

Abstract
Nitric oxide (NO), peroxynitrite, formed from NO and superoxide anion, poly (ADP-ribole) synthetase have been implicated as mediators of neuronal damage following focal ischaemia. Here we have investigated the effects of n-acetylcysteine (NAC) treatment in Mongolian gerbils subjected to cerebral ischaemia.
Treatment of gerbils with NAC (20 mg kg−1 30 min before reperfusion and 1, 2 and 6 h after reperfusion) reduced the formation of post-ischaemic brain oedema, evaluated by water content.
NAC also attenuated the increase in the brain levels of malondialdehyde (MDA) and the increase in the hippocampus of myeloperoxidase (MPO) caused by cerebral ischaemia.
Positive staining for nitrotyrosine was found in the hippocampus in Mongolian gerbils subjected to cerebral ischaemia. Hippocampus tissue sections from Mongolian gerbils subjected to cerebral ischaemia also showed positive staining for poly (ADP-ribose) synthetase (PARS). The degree of staining for nitrotyrosine and for PARS were markedly reduced in tissue sections obtained from animals that received NAC.
NAC treatment increased survival and reduced hyperactivity linked to neurodegeneration induced by cerebral ischaemia and reperfusion.
Histological observations of the pyramidal layer of CA1 showed a reduction of neuronal loss in animals that received NAC.
These results show that NAC improves brain injury induced by transient cerebral ischaemia.
Keywords: Cerebral ischaemia, gerbil, neuronal death, brain injury, brain oedema, lipid peroxidation, NAC
Introduction
Neural damage following stroke and other neurodegenerative processes is thought the stem from overexcitation attributable to a massive release of excitatory neurotransmitter glutamate acting on the N-methyl-D-aspartate (NMDA) receptor and other receptor subtypes (Lipton & Rosenberg, 1994; Meldrum & Garthwaite, 1990; Choi et al., 1956). Evidence includes findings in many animal species that glutamate receptor antagonists block neuronal damage following vascular stroke and reduce neurotoxicity elicited by treatment of cerebral cortical cultures with glutamale or NMDA (Dawson & Dawson, 1997). Neurotoxicity elicited by stimulation of NMDA receptors is mediated, at least in part, by augmentation of nitric oxide (NO) formation, as NMDA receptor activation stimulates neuronal NO synthase (nNOS) activity. Protection against NMDA neurotoxicity occurs following treatment of primary brain cultures with NOS inhibitors (Dawson et al., 1991, 1993) and cortical cultures from mice with targeted disruption of nNOS (Dawson et al., 1996). Neuronal damage following vascular stroke is markedly diminished in animals treated with NOS inhibitors (Dawson & Dawson, 1997; Iadecola, 1997) or in mice with nNOS gene distruption (Huang, 1994).
Nitric oxide is a free radical that chemically reacts with its cellular target. There are multiple potential cellular targets that NO can modify to elicit a range of activities from cellular signalling to cell death. The majority of the toxic effect of NO appear to be a result of the reaction of NO with superoxide to form the very toxic compound peroxynitrite (Beckman et al., 1990). Peroxynitrite is cytotoxic via a number of independent mechanisms including (i) the initiation of lipid peroxidation, (ii) the inactivation of a variety of enzymes (most notably, mitochondrial respiratory enzymes and membrane pumps) (Crow & Beckman, 1995) and (iii) depletion of glutathione (Phelps et al., 1995). Moreover, peroxynitrite can also cause DNA damage (Inoue & Kawanishi, 1995; Salgo et al., 1995) resulting in the activation of the nuclear enzyme poly (ADP-Ribose) synthetase (PARS), depletion of NAD and ATP and ultimately cell death (Szabó et al., 1997). Interventions, which reduce the generation or the effects of peroxynitrite and PARS activation exert beneficial effects in a variety of models of inflammation and shock including the model of cerebral ischaemia used here. These therapeutic interventions include NOS inhibitor (Iadecola, 1997), a vitamin E-like antioxidant (Cuzzocrea et al., 1999a; Calapai et al., 1993), a SOD-mimetic (Cuzzocrea et al., 1999b), a peroxynitrite decomposition catalyst (Salvemini et al., 1998), and PARS inhibitors (Cuzzocrea et al., 1997; Eliasson et al., 1997).
NAC has antioxidant property (Aruoma et al., 1989) and as a sulphydryl donor, may contribute to the regeneration of endothelium-derived relaxing factor and glutathione (Harrison et al., 1991). Increasing evidence indicates that the action of NAC is pertinent to microcirculatory blood flow and tissue oxygenation. NAC was shown to enhance oxygen consumption via increased oxygen extraction in patients 18 h after the onset of fulminant liver failure (Harrison et al., 1991). It was speculated that NAC could also exert beneficial effects on impaired nutritive blood flow in patients with severe sepsis (Harrison et al., 1991).
In the present study, we examined the protective effect of NAC against oxidative stress during brain ischaemia and reperfusion injury using both biochemical and morphological parameters as follows: the levels of lipid peroxidation, the number of polymorphonuclear leukocytes (PMNs) that infiltrate the oxidatively damaged region of the brain were detected.
