 1 mg kg−1 and in (B) P<0.05 vs (−)-quinpirole 25 μg kg−1+Δ9-THC or (−)-quinpirole 500 μg kg−1+Δ9-THC (Student-Newman-Keuls test). Data are expressed as percentage (mean±s.e.mean; n=5) of the baseline concentration. Basal values of extracellular acetylcholine concentration, prior to drug administration, were: 1.34±0.15 and 1.26±0.22 fmol μl−1 for groups treated with (−)-quinpirole at the doses of 25 and 500 μg kg−1, respectively, 1.24±0.12 fmol μl−1 for Δ9-THC group, 1.30±0.17 and 1.43±0.13 fmol μl−1 for groups treated with (−)-quinpirole 25 μg kg−1+Δ9-THC and (−)-quinpirole 500 μg kg−1+Δ9-THC, respectively, 1.15±0.09 and 1.36±0.18 fmol μl−1 for groups treated with (−)-quinpirole 25 μg kg−1+Δ9-THC+SR141716A and (−)-quinpirole 25 μg kg−1+Δ9-THC+S(−)-sulpiride 25 mg kg−1, respectively, 1.25±0.16 and 1.24±0.11 fmol μl−1 for groups treated with (−)-quinpirole 500 μg kg−1+Δ9-THC+SR141716A and (−)-quinpirole 500 μg kg−1+Δ9-THC+S(−)-sulpiride, respectively. SR141716A and S(−)-sulpiride were given 20 and 30 min after Δ9-tetrahydrocannabinol, respectively. (−)-Quinpirole and Δ9-tetrahydrocannabinol given alone had no significant effect on acetylcholine concentration. s.e. values were not more than ±9.80%.
Working memory
In line with previous results (Jentsch et al., 1997), administration of Δ9-tetrahydrocannabinol at the dose of 2.5 and 5 mg kg−1 impaired delayed alternation tasks in the T-maze (Figure 5). Following a dose of 2.5 mg kg−1, a significant inhibition of working memory was observed at 20 min after treatment, was maximal at 60 min, persisted over 180 min and disappeared at 12 h (Figure 5). A higher dose of 7.5 mg kg−1 produced no further memory impairment (Figure 5). A Δ9-tetrahydrocannabinol dose of 1 mg kg−1 had no effect on working memory (Figure 5). As shown in Figure 6, memory impairment induced by Δ9-tetrahydrocannabinol (5 mg kg−1) was antagonized not only, as expected, by the CB1 cannabinoid receptor antagonist SR141716A (0.5 mg kg−1) but also, unexpectedly, and dose-dependently, by the D2 dopamine receptor antagonist S(−)-sulpiride (5, 10 and 25 mg kg−1). The latter also suppressed the reduction of working memory induced by the effective Δ9-tetrahydrocannabinol dose of 7.5 mg kg−1 (Figure 3). At the doses used, neither SR141716A (0.5 mg kg−1) nor S(−)-sulpiride (25 mg kg−1), given alone, modified working memory (Figure 6). Conversely, administration of the D2 dopamine receptor agonist (−)-quinpirole (25 μg kg−1) markedly potentiated the effect of an ineffective Δ9-tetrahydrocannabinol dose of 1 mg kg−1 (Figure 7). A higher dose of (−)-quinpirole (500 μg kg−1) produced similar results (Figure 7). (−)-Quinpirole (25 and 500 μg kg−1) given alone failed to modify working memory (Figure 7). Finally, memory impairment produced by the combination of (−)-quinpirole (25 and 500 μg kg−1) and Δ9-tetrahydrocannabinol (1 mg kg−1) was totally suppressed by either SR141716A (0.5 mg kg−1) or S(−)-sulpiride (25 mg kg−1) (Figure 7). The administration of all drugs produced a significant effect on delayed response performance (Figure 8).
Figure 5.