Methods
Animals
Adult male Mongolian gerbils (60–70 g; Charles River; Milan; Italy) were housed in a controlled environment and provided with standard rodent chow and water. Animal care was in compliance with Italian regulations on protection of animals used for experimental and other scientific purposes (D.M. 116192) as well as with the EEC regulations (O.J. of E.C. L 358/1 12/18/1986).
Surgical procedures
The ischaemia/reperfusion injury was induced by a single 5 min bilateral occlusion of the common carotid arteries (BCO) initially (about 4–5 min) under halothane (2%) anaesthesia followed by nitrous oxygen/O2 anaesthesia. Surgery was conducted always between 1000 and 1200 h. Survival was evaluated for the animals that lived 24 h after surgery.
Experimental groups
In the treated group of animals, NAC (20 mg kg−1) was given as a intraperitoneal bolus 30 min before reperfusion and 1, 2 and 6 h after reperfusion (BCO+ NAC group). In a vehicle-treated group of gerbils, vehicle (saline) was given instead of NAC (BCO group). In separate groups of gerbils, surgery was performed in its every aspect identical to the one in the BCO group, except that the blood vessels were not occluded (time-controlled sham group; Sham). In an additional group of animals, sham surgery was combined with the administration of NAC (dose as above) (Sham+ NAC).
Measurement of nitrite/nitrate
Nitrite+nitrate production, an indicator of NO synthesis, was measured in the plasma collected at a specified time as previously described (Cuzzocrea et al., 1997). Briefly, the nitrate in the plasma was first reduced to nitrite by incubation with nitrate reductase (670 mμ.ml−1) and NADPH (160 μM) at room temperature for 3 h. The nitrite concentration in the samples was then measured by the Griess reaction, by adding 100 μl of Griess reagent (0.1% naphthylethylenediamide dihydrochloride in H2O and 1% sulphanilamide in 5% concentrated H2PO4; vol. 1 : 1) to 100 μl samples. The optical density at 550 nm (OD550) was measured using ELISA microplate reader (SLT-Labinstruments Salzburg, Austria). Nitrate concentrations were calculated by comparison with OD550 of standard saline solutions.
Histological examination
Brain biopsies were taken at the fourth day after ischaemia and reperfusion. The biopsies were fixed for 1 week in buffered formaldehyde solution (10% in phosphate buffered saline) at room temperature, dehydrated by graded ethanol and embedded in Paraplast (Sherwood Medical, Mahwah, NJ, U.S.A.). Tissue sections (thickness 7 μm) were deparaffinized with xylene, stained with haematoxylin and eosin and studied using light microscopy (Dialux 22 Leitz). The grading system of Pulsinelli (Pulsinelli et al., 1982) was used: 0=0% of the neurones damaged (normal brain), 1=1–10% of the neurones damaged, 2=11–50% of the neurones damaged and 4= infarction (necrosis of both neurones and glia). The slices were evaluated independently by two examiners.
Immunohistochemical localization of nitrotyrosine
Tyrosine nitration, an index of the nitrosylation of proteins by peroxynitrite and/or oxygen-derived free radicals, was determined by immunohistochemistry as previously described (Cuzzocrea et al., 1997). At the end of the experiment, the relevant organs were fixed in 10% buffered formaldehyde and 8 μm sections were prepared from paraffin embedded tissues. After deparaffinization, endogenous peroxidase was quenched with 0.3% H2O2 in 60% methanol for 30 min. The sections were permeabilized with 0.1% Triton X-100 in PBS for 20 min. Non-specific adsorption was minimized by incubating the section in 2% normal goat serum in phosphate buffered saline for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with avidin and biotin. The sections were then incubated overnight with 1 : 1000 dilution of primary anti-nitrotyrosine antibody or with control solutions. Controls included buffer alone or non specific purified rabbit IgG. Some sections were also incubated with the primary antibody (anti-nitrotyrosine) in the presence of excess nitrotyrosine (10 mM) to verify the binding specificity. Specific labelling was detected with a biotin-conjugated goat anti-rabbit IgG and avidin-biotin peroxidase complex. Diaminobenzidine was used as a cromogen (DBA, Milan, Italy).
Immunohistochemical localization of PARS
At the specified time following the reperfusion, brain tissues were fixed in 10% buffered formalin and 8 μm sections were prepared from paraffin embedded tissues. After deparaffinization, endogenous peroxidase was quenched with 0.3% H2O2 in 60% methanol for 30 min. The sections were permeabilized with 0.1% Triton X-100 in phosphate buffered saline for 20 min. Non-specific adsorption was minimized by incubating the section in 2% normal goat serum in phosphate buffered saline for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with avidin and biotin (DBA, Milan, Italy). The sections were then incubated overnight with 1 : 500 dilution of primary anti-poly (ADP-Ribose) antibody (DBA, Milan, Italy) or with control solutions. Controls included buffer alone or non-specific purified rabbit IgG. Specific labelling was detected with a biotin-conjugated goat anti-rabbit IgG and avidin-biotin peroxidase complex. Diaminobenzidine was used as a cromogen (DBA, Milan, Italy).