Time-course for the reduction of working memory after Δ9-tetrahydrocannabinol administration (For the number of correct entries: ANOVA main effect Δ9-THC2.5 mg kg−1 F1.8=44.41, P<0.001; ANOVA main effect of repeated measures Δ9-THC2.5 mg kg−1 F4.20=0.95, P=0.45; ANOVA main effect Δ9-THC5 mg kg−1 F1.8=62.45, P<0.0001; ANOVA main effect of repeated measures Δ9-THC5 mg kg−1 F4.20=0.93, P=0.46; ANOVA main effect Δ9-THC7.5 mg kg−1 F1.8=36.62, P<0.001; ANOVA main effect of repeated measures Δ9-THC7.5 mg kg−1 F4.20=1.06, P=0.40; For the number of incorrect entries: ANOVA main effect Δ9-THC2.5 mg kg−1 F1.8=44.11, P<0.0001; ANOVA main effect of repeated measures Δ9-THC2.5 mg kg−1 F4.20=0.95, P<0.45; ANOVA main effect Δ9-THC5 mg kg−1 F1.8=58.87, P<0.0001; ANOVA main effect of repeated measures Δ9-THC5 mg kg−1 F4.20=0.86, P=0.50; ANOVA main effect Δ9-THC7.5 mg kg−1 F1.8=36.45, P<0.001; ANOVA main effect of repeated measures Δ9-THC7.5 mg kg−1 F4.20=1.03, P=0.41). Data are expressed as number of correct and incorrect entries (mean±s.e.mean; n=5) in T-maze on 10 trials. A Δ9-tetrahydrocannabinol dose of 1 mg kg−1 had no effect on delayed alternation tasks in the T-maze. Intertrial delay was fixed at 8 s. s.e. values were not more than ±1.65%.
Figure 6.
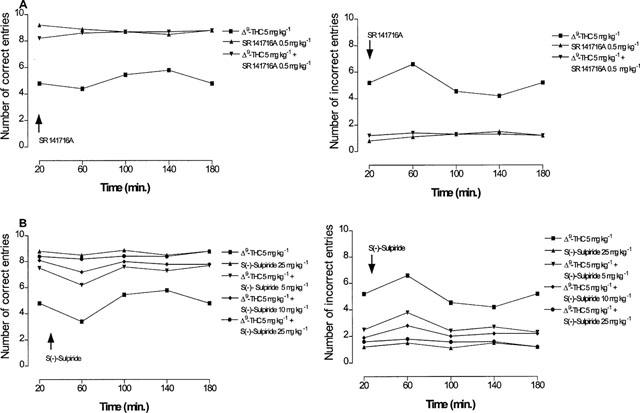
Time course for the antagonism by SR 141716A (A) and S(−)-sulpiride (B) of Δ9-tetrahydrocannabinol-induced working memory impairment. (For the number of correct entries: ANOVA main effect Δ9-THC+SR 141716A F1.8=78.53, P<0.0001; ANOVA main effect Δ9-THC+S(−)-sulpiride5 mg kg−1 F1.8=24.09, P<0.002; ANOVA main effect Δ9-THC+S(−)-sulpiride10 mg kg−1 F1.8=34.41, P<0.0005; ANOVA main effect Δ9-THC+S(−)-sulpiride25 mg kg−1 F1.8=73.14, P<0.0001; For the number of incorrect entries: ANOVA main effect Δ9-THC+SR 141716A F1.8=47.78, P<0.0001; ANOVA main effect Δ9-THC+S(−)-sulpiride5 mg kg−1 F1.8=25.77, P<0.0001; ANOVA main effect Δ9-THC+S(−)-sulpiride10 mg kg−1 F1.8=47.53, P<0.0001; ANOVA main effect Δ9-THC+S(−)-sulpiride25 mg kg−1 F1.8=16.56, P<0.004). P<0.05 vs Δ9-THC 5 mg kg−1 (Student-Newman-Keuls test). Data are expressed as number of correct and incorrect entries (means±s.e.mean; n=5) in T-maze on 10 trials. SR141716A and S(−)-sulpiride were given 20 and 30 min after Δ9-tetrahydrocannabinol, respectively. SR141716A and S(−)-sulpiride given alone had no effect on working memory. Intertrial delay was fixed at 8 s. s.e. values were not more than ±1.60%.
Figure 7.
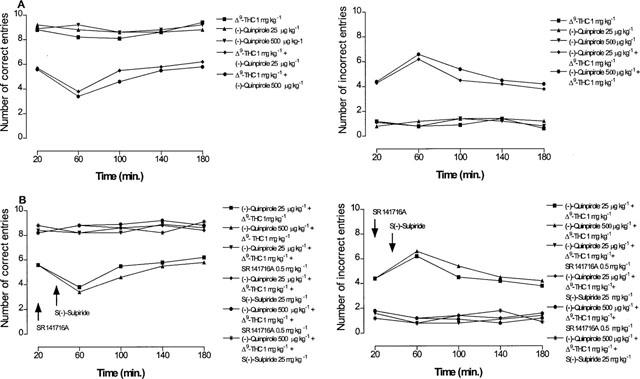
Time course for the potentiation by (−)-quinpirole on Δ9-tetrahydrocannabinol effect on working memory (A) and reversal by SR 141716A or S(−)-sulpiride (B). (For number of correct entries: ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC F1.8=46.91, P<0.001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC F1.8=53.75, P<0.0001; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+SR 141716A F1.8=50.28, P<0.0001; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+S(−)-sulpiride F1.8=55.41, P<0.0001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+SR 141716A F1.8=65.87, P<0.001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+S(−)-sulpiride F1.8=58.24, P<0.0001; For number of incorrect entries: ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC F1.8=28.42, P<0.0001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC F1.8=16.95, P<0.01; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+SR 141716A F1.8=59.25, P<0.0001; ANOVA main effect (−)-quinpirole 25 μg kg−1+Δ9-THC+S(−)-sulpiride F1.8=49.43, P<0.0001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+SR 141716A F1.8=63.31, P<0.0001; ANOVA main effect (−)-quinpirole 500 μg kg−1+Δ9-THC+S(−)-sulpiride F1.8=61.47, P<0.0001). In panel A P<0.05 vs Δ9-THC 1 mg kg−1 and in panel B P<0.05 vs (−)-quinpirole 25 μg kg−1+Δ9-THC or (−)-quinpirole 500 μg kg−1+Δ9-THC (Student-Newman Keuls test). Data are expressed as number of correct and incorrect entries (means±s.e.mean; n=5) in the T-maze on 10 trials. SR141716A and S(−)-sulpiride were given 20 and 30 min after Δ9-tetrahydrocannabinol, respectively. (−)-Quinpirole and Δ9-tetrahydrocannabinol given alone had no significant effect on working memory. s.e. values were not more than ±2.30%.
Figure 8.
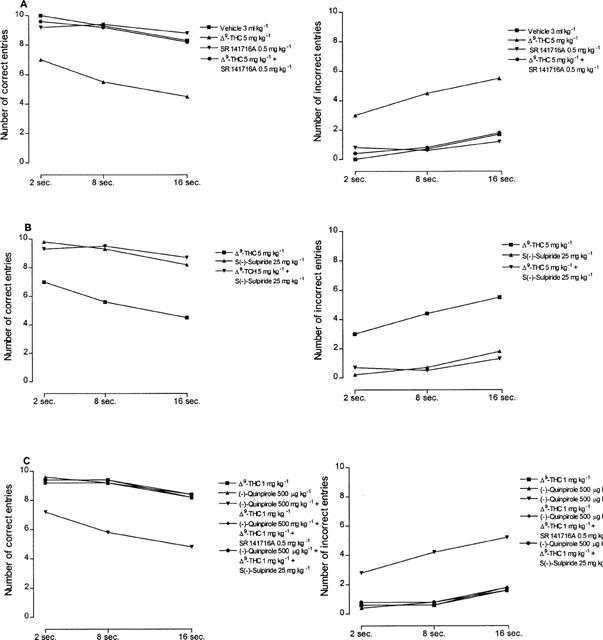
Effects of drugs treatment on delayed alternation tasks in T-maze for each of the three delay intervals used. The range delays were: 2, 8 and 16 s. Results represent the number of correct and incorrect entries (means±s.e.mean; n=5) in the T-maze on 10 trials. Δ9-tetrahydrocannabinol showed a significant effect on delayed response performance (For correct entries: ANOVA main effect F1.4=12.08, P<0.05; For incorrect entries: ANOVA main effect F1.4=16.12, P<0.05). At each delay SR141716A and S(−)-sulpiride reversed the Δ9-tetrahydrocannabinol reduction of working memory, while (−)-quinpirole potentiated the inhibition of working memory induced by Δ9-tetrahydrocannabinol (all statistical comparisons revealed significant with P<0.001). s.e. were not more than ±1.56%.
Discussion
These results confirm previous studies showing that Δ9-tetrahydrocannabinol impairs working memory (Lichtman et al., 1995; Lichtman & Martin, 1996; Mallet & Beninger, 1996) and inhibits hippocampal extracellular acetylcholine concentration (Gessa et al., 1997; 1998a; Carta et al., 1998) through the activation of CB1 cannabinoid receptors.
The major outcome of this study is that both Δ9-tetrahydrocannabinol effects are antagonized by the D2 dopamine receptor antagonist S(−)-sulpiride and potentiated by the D2 dopamine receptor agonist (−)-quinpirole.