Myeloperoxidase activity
Myeloperoxidase (MPO) activity, an indicator of polymorphonuclear leukocyte (PMN) accumulation, was determined as previously described (Mullane et al., 1988). At the specified time following the reperfusion, hippocampus were obtained and weighed. Each piece of tissue was homogenized in a solution containing 0.5% hexa-decyl-trimethyl-ammonium bromide dissolved in 10 mM potassium phosphate buffer (pH 7) and centrifuged for 30 min at 20,000×g at 4°C. An aliquot of the supernatant was then allowed to react with a solution of tetra-methyl-benzidine (1.6 mM) and 0.1 mM H2O2. The rate of change in absorbance was measured spectrophotometrically at 650 nm. MPO activity was defined as the quantity of enzyme degrading 1 μmol of peroxide min−1 at 37°C and was expressed in milliunits per gram weight of wet tissue.
Malondialdehyde (MDA) measurement
Malondialdehyde (MDA) levels in the brain tissue were determined as an indicator of lipid peroxidation (Ohkawa et al., 1979). Brain tissue, collected at the specified time, was homogenized in 1.15% KCl solution. An aliquot (100 μl) of the homogenate was added to a reaction mixture containing 200 μl of 8.1% SDS, 1500 μl of 20% acetic acid (pH 3.5), 1500 μl of 0.8% thiobarbituric acid and 700 μl distilled water. Samples were then boiled for 1 h at 95°C and centrifuged at 3000×g for 10 min. The absorbance of the supernatant was measured by spectrophotometry at 650 nm.
Locomotor activity
After 24 and 48 h of ischaemia an open field test was performed. Gerbils were placed in an open field apparatus and the number of squares crossed and the number of readings in 6 min was determined. The test was always performed between 1000 and 1200 h.
Determination of cerebral oedema
Tissue sections were assayed for water content at 24 and 48 h after injury using wet weight/dry weight ratios. Freshly dissected tissue samples were weighted on aluminium foil, dried for 24 h at 105°C, and reweighed.
The percentage of the water was calculated as follows:
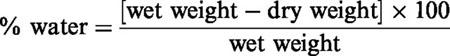 |
Materials
Cell culture medium, heparin and foetal calf serum were obtained from Sigma (Milan, Italy). Perchloric acid was obtained from Aldrich (Milan, Italy). Primary anti-nitrotyrosine antibody was from Upstate Biotech (DBA, Milan, Italy). All other reagents and compounds used were obtained from Sigma Chemical Company (Sigma, Milan, Italy).
Data analysis
All values in the figures and text are expressed as mean±standard error (s.e.m.) of the mean of n observations. For the in vivo studies n represents the number of animals studied. In the experiments involving histology or immunohistochemistry, the figures shown are representative of at least three experiments performed on different experimental days. The results were analysed by one-way ANOVA followed by a Bonferroni post-hoc test for multiple comparisons. A P-value less than 0.05 was considered significant.
Results
Effect of NAC treatment on NO production in BCO
BCO gerbils showed an increase in the plasma levels of nitrate/nitrite at 4 h after reperfusion compared with plasma collected from sham animals (Figure 1). In vivo treatment with NAC 30 min before reperfusion and 1–2 h after reperfusion reduced significantly the nitrite/nitrate formation (Figure 1A).
Figure 1.
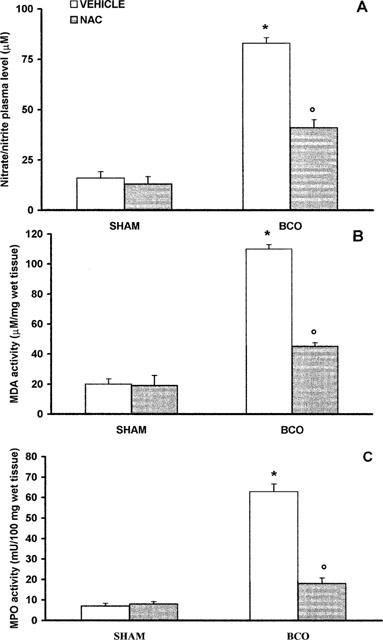
Nitrate/nitrite production (A), MDA levels (B), MPO activity (C) at 4 h after BCO. Nitrite and nitrate plasma levels in BCO gerbils were significantly increased versus sham group. MPO activity and MDA levels were significantly increased in the brain of the BCO gerbils (*P<0.01). NAC reduced the BCO-induced increase in plasma nitrite/nitrate and tissue MDA levels and MPO activity. Values are means±s.e.means of eight animals for each group. *P<0.01 versus sham. °P<0.01 versus BCO.
Effect of NAC treatment on malonaldehyde and myeloperoxidase activities in the reperfused brain
Untreated BCO gerbils showed an increase in brain MDA levels, indicative of lipid peroxidation, detected at 4 h after reperfusion (Figure 1B). The accumulation of neutrophils was investigated by measuring MPO activity in the hippocampus from BCO animals at 4 h after reperfusion. BCO animals showed a significant increase in MPO activity (Figure 1C). NAC treatment at 30 min before reperfusion and 1–2 h after reperfusion significantly reduced (P<0.01) the MPO activity as well as the MDA levels (Figure 1B,C).
Effect of NAC treatment on brain oedema
Brain oedema, measured through the evaluation of water content in the cortex and hippocampus, was observed at 24 and 48 h after reperfusion in BCO gerbils (Figure 2A). Administration of NAC at 30 min before reperfusion and 1–2 h after reperfusion significantly reduced the oedema formation in the two areas (Figure 2A).
Figure 2.
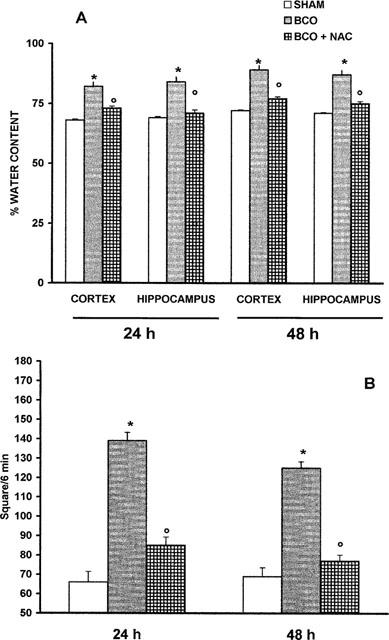
Effects of administration of NAC on water content (A) in the cortex and hippocampus and on locomotor activity (B) at 24 and 48 h after reperfusion. Values are means±s.e.means of eight animals for each group. *P<0.01 versus sham. °P<0.01 versus BCO.
Effect of NAC treatment on locomotor activity
In BCO animals an increase in locomotor activity, evaluated in the open-field apparatus, was observed 24 and 48 h following reperfusion (Figure 2B). There was no significant increase in the locomotor activity of BCO gerbils treated with NAC at 30 min before reperfusion and 1–2 h after reperfusion (Figure 2B).
Effect of NAC treatment on nitrotyrosine formation and PARS activation
At the 4th day after reperfusion brain sections were taken from sham or shocked gerbils in order to determine the immunohistological staining for nitrotyrosine. Immunohistochemical analysis, using a specific anti-nitrotyrosine antibody, revealed a positive staining in dorsal hippocampus from BCO animals (Figure 3A). NAC treatment at 30 min before reperfusion and 1–2 h after reperfusion significantly reduced the degree of immunostaining for nitrotyrosine in the reperfused brain (Figure 3).
Figure 3.
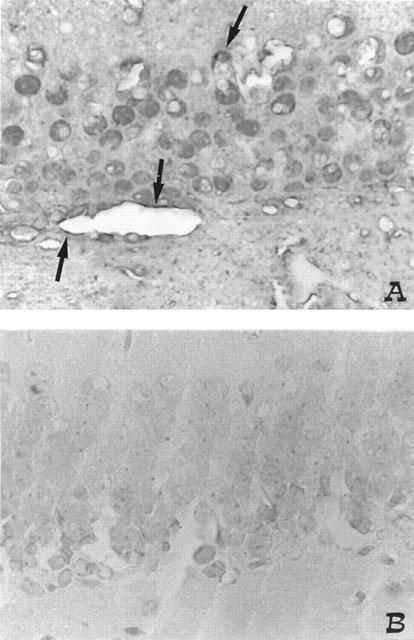
Effect of NAC on nitrotyrosine formation: 4 days following BCO, positive staining for nitrotyrosine was observed in neurones and vessels endothelium (see arrows) (A). There was a marked reduction in the immunostaining in the brain of BCO gerbils treated with NAC (B). Original magnification: ×125. Figure is representative of at least three experiments performed on different experimental days.
Immunohistochemical analysis, using a specific anti-PARS antibody, revealed a positive staining in dorsal hippocampus from BCO animals (Figure 4A). NAC treatment at 30 min before reperfusion and 1–2 h after reperfusion significantly reduced the degree of immunostaining for PARS in the reperfused brain (Figure 4B). There was no staining for either nitrotyrosine or PARS in the brain sections of sham animals (data not shown).
Figure 4.
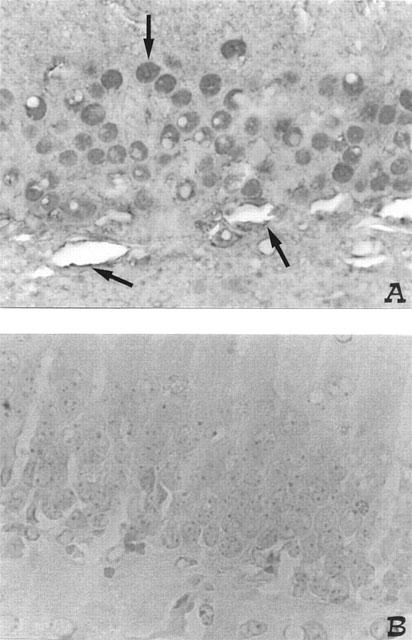
Effect of NAC on PARS immunostaining: 4 days following BCO, PARS immunoreactivity was observed in neurones and vessels endothelium (see arrows) (A). In the brain of BCO gerbils treated with NAC (B), no positive staining was found. Original magnification: ×125. Figure is representative of at least three experiments performed on different experimental days.
Brain sections obtained 48 h after reperfusion from BCO gerbils showed similar positive staining for nitrotyrosine and PARS in comparison with sections collected at 4 days after reperfusion (data not shown). In addition, NAC treatment significantly reduced nitrotyrosine and PARS staining at 48 h after reperfusion (data not shown) as well as 4 days after reperfusion (Figures 3B and 4B).