The results confirm previous studies indicating that Δ9-tetrahydrocannabinol effects on the dopamine system are very complex. Several works have demonstrated that the increase in dopamine release induced by Δ9-tetrahydrocannabinol is strain (Chen et al., 1991) and brain area dependent (Chen et al., 1993). Conversely, other studies have shown that striatal dopamine release is unaffected by Δ9-tetrahydrocannabinol (Castaneda et al., 1991). However, our results indicate that memory impairment and reduction of extracellular acetylcholine concentration are produced by the concomitant activation of both the CB1 cannabinoid and the D2 dopamine receptors, the latter most likely being activated by endogenous dopamine released following Δ9-tetrahydrocannabinol administration. In fact, stimulation of either receptors alone would be insufficient to produce similar effects. Our findings are also in accord with our recent results showing that analgesic (Carta et al., 1999) and hypothermic (Nava et al., 2000) effects induced by Δ9-tetrahydrocannabinol are potentiated by (−)-quinpirole and (+)-bromocriptine and reversed by S(−)-sulpiride and S(−)-raclopride.
The mechanism underlying how the D2 dopamine receptor stimulation enables the onset of Δ9-tetrahydrocannabinol effects is not clear. Since CB1 cannabinoid and D2 dopamine receptors are both coupled to adenylate cyclase via a pertussis toxin-sensitive G-protein (Sibley & Monsma, 1992; Pertwee, 1997) and may be co-localized in the same brain areas (Sibley & Monsma, 1992; Matsuda et al., 1993), we might suppose that the concomitant activation of both receptors produce a cellular degree of cyclic AMP inhibition enough for Δ9-tetrahydrocannabinol effects to occur. In other words, the detrimental effects of Δ9-tetrahydrocannabinol both on working memory and hippocampal extracellular acetylcholine concentration would be due to cellular cyclic AMP reduction within definite brain areas controlling cognitive processes. In line with this hypothesis, recent studies have shown that improvement of working memory function is produced in aged monkeys by perfusing the prefrontal cortex with low doses of the D1 dopamine receptor agonists A77639 and SKF81297 (Cai & Arnsten, 1997); a treatment that should cause cellular cyclic AMP accumulation, since D1 dopamine receptors are positively linked to adenylate cyclase (Monsma et al., 1990). However, a recent observation by Glass & Felder (1997) might offer an alternative explanation for the mechanisms involved. These authors found in primary cultures of striatal neurons that the concomitant stimulation of CB1 cannabinoid and D2 dopamine receptors results in the accumulation of cellular cyclic AMP, in contrast to the decrease normally observed with activation of either receptors alone (Sibley & Monsma, 1992; Pertwee, 1997). In line with this hypothesis, we might suggest that in vivo Δ9-tetrahydrocannabinol activating dopamine neurons (Diana et al., 1998; Gessa et al., 1998b) and probably releasing endogenous dopamine (Chen et al., 1990a) might stimulate both CB1 cannabinoid and D2 dopamine receptors. The concurrent activation of both receptors might produce an accumulation of cellular cyclic AMP in neurons where these receptors are co-localized.
Our results leave the important issue concerning the correlation between extracellular acetylcholine concentration reduction and working memory impairment after Δ9-tetrahydrocannabinol administration unresolved. The failure of physostigmine, a cholinesterase inhibitor, to improve Δ9-tetrahydrocannabinol cognitive deficits (Lichtman & Martin, 1996) and the inability of SR141716A to prevent the impairment of working memory induced by scopolamine (Lichtman & Martin, 1996), a muscarinic antagonist, suggest that the negative effects of cannabinoids on working memory are not directly mediated by the cholinergic system (Lichtman & Martin, 1996). Indeed, Childers & Deadwyler (1996) have shown that cannabinoids modulate conductance at a voltage-dependent K+ channel in the hippocampus via a cyclic AMP dependent process without cholinergic neuronal mediation. These data suggest that cannabinoid and cholinergic systems do not affect memory through a common serial pathway. Moreover, several works have shown that cannabinoid ligands and endogenous cannabinoids can directly block the cellular processes associated with memory formation (Collins et al., 1995; Norwicky et al., 1987; Stella et al., 1997; Terranova et al., 1995). This evidence, including our results suggesting that memory loss occurred within 20 min after Δ9-tetrahydrocannabinol administration, whereas the fall in acetylcholine concentration was observed up to 80 min after treatment, support the possibility that these two effects might be separated and controlled by different neurochemical systems, as suggested by Lichtman & Martin (1996). Conversely, the fact that the D2 dopamine receptor antagonist, S(−)-sulpiride modified Δ9-tetrahydrocannabinol-effects on hippocampal acetylcholine concentration and working memory might suggest that the two phenomena, although not directly correlated, may be controlled by similar mechanisms. On the other hand, several studies have demonstrated that a modulatory system such as the endogenous opiod system may control Δ9-tetrahydrocannabinol effects on the dopamine system (Chen et al., 1990b; Tanda et al., 1997). Specifically, the opiod antagonist naloxone has been shown to prevent the increase of dopamine release induced by Δ9-tetrahydrocannabinol in the shell of nucleus accumbens (Tanda et al., 1997). In light of this evidence, we may also suppose that the opiod endogenous system controls with an indirect mechanism with several steps the delayed effects of Δ9-tetrahydrocannabinol on extracellular acetylcholine concentration. In other words, the cannabinoid and cholinergic systems may induce the inhibition of hippocampal acetylcholine concentration through the mediation of a third receptor system. This possibility may explain the delayed effects on inhibition of hippocampal acetylcholine concentration observed after Δ9-tetrahydrocannabinol treatment. The above data, coupled with the evidence that cannabinoids inhibit the release of acetylcholine (Carta et al., 1999; Gessa et al., 1998a; Gifford et al., 1996; 1997), norepinephrine (Schlicker et al., 1997) and glutamate (Shen et al., 1996) from the hippocampus, suggest that the memory deficit induced by cannabinoids is a complex process involving different neurotransmitters.
The Δ9-tetrahydrocannabinol effect on working memory is an on-off response. In fact, all effective Δ9-tetrahydrocannabinol doses cause a similar degree of inhibition. In particular, the evidence showing that the deficit of working memory is an on-off response, coupled with results showing that the reduction of extracellular acetylcholine concentration is a dose-related response suggests that the two phenomena are not directly correlated and that several mechanisms and/or neuronal circuits could be involved in both of the inhibitory effects induced by Δ9-tetrahydrocannabinol. Specifically, the inhibition of acetylcholine concentration could represent only one aspect of the detrimental effects induced by Δ9-tetrahydrocannabinol, while the deficit found in working memory could be expression of a neurotransmission block into and within the hippocampus. This hypothesis could explain the on-off response observed in working memory deficit and at the same time the marked time course discrepancy between inhibition of acetylcholine concentration and loss of memory. Moreover, we may exclude that the on/off response observed in the inhibition of working memory induced by Δ9-tetrahydrocannabinol is caused from deficits in locomotory activity or motivation. Indeed, several studies have excluded that the deficit of working memory induced by cannabinoids may be correlated to a possible reduction of food reinforcement or hunger (Lichtman et al., 1995; Lichtman & Martin, 1996). In fact, the sunflower seed at the end of each trial was always consumed whenever an arm was selected regardless of drug treatment. Moreover, the evidence showing that intrahippocampal administration of CP55,940 [1α,2β-(R)5α]-(−)-5-(1,1-dimethylheptyl)-2 - [5 - hydroxy -2-(3- hydroxypropil)cyclohexyl]phenol, a synthetic cannabinoid agonist, impairs choice accuracy in radial maze without retarding the time required to complete the maze (Lichtman et al., 1995), indicates that the memory deficits are dissociated from locomotory activity.
The findings showing that D2 dopamine receptor antagonists reverse, in a dose-dependent manner, hippocampal acetylcholine concentration and working memory deficit, are suggestive that D2 dopamine receptors could exert a control on both Δ9-tetrahydrocannabinol effects.
Previous results from our laboratory have shown that Δ9-tetrahydrocannabinol reduces acetylcholine concentration in the prefrontal cortex as well as in the hippocampus (Gessa et al., 1998a) suggesting that similar mechanisms control cholinergic transmission in both areas. However, as for hippocampal extracellular acetylcholine concentration, further investigations are needed in order to clarify the correlation between acetylcholine reduction in the prefrontal cortex and memory impairment.
Irrespective of the mechanisms and brain areas involved, the finding that S(−)-sulpiride reverses Δ9-tetrahydrocannabinol-induced amnesia suggests that D2 dopamine receptor antagonists should be clinically tested as a potential treatment for memory deficits produced during marijuana intoxication. On the other hand, our results raise the relevant concern whether Δ9-tetrahydrocannabinol-induced memory impairment might be potentiated by drugs of abuse, such as cocaine, amphetamine, alcohol and ecstasy which are all able to increase the release of endogenous dopamine and whether D2 dopamine receptors play a permissive role in the pharmacological effects of cannabinoids besides cognition.