Histological change
The microscopic observation of hippocampal sections showed that BCO produced neuronal loss in the CA1 area at 4 days after reperfusion (Figure 5A). NAC treatment at 30 min before reperfusion and 1–2 h after reperfusion significantly prevent the neuronal loss (Figure 5B). The evaluation of sections from BCO animals treated with NAC at 30 min before reperfusion and 1–2 h after reperfusion totalized a significantly lesser score in comparison with that reached by BCO animals treated with vehicle (Figure 6).
Figure 5.
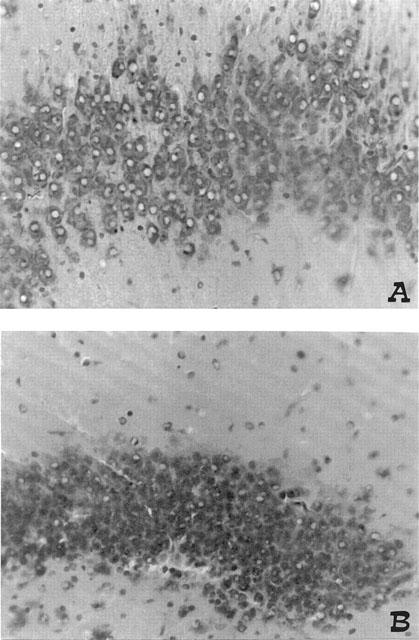
Neuroprotective effects of melatonin on the pyramidal cell layer in the central area of CA1 sector of hippocampus. (A) vehicle ischaemia, (B) NAC-ischaemia. Treatment with NAC produced a significant attenuation of neurodegeneration in the pyramidal cell layer of CA as well as prevents the loss of neurones in comparison with vehicle-ischaemia. Original magnification: ×200. Figure is representative of at least three experiments performed on different experimental days.
Figure 6.
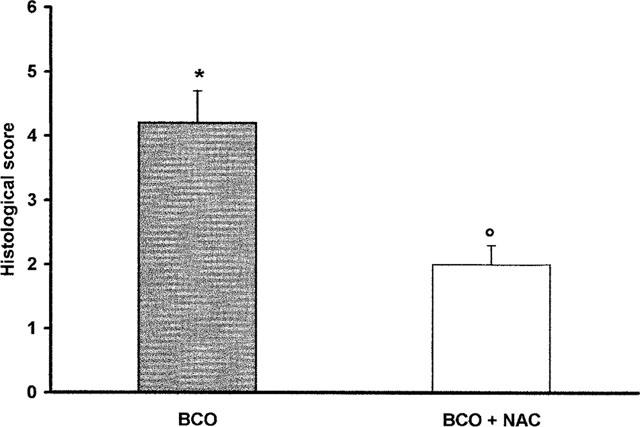
Effects of NAC on histological score obtained by the microscopic observation of CA1, area of post-ischaemic animals. Values are means±s.e.means of eight animals for each group. *P<0.01 versus sham. °P<0.01 versus BCO.
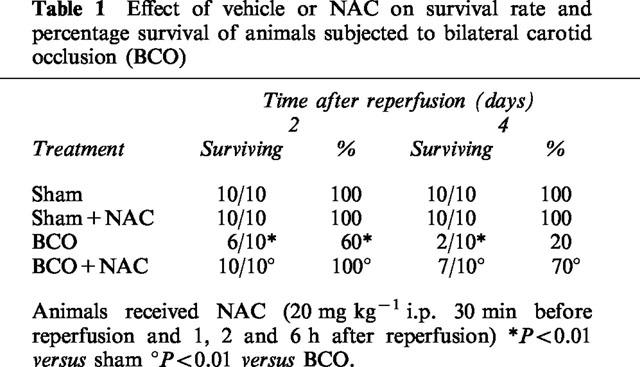
All sham animals survived the entire observation period. In contrast, BCO animals showed a profound shock state characterized by a 80% lethality (Table 1). NAC given at 30 min before and 1, 2 and 6 h after reperfusion significantly reduced BCO-induced mortality (Table 1).
Table 1.
Effect of vehicle or NAC on survival rate and percentage survival of animals subjected to bilateral carotid occlusion (BCO)
Discussion
Loss of blood flow to the brain results in neuronal injury due to both the cessation of blood flow leading to oxygen and nutrient deprivation and the initiation of secondary mechanisms (Dawson & Dawson, 1997). This neurotoxic cascade involves derangement in normal metabolic and physiological functions as well as recruitment of cell death processes. Thus, both restoration of blood supply and control of secondary neurotoxic cascades are necessary to limit ischaemic neuronal damage. Numerous transmitter and second messenger pathways are inappropriately activated after the initial ischaemic event (Meldrum & Garthwaite, 1990). A major pathway leading toward neuronal injury involves elevation of extracellular glutamate and activation of glutamate receptors, with a subsequent increase in intracellular calcium, resulting in a generation of free radicals and NO (Dawson & Dawson, 1996). In addition to NO, peroxynitrite is also generated in reperfused brain (Fagni et al., 1994; Cazevieille et al., 1993; Guasekar et al., 1995).
The biological activity and decomposition of peroxynitrite is very much dependent on the cellular or chemical environment (presence of proteins, thiols, glucose, the ratio of NO and superoxide, carbon dioxide levels and other factors), and these factors influence its toxic potential (Beckman et al., 1990; Rubbo et al., 1994; Villa et al., 1994). Here we demonstrate that NAC reduces (i) the brain malondialdehyde levels, (ii) brain oedema, (iii) myeloperoxidase activity, (iv) survival and locomotor hyperactivity, (v) hippocampal neuronal loss. All of these findings support the view that NAC attenuates the degree of brain injury caused by temporary bilateral carotid occlusion in the Mongolian gerbil. What, then, is the mechanism by which NAC protects the brain against this injury.