Acknowledgments
We thank Dr C. De-Fraja and Mr H. Kim for critical reading of the manuscript. We are grateful to Sanofi Recherche, Montpellier, France, for the supply of SR 141716A. This study was carried out in accordance with the Declaration of Helsinki and the Guide for the Care and Use of Laboratory Animals.
Abbreviations
- A77636
(−)-(1R 3S)-3-adamantyl-1-(aminomethyl)-3,4-dihydro-5,6-dihydroxy-1H-2-benzopyran hydrochloride
- CP55,940
[1α,2β-(R)5α]-(−)-5-(1,1-dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropil)cyclohexyl]phenol
- HA966
3-amino-1-hydroxy-2-pyrrolidone
- SCH23390
R(+)-7-Chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine
- SKF81297
R(+)-6-Chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide
- SR141716A
N-(piperidine-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
References
- ARNSTEN A.F.T., CAI X., MURPHY B.L., GOLDMAN-RAKIC P.S. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–151. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- ARNSTEN A.F.T., GOLDMAN-RAKIC P.S. Noise stress impairs prefrontal cortical cognitive function in monkeys: evidence for a hyperdopaminergic mechanism. Arch. Gen. Psychiatry. 1998;55:362–368. doi: 10.1001/archpsyc.55.4.362. [DOI] [PubMed] [Google Scholar]
- BRODKIN J., MOERSCHBAECHER J.M. SR 141716A antagonizes the disruptive effects of cannabinoid ligands on learning in rats. J. Pharmacol. Exp. Ther. 1997;282:1526–1532. [PubMed] [Google Scholar]
- CAI X., ARNSTEN A.F.T. Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J. Pharmacol. Exp. Ther. 1997;283:183–189. [PubMed] [Google Scholar]
- CARTA G., GESSA G.L., NAVA F. D2 dopamine receptor antagonist prevents Δ9-tetrahydrocannabinol-induced antinociception in rat. Eur. J. Pharmacol. 1999;384:153–156. doi: 10.1016/s0014-2999(99)00696-2. [DOI] [PubMed] [Google Scholar]
- CARTA G., NAVA F., GESSA G.L. Inhibition of hippocampal acetylcholine release after acute and repeated Δ9-tetrahydrocannabinol in rats. Brain Res. 1998;809:1–4. doi: 10.1016/s0006-8993(98)00738-0. [DOI] [PubMed] [Google Scholar]
- CASTANEDA E., MOSS D.E., ODDIE S.D., WHISHAW I.Q. THC does not affect striatal dopamine release: microdialysis in freely moving rats. Pharmacol. Biochem. Behav. 1991;40:587–591. doi: 10.1016/0091-3057(91)90367-b. [DOI] [PubMed] [Google Scholar]
- CHEN J., MARMUR R., PULLES A., PAREDES W., GARDNER E.L. Ventral tegmental microinjection of delta-9-tetrahydrocannabinol enhances ventral tegmental somatodendritic dopamine levels but not forebrain dopamine levels: Evidence for local neural action by marijuana's psychoactive ingredient. Brain Res. 1993;621:65–70. doi: 10.1016/0006-8993(93)90298-2. [DOI] [PubMed] [Google Scholar]
- CHEN J., PAREDES W., LOWINSON J.H., GARDNER E.L. Delta9-tetrahydrocannabinol enhances presynaptic dopamine efflux in medial prefrontal cortex. Eur. J. Pharmacol. 1990a;190:259–262. doi: 10.1016/0014-2999(90)94136-l. [DOI] [PubMed] [Google Scholar]
- CHEN J., PAREDES W., LI J., SMITH D., LOWINSON J., GARDNER E.L. Δ9-Tetrahydrocannabinol produces naloxone-blockable enhancement of presynaptic basal dopamine efflux in nucleus accumbens of conscious, freely-moving rats as measured by intracerebral microdialysis. Psychopharmacology. 1990b;102:156–162. doi: 10.1007/BF02245916. [DOI] [PubMed] [Google Scholar]
- CHEN J., PAREDES W., LOWINSON J.H., GARDNER E.L. Strain-specific facilitation of dopamine efflux by Δ9-tetrahydrocannabinol in the nucleus accumbens of rat: an in vivo microdialysis study. Neurosci. Lett. 1991;129:136–140. doi: 10.1016/0304-3940(91)90739-g. [DOI] [PubMed] [Google Scholar]
- CHILDERS S.R., DEADWYLER S.A. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem. Pharmacol. 1996;52:819–827. doi: 10.1016/0006-2952(96)00419-4. [DOI] [PubMed] [Google Scholar]