NAC exerts its effect both as a source of sulphydryl groups (repletion of intracellular reduced glutatione) and through a direct reaction with hydroxyl radical (Galley et al., 1997). Recently, a number of studies in animals suggest benefits from acetylcysteine in the context of systemic inflammatory response syndrome caused by severe sepsis model. In a pig gram-negative sepsis model, an infusion of acetylcysteine reduced pulmonary capillary leak without reducing mortality (Groenveld et al., 1990).
There are a number of sites where NAC can interfere with the inflammatory process. Here we demonstrated that NAC inhibits NO production. This effect is thought to be due to removal of superoxide anion and potentiation of feedback inhibition on nitric oxide synthase by nitric oxide via inactivation of its haeme centre (Buga et al., 1993) although suppression of the cytokine-mediated induction of nitric oxide synthase may also provide an explanation (Galley et al., 1996).
In addition, the levels of MDA, which is the product of lipid peroxidation, were significantly increased by ischaemia-reperfusion. This observation is in agreement with Calapai and colleagues that have found elevated levels of lipid peroxidation products in the same experimental model (Calapai et al., 1993). NAC treatment abolishes the increase in lipid peroxidation products, probably in part by scavenging the very reactive .OH and ROO..
Reduction of lipid peroxidation was also paralleled with the inhibition of nitrotyrosine immunoreactivity. Nitrotyrosine formation, along with its detection by immunostaining, was initially proposed as a relatively specific means for detection of the ‘footprint' of peroxynitrite (Beckman, 1996). Recent evidence indicate, however, that certain other reactions can also induce tyrosine nitration; e.g., the reaction of nitrite with hypochlorous acid and the reaction of myeloperoxidase with hydrogen peroxide can lead to the formation of nitrotyrosine (Eiserich et al., 1998). Increased nitrotyrosine staining is considered, therefore, as an indication of ‘increased nitrosative stress' rather than specific marker of peroxynitrite. This effect may be related to a direct scavenging effect of NAC on peroxynitrite.
Additional protective effects of NAC may lie within the ability of this compound to reduce oxyradical-related oxidant processes by either directly interfering with the oxidants, or up-regulating antioxidant systems such as superoxide dismutase (Groenveld et al., 1990) or enhancing the catalytic activity of glutathione peroxidase (Schillier et al., 1993). Therefore oxygen radical scavengers, administered before or at the onset of sepsis, were shown to improve the survival in animal models of sepsis (Pouwell et al., 1991). NAC has antioxidant property (Aruoma et al., 1989) and as a sulphydryl donor, may contribute to the regeneration of endothelium-derived relaxing factor and glutathione (Harrison et al., 1991). Increasing evidence indicates that the action of NAC is pertinent to microcirculatory blood flow and tissue oxygenation. NAC was shown to enhance oxygen consumption via increased oxygen extraction in patients 18 h after the onset of fulminant liver failure (Harrison et al., 1991). It was speculated that NAC could also exert beneficial effects on impaired nutritive blood flow in patients with severe sepsis (Harrison et al., 1991).
Reactive oxygen species (ROS) and peroxynitrite produce cellular injury and necrosis via several mechanisms including peroxidation of membrane lipids, protein denaturation and DNA damage. ROS produce strand breaks in DNA which triggers energy-consuming DNA repair mechanisms and activates the nuclear enzyme PARS resulting in the depletion of its substrate NAD in vitro and a reduction in the rate of glycolysis. As NAD functions as a cofactor in glycolysis and the tricarboxylic acid cycle, NAD depletion leads to a rapid fall in intracellular ATP. This process has been termed ‘the PARS Suicide Hypothesis'. There is recent evidence that the activation of PARS may also play an important role in ischaemia and reperfusion injury (Cuzzocrea et al., 1997; Zingarelli et al., 1997; Thiemermann et al., 1997). We demonstrate here that NAC attenuates the increase in PARS activity caused by temporary bilateral carotid occlusion in the brain.
In our study, an increase of the activity of myeloperoxidase, an enzyme specific to granulocyte lysosomes, a parameter directly related to the absolute number of polymorphonuclear cells (PMN cells), correlated well with morphological alterations in the brain at histological examination. Activation and accumulation of PMNs is one of the initial events of tissue injury, which triggers the release of oxygen free radicals, arachidonic acid metabolites and lysosomal proteases, with subsequent tissue injury (Fantone & Ward, 1982).
We found that early treatment with NAC prevented the PMN infiltration into the brain as demonstrated by a significant reduction in MPO activity.
Taken together, the results of the present study, support the view that NAC can exert a protective effect against brain injury caused by temporary bilateral carotid occlusion in the Mongolian gerbils. We speculate that the observed anti-inflammatory effects of NAC may be dependent upon a combination of the following pharmacological properties of this agent: (1) NAC scavenges and inactivates superoxide anions and NO, which would prevent the formation of peroxynitrite. This, in turn, prevents the activation of PARS and the associated tissue injury. (2) In addition to superoxide anions, NAC also scavenges other ROS including hydroxyl radicals. (3) In addition, NAC reduces the recruitment of PMNs into the inflammatory site. These results support the view that the overproduction of reactive oxygen or nitrogen free radicals contributes to a stroke. Finally, we propose that NAC, may be useful in the therapy of conditions associated with local or systemic inflammation.