- COLLINS D.R., PERTWEE R.G., DAVIES S.N. Prevention by the cannabinoid antagonist SR141716A, of cannabinoid-mediate blockade of long-term potentiation in the rat hippocampal slice. Br. J. Pharmacol. 1995;115:869–870. doi: 10.1111/j.1476-5381.1995.tb15889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAMSMA G., WESTERINK B.H.C.A microdialysis and automated on-line analysis approach to study central cholinergic transmission in vivo Microdialysis in Neuroscience 1991Elsevier, Amsterdam; 237–252.ed. Robinson T.E., Justice J. pp [Google Scholar]
- DESIMONE R. Is dopamine a missing link. Nature. 1995;376:549–550. doi: 10.1038/376549a0. [DOI] [PubMed] [Google Scholar]
- DIANA M., MELIS M., GESSA G.L. Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur. J. Neurosci. 1998;10:2825–2830. doi: 10.1111/j.1460-9568.1998.00292.x. [DOI] [PubMed] [Google Scholar]
- DURSTEWITZ D., KELC M., GUNTURKUN O. A neurocomputational theory of the dopaminergic modulation of working memory functions. J. Neurosci. 1999;19:2807–2822. doi: 10.1523/JNEUROSCI.19-07-02807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GESSA G.L., CASU M.A., CARTA G., MASCIA M.S. Cannabinoids decrease acetylcholine release in the medial prefrontal cortex and hippocampus, reversal by SR 141716A. Eur. J. Pharmacol. 1998a;355:119–124. doi: 10.1016/s0014-2999(98)00486-5. [DOI] [PubMed] [Google Scholar]
- GESSA G.L., MASCIA M.S., CASU M.A., CARTA G. Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR141716A. Eur. J. Pharmacol. 1997;327:R1–R2. doi: 10.1016/s0014-2999(97)89683-5. [DOI] [PubMed] [Google Scholar]
- GESSA G.L., MELIS M., MUNTONI A.L., DIANA M. Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur. J. Pharmacol. 1998b;341:39–44. doi: 10.1016/s0014-2999(97)01442-8. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R., Jr Electrically evoked acetylcholine release from hippocampal slice is inhibited by the cannabinoid receptor agonist, WIN55,212-2 and is potentiated by the cannabinoid antagonist, SR 141716A. J. Pharmacol. Exp. Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- GIFFORD A.N., SAMIIAN L., GATLEY S.J., ASHBY C.R., Jr Examination of the effect of cannabinoid receptor agonist, CP55,940 on electrically evoked transmitter release from rat brain slices. Eur. J. Pharmacol. 1997;324:187–192. doi: 10.1016/s0014-2999(97)00082-4. [DOI] [PubMed] [Google Scholar]
- GLASS M., FELDER C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL W., SOLOWIJ N. Adverse effects of cannabis. Lancet. 1998;352:1611–1616. doi: 10.1016/S0140-6736(98)05021-1. [DOI] [PubMed] [Google Scholar]
- IMPERATO A., ANGELUCCI L., CASOLINI P., ZOCCHI A., PUGLISI-ALLEGRA S. Repeated stressful experiences differently affect limbic dopamine release during and following stress. Brain Res. 1992;577:194–199. doi: 10.1016/0006-8993(92)90274-d. [DOI] [PubMed] [Google Scholar]
- JENTSCH J.D., ANDRUSIAK E., TRAN A., BOWERS M.B., Jr, ROTH R.H. Delta9-tetrahydrocannabinol increases prefrontal cortical catecholaminergic utilization and impairs spatial working memory in the rat: blockade of dopaminergic effects with HA966. Neuropsychopharmacology. 1997;16:426–432. doi: 10.1016/S0893-133X(97)00018-3. [DOI] [PubMed] [Google Scholar]
- LEVY R., GOLDMAN-RAKIC P.S. Association of storage and processing functions in the dorsolateral prefrontal cortex of the non human primate. J. Neurosci. 1999;19:5149–5158. doi: 10.1523/JNEUROSCI.19-12-05149.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LICHTMAN A.H., DIMEN K.R., MARTIN B.R. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology. 1995;119:282–290. doi: 10.1007/BF02246292. [DOI] [PubMed] [Google Scholar]
- LICHTMAN A.H., MARTIN B.R. Δ9-Tetrahydrocannabinol impairs spatial memory through a cannabinoid receptor mechanism. Psychopharmacology. 1996;126:125–131. doi: 10.1007/BF02246347. [DOI] [PubMed] [Google Scholar]
- MALLET P.E., BENINGER R.J. The cannabinoid CB1 receptor antagonist SR 141716A attenuates the memory impairment produced by delta9-tetrahydrocannabinol or anandamide. Psychopharmacology. 1998;140:11–19. doi: 10.1007/s002130050733. [DOI] [PubMed] [Google Scholar]