Acknowledgments
We gratefully acknowledge Zambon Italia, Bresso, (Mi), Italy for the generous support to this study. We also thank Fabio Giuffrè and Carmelo La Spada for their excellent technical assistance during this study, Mrs Caterina Cutrona for secretarial assistance and Miss Valentina Malvagni for editorial assistance with the manuscript.
Abbreviations
- ecNOS
constitutive endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- MPO
myeloperoxidase
- NAC
n-acetylcysteine
- NO
nitric oxide
- NOS
nitric oxide synthase
- PARS
poly (ADP-ribose) synthetase
- PBS
phosphate-buffer saline
- PMS
polymorphonuclear cell
References
- ARUOMA O.I., HALLIWELL B., HOEY B.M. The antioxidant action of n-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxyde, and hypochlorous acid. J. Free Rad. Biol. Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.A., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: implication for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUGA G.M., GRISCAVAGE J.M., ROGERS N.E., INGNARRO L.J. Negative feedback inhibition of endothelial cell function by nitric oxide. Circulation Reserch. 1993;73:808–812. doi: 10.1161/01.res.73.5.808. [DOI] [PubMed] [Google Scholar]
- CALAPAI G., SQUADRITO F., RIZZO A., CRISAFULLI C., CAMPO G.M., MARCIANO M.C., MAZZAGLIA G., SCURI R.A. New antioxidant drug limits brain damage induced by transient cerebral ischaemia. Drugs Exptl Clin. Res. 1993;XIX:159–164. [PubMed] [Google Scholar]
- CAZEVIEILLE C., MULLER A., MEYNIER F., BONNE C. Superoxide and nitric oxide cooperation in hypoxia/reoxigenation-induced neuron injury. Free Radical Biol. Med. 1993;14:389–395. doi: 10.1016/0891-5849(93)90088-c. [DOI] [PubMed] [Google Scholar]
- CHOI D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1956;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- CROW J.P., BECKMAN J.S. The role of peroxynitrite in nitric oxide-mediated toxicity. Curr. Top. Microbiol. Immunol. 1995;196:57–73. doi: 10.1007/978-3-642-79130-7_7. [DOI] [PubMed] [Google Scholar]
- CUZZOCREA S., COSTANTINO G., MAZZON E., CAPUTI A.P. Beneficial effects of raxofelast (IRFI 016), a new hydrophilic vitamin e-like antioxidant, in carrageenan-induced pleurisy. Br. J. Pharmacol. 1999a;126:407–414. doi: 10.1038/sj.bjp.0702275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUZZOCREA S., ZINGARELLI B., COSTANTINO G., CAPUTI A.P. Protective effect of melatonin in a non septic shock model induced by zymosan in the rat. J. Pineal Res. 1998;25:24–33. doi: 10.1111/j.1600-079x.1998.tb00382.x. [DOI] [PubMed] [Google Scholar]
- CUZZOCREA S., ZINGARELLI B., COSTANTINO G., CAPUTI A.P. Beneficial effects of Mn(III)tetrakis (4-benzoic acid) porphyrin (MnTBAP), a superoxide dismutase mimetic, in carrageenan-induced pleurisy. Free Radic. Biol. Med. 1999b;26:25–33. doi: 10.1016/s0891-5849(98)00142-7. [DOI] [PubMed] [Google Scholar]
- CUZZOCREA S., ZINGARELLI B., COSTANTINO G., SZABÓ C., SALZMAN A.L., CAPUTI A.P. Beneficial effects of 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase in a rat model of splanchnic artery occlusion and reperfusion. Br. J. Pharmacol. 1997;121:1065–1074. doi: 10.1038/sj.bjp.0701234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWSON T.M., DAWSON V.L.Protection of the brain from ischemia in cerebrovascular disease 1997Philadelphia: Lippincott-Raven; 319–325.ed. Batjer H.H pp [Google Scholar]
- DAWSON V.L, DAWSON T.M. Free radicals and neuronal cell death. Cell Death Differ. 1996;3:71–78. [PubMed] [Google Scholar]
- DAWSON V.L., DAWSON T.M., BARTLEY D.A., UHL G.R., SNYDER S.H. Mechanisms of nitric oxide-mediated in primary brain cultures. J. Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWSON V.L., DAWSON T.M., LONDON E.D., BREDT D.S., SNYDER S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWSON V.L, , KIZUSHI V.M., HUANG P.L, , SNYDER S.H., DAWSON T.M. Resistance to neurotoxicity In cortical cultures from neuronal nitric oxide synthase deficient mice. J. Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EISERICH J.P., HRISTOVA M., CROSS C.E., JONES A.D., FREEMAN B.A., HALLIWELL B., VAN DER VLIET A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- ELIASSON M.J.L., SAMPEI K., MANDIR A.S., HURN P.D., TRAYSTMAN R.J., BAO J., PIEPER A., WANG Z.Q., DAWSON T.M., SNYDER S.H., DAWSON V.L. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nature Med. 1997;10:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- FAGNI L., LAFON-CAZAL M., ROUNDOULIN G., MANZONI O., LERNER-NATOLI M., BOCKAERT J. The role of free radicals in NMDA-dependent neurotoxicity. Prog. Brain Res. 1994;103:181–390. doi: 10.1016/s0079-6123(08)61152-8. [DOI] [PubMed] [Google Scholar]