- MALLET P.E., BENINGER R.J. The endogenous cannabinoid receptor agonist anandamide impairs memory in rats. Behav. Pharmacol. 1996;7:276–284. [Google Scholar]
- MATSUDA L.A., BONNER T.I., LOLAIT S.J. Localization of cannabinoid receptor mRNA in rat brain. J. Comp. Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- MONSMA F.J., Jr, MAHAN L.C., MCVITTIE L.D., GERFEN C.R., SIBLEY D.R. Molecular cloning and expression of a D1 dopamine receptor linked to adenyl cyclase activation. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6723–6727. doi: 10.1073/pnas.87.17.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORROW B.A., LEE E.J.K., TAYLOR J.R., ELSWORTH J.D., NYE H.E., ROTH R.H. (S)-(−)-HA 966, a gamma-hydroxybutyrate-like agent, prevents enhanced mesocorticolimbic dopamine metabolism and behavioural correlates of restraint stress, conditioned fear and cocaine sensitization. J. Pharmacol. Exp. Ther. 1997;283:712–721. [PubMed] [Google Scholar]
- MURPHY B.L., ARNSTEN A.F., GOLDMAN-RAKIC P.S., ROTH R.H. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAVA F., CARTA G., GESSA G.L.Permissive role of dopamine D2 receptors in the hypothermia induced by Δ9-tetrahydrocannabinol in rats Pharmacol. Biochem. Behav. 2000. in press [DOI] [PubMed]
- NORWICKY A.V., TEYIER T.J., VARDARIS R.M. The modulation of long-term potentiation by delta-9-tetrahydrocannabinol in the rat hippocampus, in vitro. Brain Res. Bull. 1987;19:663. doi: 10.1016/0361-9230(87)90052-9. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. Academic Press, London; 1986. The rat brain in stereotaxic coordinate. [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- POPE H.G., Jr, GRIBER A.J., JURGELUN-TODD D. The residual neuropsychological effects of cannabis: the current status of research. Drug Alcohol Depend. 1995;38:25–34. doi: 10.1016/0376-8716(95)01097-i. [DOI] [PubMed] [Google Scholar]
- SAWAGUCHI T., GOLDMAN-RAKIC P.S. D1 dopamine receptors in prefrontal cortex involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., ZENTER J., GOETHERT M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- SEAMANS J.K., FLORESCO S.B., PHILLIPS A.G. D1 receptor modulation of hippocampal-prefrontal cortical circuits integrating spatial memory with executive functions in the rats. J. Neurosci. 1998;18:1613–1621. doi: 10.1523/JNEUROSCI.18-04-01613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN M,. , PISER T.M., SEYBOLD V.S., THAYER S.A. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J. Neurosci. 1996;72:169–177. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIBLEY D.R., MONSMA F.J. Molecular biology of dopamine receptors. Trends Pharmacol. Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- STELLA N., SCHWEITZER P., PIOMELLI D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- TANDA G., PONTIERI F.E., DI CHIARA G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common μ1 opiod receptor mechanism. Science. 1997;276:2048–2050. doi: 10.1126/science.276.5321.2048. [DOI] [PubMed] [Google Scholar]
- TERRANOVA J.P., MICHAUD J.D., LE FUR G., SOUBRIE P. Inhibition of long-term potentiation in rat hippocampal slice by anandamide and WIN 55,212-2: Reversal by SR141716A, a selective antagonist of CB1 cannabinoid receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- THOMAS H. Psychiatric symptoms in cannabis users. Br. J. Psychiatry. 1993;163:141–149. doi: 10.1192/bjp.163.2.141. [DOI] [PubMed] [Google Scholar]
- WATANABE M., KODAMA T., HIKOSAKA K. Increase of extracellular dopamine in primate prefrontal cortex during a working memory task. J. Neurophysiol. 1997;78:2795–2798. doi: 10.1152/jn.1997.78.5.2795. [DOI] [PubMed] [Google Scholar]
- WILLIAMS G.V., GOLDMAN-RAKIC P.S. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- ZAHRT J., TAYLOR J.R., MATHEW R.G., ARNSTEN A.F.T. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J. Neurosci. 1997;17:8528–8535. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]