- FANTONE J.C., WARD P.A. A review: role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am. J. Pathol. 1982;107:395–418. [PMC free article] [PubMed] [Google Scholar]
- GALLEY H.F., HOWDLE P.D., WALKER B.E. The effect of intravenous antioxidants in patients with septic shock. Free Rad. Biol. Med. 1997;23:768–774. doi: 10.1016/s0891-5849(97)00059-2. [DOI] [PubMed] [Google Scholar]
- GALLEY H.F., RICHARDSON N., HOWDLE P.D., WALKER B.E., WEBSTER N.R. Regulation of nitric oxide synthase activity in cultured human endothelial cells: effect of antioxidants. Free Rad. Biol. Med. 1996;21:97–101. doi: 10.1016/0891-5849(95)02216-3. [DOI] [PubMed] [Google Scholar]
- GROENVELD A.B.J., DEN HOLLANDER W., STRAUB J. Effects n-acetylcysteine and terbutaline treatment on hemodynamics and regional albumin extravasation in porcine septic model. Circ. Shock. 1990;30:185–205. [PubMed] [Google Scholar]
- GUASEKAR P.G., KANTASAMY A.G., BOROWITZ J.L., ISOM G.E. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: Implication for cell death. J. Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- HARRISON P.M., WENDON Y.A., GIMSON A.E.S. Improvement by acetylcisteine of hemodinamics and oxygen transport in fulminant hepatic failure. N. Engl. J. Med. 1991;324:1852–1857. doi: 10.1056/NEJM199106273242604. [DOI] [PubMed] [Google Scholar]
- HUANG L. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- IADECOLA C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- INOUE S., KAWANISHI S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett. 1995;371:86–88. doi: 10.1016/0014-5793(95)00873-8. [DOI] [PubMed] [Google Scholar]
- LIPTON S.A., ROSENBERG P.A. Excitatory Amino acids as a final common pathway for neurologic disorders. N. Eng. Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- MELDRUM L., GARTHWAITE J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- MULLANE K.M., WESTLIN W., KRAEMER R. Activated neutrophils release mediators that may contribute to myocardial injury and dysfunction associated with ischemia and reperfusion. Ann. N.Y. Acad. Sci. 1988;524:103–121. doi: 10.1111/j.1749-6632.1988.tb38534.x. [DOI] [PubMed] [Google Scholar]
- OHKAWA H., OHISHI N., YAGI K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- PHELPS D.T., FERRO T.J., HIGGINS P.J., SHANKAR R., PARKER D.M., JOHNSON M. TNF-alpha induces peroxynitrite-mediated depletion of lung endothelial glutathione via protein kinase C. Am. J. Physiol. 1995;269:L551–559. doi: 10.1152/ajplung.1995.269.4.L551. [DOI] [PubMed] [Google Scholar]
- POUWELL R.J., MACHIEDO G.W., RUSH B.J. Effect of oxygen-free radical scavengers on survival in sepsis. Am. Surg. 1991;57:86–88. [PubMed] [Google Scholar]
- PULSINELLI W.A, , BRIERIEY J.B., PLUM F. Temporal profile of neuronal damage in a model of transient forebrain ischaemia. Ann. Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- RUBBO H., RADI R., TRUJILLO M., TELLERI R., KALYANARAMAN B., BARNES S., KIRK M., FREEMAN B.A. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidised lipid derivatives. J. Biol. Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- SALGO M.G., BERMUDEZ E., SQUADRITO G., PRYOR W. DNA damage and oxidation of thiols peroxynitrite causes in rat thymocytes. Arch. Biochem. Biophys. 1995;322:500–505. doi: 10.1006/abbi.1995.1493. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., WANG Z.Q., STERN M.K., CURRIE M.G., MISKO T.P. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc. Natl. Acad. Sci. U.S.A. 1998;95:2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHILLIER H.J., REILLY P.M., BULKLEY G.B. Antioxidant therapy. Crit. Care Med. 1993;21:92–102. [PubMed] [Google Scholar]
- SZABÓ C., CUZZOCREA S., ZINGARELLI B., O'CONNOR M., SALZMAN A.L. Endothelial dysfunction in endotoxic shock: importance of the activation of poly (ADP ribose synthetase (PARS) by peroxynitrite. J. Clin. Invest. 1997;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THIEMERMANN C., BOWES J., MYNNY F.P., VANE J.R. Inhibition of the activity of poly(ADP ribose) synthase reduces ischaemia-reperfusion injury in the heart and skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLA L.M., SALAS E., DARLEY-USMAR M., RADOMSKI M.W., MONCADA S. Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12383–12387. doi: 10.1073/pnas.91.26.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZINGARELLI B., CUZZOCREA S., ZSENGELLER Z., SALZMAN A.L., SZABÓ C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of Poly (ADP-Ribose) synthetase. Cardiovasc. Res. 1997;36:205–215. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]